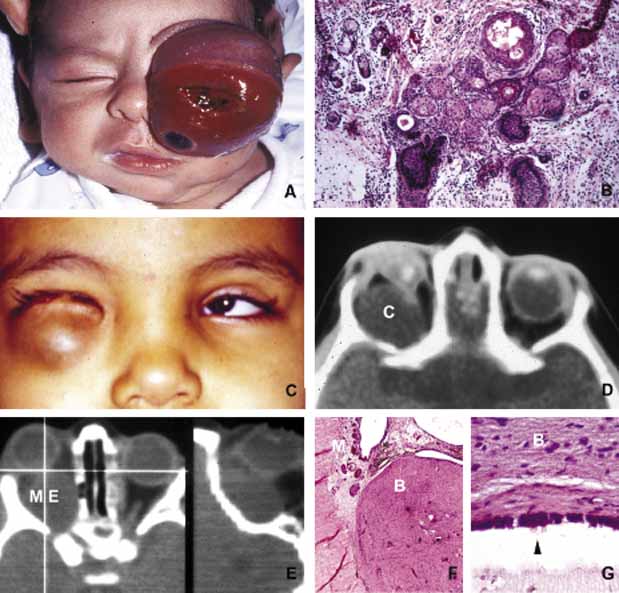
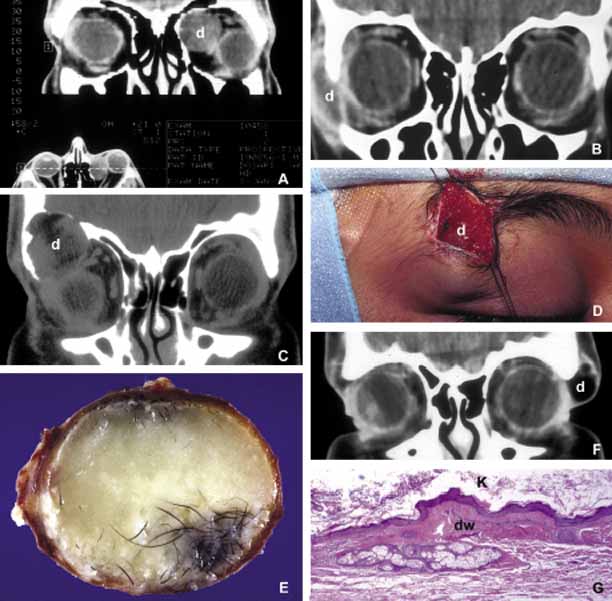
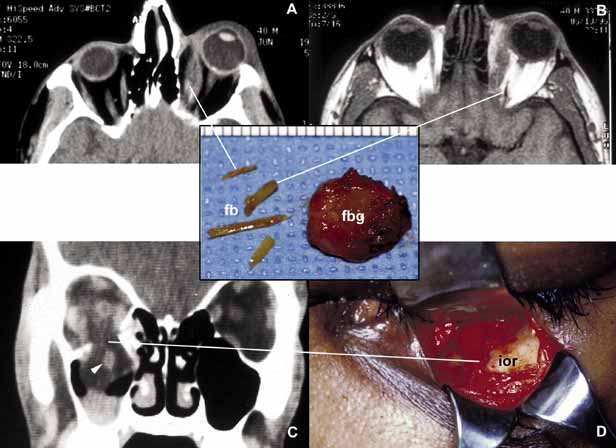
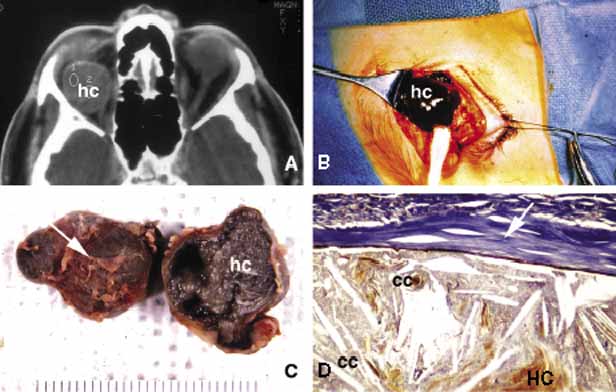
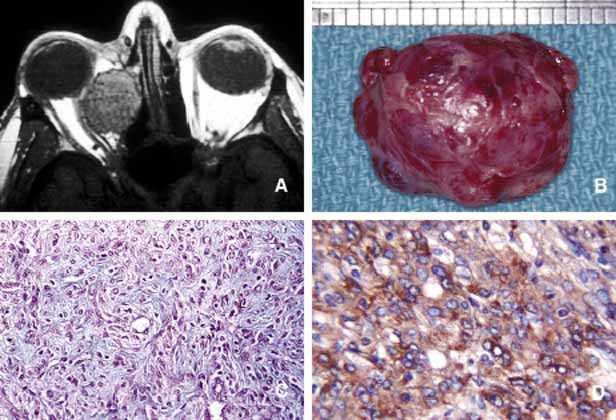
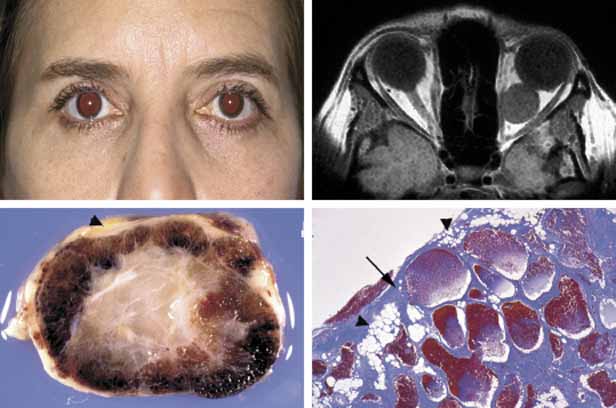
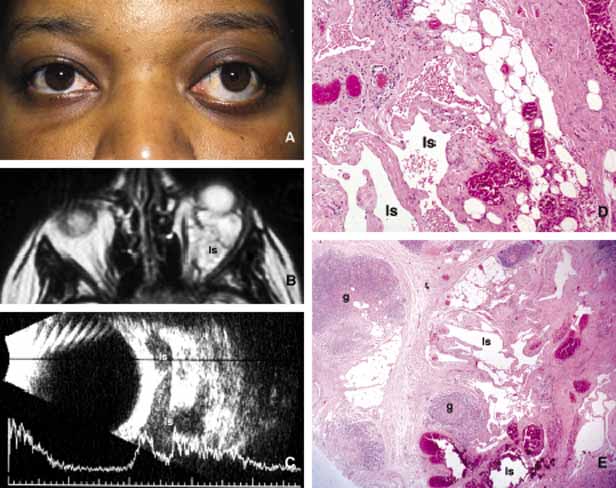
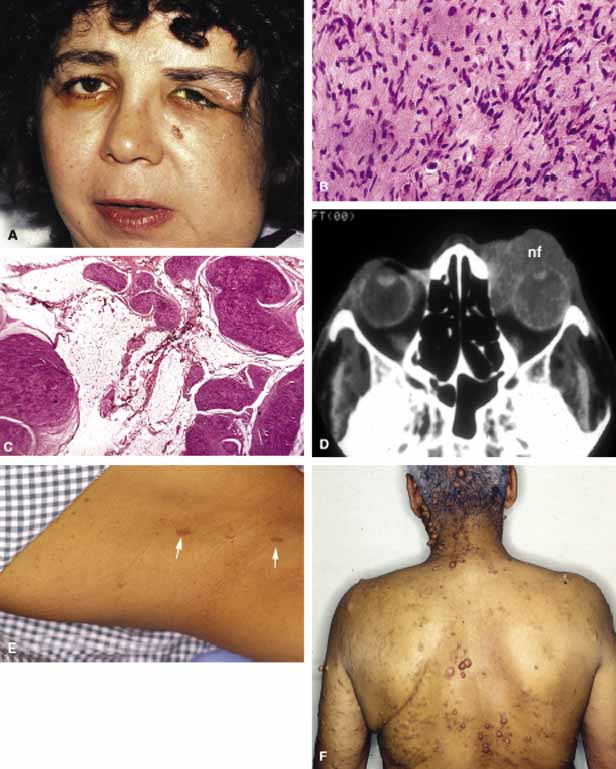
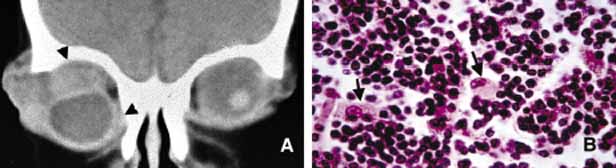
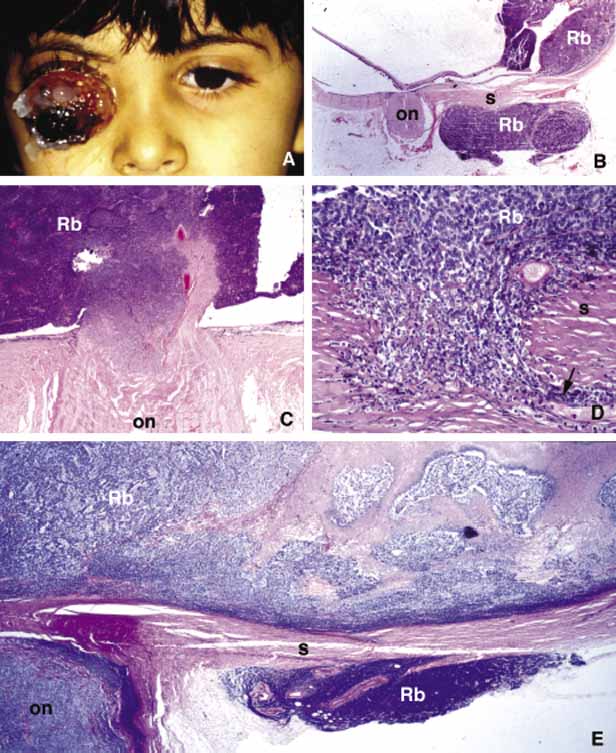
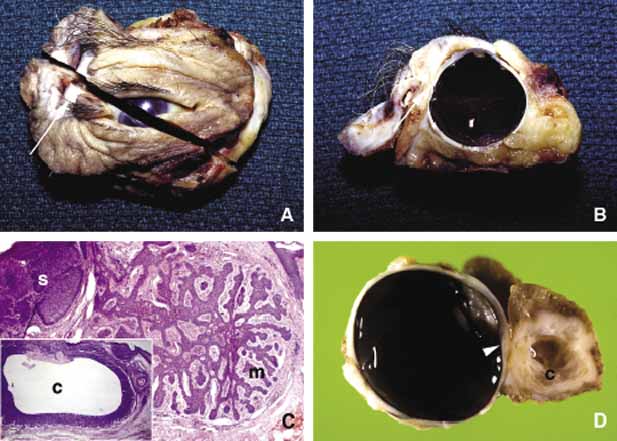
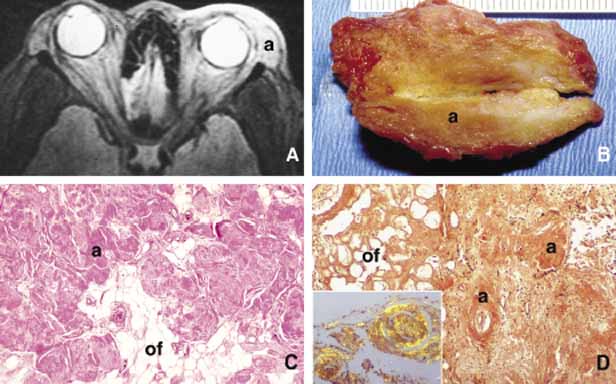
1. Ozanics V, Jakobiec F: Prenatal development of the eye and its adnexa. In Jakobiec. Ocular anatomy, embryology and teratology. Philadelphia: Harper & Row, 1982:11–96 2. Barishak YR: Embyology of the eye and its adnexa. Karger: Basel, Switzerland, 2001:15 3. Barkovich AJ: Pediatric neuroimaging, 3rd edition. New York: Raven, 2000 4. Cohen MM Jr : Craniosynostosis update. Am J Med Genet Suppl 4:99–148, 1987 5. Chumas PD, Cinalli G, Arnaud E, et al: Classification of previously unclassified cases of craniosynostosis. J Neurosurg 86:177–181, 1997 6. Casper DS, Chi TL, Trokel SL, (eds): Orbital disease, imaging and analysis. Stuttgart: Thieme, 1993 7. Beard C: Congenital hereditary abnormalities of the eyelids, lacrimal system and
orbit. Transcript of New Orleans Academy of Ophthlamology. St. Louis: Mosby, 1968 8. Hertle RW, Quinn GE, Katowitz JA: Ocular and adnexal findings in patients with facial microsomias. Ophthalmology 99:114–119, 1992 9. Barkovich AJ: Neuroimaging manifestations and classification of congenital muscular dystrophies. AJNR Am J Neuroradiol 19:1389–1396, 1998 10. Waugh MC, Chong WK, Sonksen P: Neuroimaging in children with congenital disorders of the peripheral visual
system. Dev Med Child Neurol 40:812–819, 1998 11. DePotter P, Shields JA, Shields CL: MRI of the eye and orbit. Philadelphia: Lippincott, 1995 12. Suwanwela C, Suwanwela N: A morphological classification of sincipital encephalomeningoceles. J Neurosurg 36:201, 1972 13. Newman NJ, Miller NR, Green WR: Ectopic brain in the orbit. Ophthalmology 93:268, 1986 14. Grossniklaus HE, Wojno T: Orbital aberrant fibroglial tissue associated with microphthalmos. J Ped Ophthalmol Strabismus 31:338–340, 1994 15. Brown HH, Kersten RC, Kulwin DR: Lipomatous hamartoma of the orbit. Arch Ophthalmol 109:240, 1991 16. Nicholson DH, Green WR: Pediatric Ocular Tumors. New York: Masson Publishing, 1981 17. Shields JA, Augsburger JJ, Donoso LA: Orbital dermoid cyst of conjunctival origin. Am J Ophthalmol 101:726, 1986 18. Smirniotopoulos JG, Chiechi MV: Teratomas, dermoids and epidermoids of the head and neck. Radiographs 15:1437–1455, 1995 19. Rochels R, Nover A, Hackelbusch R: Echographische Befunde und Differential diagnostik bei (peri-orbitalen
Dermoidzyste). (Echographic findings and differential
diagnosis in (peri-orbital dermoid cysts) Klin Monatsbl Augenheilkd 188:101–104,l 1986 20. Sathananthan N, Moseley IF, Rose GE, et al: the frequency and clinical significance of bone involvement in outer canthus
dermoid cysts. Br J Ophthalmol 77:789–794, 1993 21. Kivela T, Tarkkanen A: Orbital germ cell tumors revisited: a clinicopathological approach to classification. Surv Ophthalmol 38:541–554, 1994 22. Lacey NA, McWilliams S, Jan W, et al: Case of the month: congenital unilateral proptosis. Br J Radiol 75:191–192, 2002 23. Soares EJC, Lopes KDS, Andrade JDG, et al: Orbital malignant teratoma. A case report. Orbit 2:235–242, 1983 24. Itani K, Traboulsi EI, Karim FWA, et al: Conservative surgery in orbital teratoma. Orbit 5:61–65, 1986 25. Hötte HH: Orbital fractures. Springfield: Charles Thomas, 1970 26. Green RP, Peters DR, et al: Force necessary to fracture the orbital floor. Ophthalmic Plast Reconst Surg 6:211, 1990 27. Fujino T, Sato TB: Mechanism of orbital blow-out fracture: experimental study by three-dimensional
eye model. Orbit 6:237, 1987 28. Karcioglu ZA, Nasr AM: Diagnosis and management of orbital inflammation and infections secondary
to foreign bodies: a clinical review. Orbit 17:247–267, 1998 29. Nasr AM, Haik BG, Fleming JC, Hussain H, Karcioglu ZA: Penetrating orbital injury with organic foreign bodies: a clinical review. Ophthalmology 106:523–532, 1999 30. Wojno TH: The incidence of extraocular muscle and cranial nerve palsy in orbital
floor blow-out fractures. Ophthalmology 94:682, 1987 31. Ehlinger P, Peeters L, Kockx M, et al: Hematic cyst of the orbit. Acta Stomatol Belg 90:177–179, 1993 32. Law FW: Spontaneous orbital hemorrhage. Br J Ophthalmol 55:556–558, 1971 33. Cameron JD, Letson RD, Summers CG: Clinical significance of hematic cysts of the orbit. Ophthalmic Plast Reconst Surg 4:95–99, 1988 34. Shapiro A, Tso MOM, Putterman AM, et al: A clinicopathologic study of hematic cysts of the orbit. Am J Ophthalmol 102:237–241, 1986 35. Wolter JR, Fralick FB, Tanton JH: Late results of orbital hemorrhage simulating orbital neoplasm: a false
aneurysm and a blood cyst with foreign body granuloma of the orbit. Am J Ophthalmol 62:528–532, 1966 36. Goldberg SH, Sassani JW, Parnes RE: Traumatic intraconal hematic cysts of the orbit. Arch Ophthalmol 110:378–380, 1992 37. Parke DW, Font RL, Boniuk M, et al: Cholesteatoma of the orbit. Arch Ophthalmol 100:612–616, 1982 38. Hill CA, Moseley IF: Imaging of orbital frontal cholesterol granuloma. Clin Radiology 46:237–242, 1992 39. Dobben GD, Fillet B, Mafee MF, et al: Orbital subperiosteal hematoma, cholesterol granuloma and infection: evaluation
with MR imaging and CT. Radiol Clin North Am 36:1185–1200, 1998 40. Seabold JE, Simonson TM, Weber PC, et al: Cranial osteomyelitis: diagnosis and followup with In-111 white
blood cell and Tc-99 m methylene diphosphonate bone SPECT, CT and
MR imaging. Radiology 196:779–788, 1995 41. Ormerod LD, et al: Ophthalmic manifestations of maxillary sinus mucoceles. Ophthalmology 94:1013, 1987 42. Friedman DP, Rao VM, Flanders AE: Lesions causing a mass in the medial canthus of the orbit: CT and MR features. Am J Roentgenol 160:1095–1099, 1993 43. Hasso AN, Lambert D: Magnetic Resonance imaging of the paranasal sinuses and nasal cavities. Top Magn Reson Imaging 6:209–223, 1994 44. DeSouza CE, et al: Mucoceles, proptosis, and transorbital frontoethmoidectomy. Orbit 7:167, 1988 45. Sagerman RH, Chung CT, Alberti WE: Radiosensitivity of ocular and orbital structures. In Albert WE, Sagerman RH (eds): Radiotherapy of intraocular and orbital tumors. Berlin: Springer-Verlag, 1993:375–385 46. Reim M, Kuckelkom R: Chemical and thermal lesions in the orbital region. Bull Soc Belge Ophthalmol 245:21–28, 1992 47. Ervin-Mulvey LD, Nelson LB, Freeley DA: Pediatric eye trauma. Pediatr Clin North Am 30:1167–1183, 1983 48. Leonard TJK, Moseley IF, Sanders MD: Ophthalmoplegia in carotid cavernous sinus fistulas. Br J Ophthalmol 68:128–134, 1984 49. Coskun O, Hamon M, Catrous G, et al: Carotid-cavernous fistulas: diagnosis with spiral CT angiography. ANJR Am J Neuroradiol 21:712–716, 2000 50. Howard GM, Jakobiec FA, Michelsen WJ: Orbital arteriovenous malformation with secondary capillary angiomatosis
treated by embolization with silastic liquid. Ophthalmology 90:1136–1139, 1983 51. Keltner JL, Satterfield D, Dublin AB, et al: Dural and carotid cavernous sinus fistulas. Ophthalmology 94:1585–1600, 1987 52. Lieb WE, Muller-Forell WS, Wichmann W: Ophthalmologic imaging methods. In, Muller-Forell WS (ed): Imaging of orbital and visual pathway pathology. Berlin: Springer 2002:9–10 53. Lieb WE, Merton DA, Shields JA: Color Doppler imaging in the demonstration of an orbital varix. Br J Ophthalmol 74:305–308, 1990 54. Bilaniuk LT: Orbital vascular lesions. Role of imaging. Radiol Clin North Am 37:169–183, 1999 55. Ambati BK, Ambati J, Azar N, et al: Periorbital and orbital cellulitis before and after the advent of Haemophilus
influenzae type B vaccination. Ophthalmology 107:1450–1453, 2000 56. Givner LB: Periorbital versus orbital cellulitis. Pediatr Infect Dis J 21:1157–1158, 2002 57. Hornblass A, Herschorn BJ, Stern K, et al: Orbital abscess. Surv Ophthalmol 29:169, 1984 58. Mohammad AEA, Al-Hussaini MK, Karoosh SS, El-Moneim MTA, Carey BJ, Karcioglu ZA: Tuberculosis of the Orbit and Lacrimal Gland. Orbit 11:199–204, 1992 59. Khalil M, Lindley S, Matouk E: Tuberculosis of the orbit. Ophthalmology 108:1624–1627, 1985 60. Al-Malki FA, Issa TM, Riley F, Karcioglu ZA: Nasolacrimal tuberculosis in a patient with conjunctivodacryocystorhinostomy. Ophthal Plast Reconst Surg 15:213–216, 1999 61. Klapper SR, Partrinely JR, Kaplan SL, Font RL: Atypical mycobacterial infection of the orbit. Ophthalmology 102:1536–1541, 1995 62. Barnes PF, Bloch AB, Davidson PT, Snider DE Jr : Tuberculosis in patients with human immunodeficiency virus infection [see
comments] [Review]. N Engl J Med 324:1644–1650, 1991 63. Ferry AP, Abdebi S: Diagnosis and management of rhino-orbital cerebral mucormycosis (phycomycosis): A report of 16 personally observed cases. Ophthalmology 90:1096, 1983 64. Green WR, Font RL, Zimmerman LE: Apsergillosis of the orbit: Report of ten cases and review of the literature. Arch Ophthalmol 82:302,1969 65. Karcioglu ZA, Caldwell DR: Frozen section diagnosis in ophthalmic surgery. Surv Ophthalmol 28:323, 1984 66. Margo CE, Hames LM, Ocular syphilis. Surv Ophthalmol 37:203–220, 1992 67. Dana M, Hochman MA, Viana MAG, et al: Ocular manifestations of leprosy in a noninstitutional community in the
United States. Arch Ophthalmol 112:626–629, 1994 68. Ffythe T: The continuing challenge of ocular leprosy. Br J Ophthalmol 75:123–124, 1991 69. Winward KE, Smith JL, Culbertson WW, et al: Ocular Lyme borreliosis. Am J Ophthalmol 108:651–657, 1989 70. Lesser RL, Kornmehl EW, Pachner AR, et al: Neuro-ophthalmologic manifestations of Lyme disease. Ophthalmology 97(6):699–706, 1990 71. Zangwill K, et al: Cat scratch disease in Connecticut. N Engl J Med 329:8, 1993 72. Carithers HA: Oculoglandular disease of Parinaud. Am J Dis Child 132:1195, 1978 73. Blanksma L, Slijper J: Actinomycotic dacryocystitis. Ophthalmologica 176:145–49, 1977 74. Karcioglu ZA: Actinomycosis in porous polyethylene orbital implant. Graefes Arch Clin Exp Ophthalmol 235:448–451, 1997 75. Kersten RC, Shoukrey NM, Tabarra KF: Orbital myiasis. Ophthalmology 93:1228, 1986 76. Walrath JD, Lalin SC, Leib ML: Cysticercosis isolated to the orbit. Ophthal Plast Reconstr Surg 19:243–244, 2003 PMID: 12918564. 77. Rauniyar RK, Thakur SK, Panda A: CT in the diagnosis of isolated cysticercal infestation of extraocular
muscle. Clin Radiol 58:154–156, 2003 78. Betharia SM, Sharma V, Pushker N: Ultrasound findings in orbital hydatid cysts. Am J Ophthalmol 135:568–570, 2003 79. Kiratli H, Bilgic S, Ozturkmen C, et al: Intramuscular hydatid cyst of the medial rectus muscle. Am J Ophthalmol 135:98–99, 2003 80. Gokcek C, Gokcek A, Akif Bayar MN, et al: Orbital hydatid cyst: CT and MRI. Neuroradiology 39:512–515, 1997 81. Awad MAN, Ray CJ, Karcioglu ZA: Echinococcus cysts of the orbit and substernum. Am J Ophthalmol 118:676–678, 1994 82. Unsold R, Greeven G: Inflammatory diseases of the orbit. Berlin: Springer Verlag, 2000 83. Bartley GB, Fatourechi V, et al: Clinical features of Graves' ophthalmopathy in an incidence cohort. Am J Ophthalmol 121:284, 1996 84. Lucarelli MJ, Shore JW: Management of thyroid optic neuropathy. International Ophth Clinics 36:179, 1996 85. Bartley GB, Gorman CA: Diagnostic criteria for Graves' ophthalmolopathy. Am J Ophthalmol 119:792, 1995 86. Leone CR, Lloyd WC: Treatment protocol for orbital inflammatory disease. Ophthalmology 92:1325, 1985 87. Kennerdell JS, Dresner SC: The nonspecific orbital inflammatory syndromes. Surv Ophthalmol 29:93–103, 1984 88. McGovern FH: The Tolusa-Hunt syndrome. Va Med 107:298–299, 1980 89. Johns ME, Wong RT: Painful ophthalmoplegia. The Tolusa-Hunt syndrome. Arch Otolaryngol 104:357–358, 1978 90. Jabs DA, Johns CJ: Ocular involvement in chronic sarcoidosis. Am J Ophthalmol 102:297–301, 1986 91. Collison MJT, et al: Involvement of orbital tissues by sarcoid. Am J Ophthalmol 102:302, 1986 92. Faris RL: Sjögren's syndrome. In Gold DH, Weingeist GA (eds): The eye in systemic disease. Philadelphia: Lippincott, 1990:70–71 93. Friedlaender MH: Ocular manifestations of Sjögren's syndrome: keratoconjunctivitis
sicca. Rheum Dis Clin North Am 18:591–608, 1992 94. Robin JB, Schanzlin DJ, Meisler DM, et al: Ocular involvement in Wegener's granulomatosis. Surv Oiphthalmol 30:127–140, 1985 95. Bullen CL, Liesegang TJ, McDonald TJ, et al: Ocular complications of Wegener's granulomatosis. Ophthalmology 90:279–290, 1983 96. Power WJ, Rodriguez A, et al: Disease relapse in patients with ocular manifestations of Wegener's
granulomatosis. Ophthalmology 102:154, 1995 97. Utresky SH, Kennerdell JS, Gupta JP: Graves' ophthalmolopathy in childhood and adolescence. Arch Ophthalmol 98:1963, 1980 98. Bradley E: Graves' ophthalmolopathy. Current Opinion in Ophthalmology 12:347–351, 2001 99. Balazs C, Bokk A, Molnar I: Graves' ophthalmolopathy, eye muscle antibodies and HLA antigens. Exp Clin Immunogen 6:190, 1989 100. Hufnagel TJ, Hickey WF, Cobbs WH, et al: Immunohistochemical and ultrastructural studies on the exenterated orbital
tissue of a patient with Graves' disease. Ophthalmology 91:1411, 1984 101. McKenzie JM: Humoral factors in the pathogenesis of Graves' disease. Physiol Rev 48:252, 1968 102. Rootman J: Diseases of the Orbit. Philadelphia: Lippincott, 1988:241–28 103. Peyster GR, Hoover E: Computerized tomography in orbital disease and neuro-ophthalmology. Chicago: Year Book Medical Publishers, 1984:97–114. 104. Yazici B, Yazici Z, Gelisken O: An unusual case: bilateral orbital varices. Acta Ophthalmol Scand 77:453–455, 1999 105. Srivastava S, Newman J: Pseudo-pseudotumor. Surv Ophthalmol 45:135–138, 2000 106. Karcioglu ZA, Haik GB: Tissue diagnosis: Orbit. In Karcioglu ZA (ed): Laboratory Diagnosis in Ophthalmology. New York: McMillan, 1987:27–29 107. Kline LB: The Tolosa-Hunt syndrome. Surv Ophthalmol 27:79–85, 1982 108. Karcioglu ZA, Aden LB, Cruz AA, et al: Orbital invasion with prolactinoma: A clinical review of 4 patients. Ophthal Plast Reconst Surg 18:64–71, 2002 109. Mesmer EP, Font RL, McCrary JA, et al: Epitheloid angiosarcoma of the orbit presenting as Tolosa-Hunt syndrome. Ophthalmology 90:1414–1421, 1983 110. Reich JM: What is sarcoidosis? Chest 124:367–371, 2003 111. Keicho N, Kitamura K, Takaku F, et al: Serum concentration of soluble interleukin-2 receptor as a sensitive
parameter of disease activity in sarcoidosis. Chest 98:112, 1990 112. Thomas PD, Hunninghake GW: Current concepts of the pathogenesis of sarcoidosis. Am Rev of Resp Diseases 135:747, 1987 113. Poulter LW: Immune aspects of sarcoidosis. Postgraduate Medical Journal 64:536–543, 1988 114. Reich JM: Deciphering histoplasmosis, systemic noncaseating granuloma, and sarcoisosis
chest. 113:1143, 1998 115. Wheat LJ, French ML, Wass JL: Sarcoidlike manifestations of histoplasmosis. Arch Intern Med 149:2421–2426, 1989 116. Bronson LJ, Fisher YL: Sarcoidosis of the paranasal sinuses with orbital extensions. Arch Ophthalmol 94:243–244, 1976 117. Power WJ, Neves RA, et al: The value of combined serum angiotensin-converting enzyme and gallium
scan in diagnosing ocular sarcoidosis. Ophthalmology 102:2007, 1995 118. Karcioglu ZA, Brear R: Conjunctival biopsy in sarcoidosis. Am J Ophthalmol 99:68, 1985 119. Obenauf CD, Shaw HE, Sydnor CF, et al: Sarcoidosis and its ophthalmic manifestations. Am J Ophthlamol 86:648–655, 1978 120. Talal N: Sjögren's Syndrome: Historical overview and clinical spectrum
of disease. Rheum Dis Clin North Am 18:507–515, 1992 121. Callen JP, Mahl CF: Oculocutaneous manifestations observed in multisystem disorders. Dermatol Clin 10:709–716, 1991 122. Alexander EL: Neurologic disease in Sjögren's syndrome: mononuclear inflammatory
vasculopathy affecting central/peripheral nervous system and muscle. Rheum Dis Clin North Am 19:869–908, 1993 123. Provost TT, Watson R: Cutaneous manifestations of Sjögren's syndrome. Rheum Dis Clin North Am 18:609–616, 1992 124. Spencer WH, Zimmerman LE: Conjunctiva. In Spencer WH (ed): Ophthalmic pathology. Philadelphia: WB Saunders, 1986:109–228 125. Jakobiec FA, Font RL: Orbit. In Spencer WH (ed): Ophthalmic pathology. Philadelphia: WB Saunders, 1986:2459–2860 126. Fox RI: Sjögren syndrome. In Sullivan DA, Dartt DA, Meneray MA (eds): Lacrimal gland, tear fluid, dry eye syndromes. New York: Plenum Press, 1998:891–902 127. Rosenbaum JT, Bennett RM: Chronic anterior and posterior uveitis and primary Sjögren's
syndrome. Am J Ophthalmol 104:346–352, 1987 128. Wegener F: Uber eine eigenartige rhinogene Granulomotose mit besonderer. Beteiligung
des Arteriensystems und der nieren. Beitr Pathol Anat 102:36–68, 1939 129. Straatsma BR: Ocular manifestations of Wegener's granulomatosis. Am J Ophthalmol 44:789–799, 1957 130. Greenberger MH: Central retinal artery closure in Wegener's granulomatosis. Am J Ophthalmol 63:515–516, 1967 131. Howell SB, Epstein WV: Circulating immune complexes in Wegener's granulomatosis. Am J Medicine 60:259–268, 1976 132. Falk RJ, Jennette JC: Wegener's granulomatosis, systemic vasculitis, and anti-neutrophil
cytoplasmic antibodies. Annu Rev Med 42:459–469, 1991 133. Fauci AS, Wolff SM: Wegener's granulomatosis: studies in 18 patients and a review of the
literature. Medicine (Baltimore) 52:535–561, 1973 134. Coutu RE, Klein M, Lessell S, et al: Limited form of Wegener's granulomatosis. Eye involvement as a major
sign. JAMA 233:868–871, 1975 135. Rao NV, Wehner NG, Marshall BC, Gray WR, et al: Characterization of proteinase 3 (PR3), a neutrophil serine protease. Structural
and functional. J Biol Chem 266:9540–9548, 1991 136. Ludemann G, Gross W: Autoantibodies against cytoplasmic structures of neutrophil granulocytes
in Wegener's granulomatosis. Clin Exp Immunol 69:350–357, 1987 137. Gans ROB, Goldschmeding R, Donker AJM, et al: Neutrophil cytoplasmic autoantibodies and Wegener's granulomatosis. Lancet 1:269–270, 1989 138. Jennette JC, Ewert BH, Falk RJ: Do antineutrophil cytoplasmic autoantibodies cause Wegener's granulomatosis
and other forms of necrotizing vasculitis? Controv Clin Rheum 19:1–14, 1993 139. Egner W, Chapel HM: Titration of antibodies against neutrophil cytoplasmic antigens is useful
in monitoring disease activity in systemic vasculitides. Clin Exp Immunol 82:244–249, 1990 140. Gross WL, Ludemann G, Kiefer G: Anti-cytoplasmic antibodies in Wegener's granulomatosis. Lancet 1:806, 1986 141. Soukiasian SH, Foster CS, Niles JL, et al: Diagnostic value of antineutrophil cytoplasmic antibodies in scleritis
associated with Wegener's granulomatosis. Ophthalmology 99:125–132, 1992 142. Kalina PH, Lie JT, Campbell RJ, et al: Diagnostic value and limitations of orbital biopsy in Wegener's granulomatosis. Ophthalmology 99:120–124, 1992 143. Jordan DR, Addison DA: Wegener's granulomatosis. Eyelid and conjunctival manifestations as
the presenting feature in two individuals. Ophthalmolgy 101:602–607, 1994 144. Devaney KO, Travis WD, Hoffman G, et al: Interpretation of head and neck biopsies in Wegener's granulomatosis. A
pathologic study of 126 biopsies in 70 patients. Am J Surg Pathol 14:555–564, 1990 145. Del Buono EA, Flint A: Diagnostic usefulness of nasal biopsy in Wegener's granulomatosis. Hum Pathol 22:107–110, 1991 146. Travis WD, Hoffman GS, Leavtitt RY, et al: Surgical pathology of the lung in Wegener's granulomatosis. Review
of 87 open lung biopsies from 67 patients. Am J Surg Pathol 15:315–333, 1991 147. Fauci AS, Haynes BF, Katz P, et al: Wegener's granulomatosis: prospective clinical and therapeutic experience
with 85 patients for 21 years. Ann Intern Med 98:76–85, 1983 148. Koorneef L, Melief CJM, Peterse HL, et al: Wegener's granulomatosis of the orbit. Orbit 2:1–10, 1983 149. Specks U, DeRemee RA: Granulomatous vasculitis. Wegener's granulomatosis and Churg-Strauss
syndrome. Rheum Clin North Am 16:377–397, 1990 150. Foster CS: Immunosuppressive therapy in external ocular inflammatory disease. Ophthalmology 87:140–150, 1980 151. Chang WJ, Shields CL, Shields JA, et al: Bilateral orbital involvement with massive allergic fungal sinusitis. Arch Ophthalmol 114:767–768, 1996 152. Dunlop IS, Billson FA: Visual failure in allergic aspergillus sinusitis: case report. Br J Ophthalmol 72:127–130, 1988 153. Torres C, El Naggar AK, Sim SJ, et al: Allergic fungal sinusitis: a clinicopathological study of 16 cases. Human Pathol 27:793–799, 1996 154. Michaels L, Lloyd G, Phelps T: Origin and spread of allergic fungal disease of the nose and paranasal
sinuses. Clin Otolaryngol Allied Sci 25:518–525, 2000 155. Ordonez NG: Application of immunocytochemistry in the diagnosis of soft tissue sarcomas: a
review and update. Adv Anat Pathology 5:67–85, 1998 156. Folpe AL, Gown AM: Immunohistochemistry for analysis of soft tissue tumors. In Weiss SW, Goldblum JR (eds): Soft tissues tumors, 4th edition. St. Louis: Mosby, 2001 157. Swanson SA, Brooks JJ: Proliferation markers Ki-67 and p105 in soft tissue lesions: correlation
with DNA flow cytometric characteristics. Am J Pathol 137:1491–1500, 1990 158. Hoos A, Stojadonovic A, Mastorides S, et al: High Ki-67 proliferative index predicts disease specific survival
in patients with high risk soft tissue sarcoma. Cancer 92:869–874, 2001 159. Karcioglu ZA, Haik BG: Tissue Diagnosis: Orbit. In Karcioglu ZA (ed): Laboratory Diagnosis in Ophthalmology. New York: MacMillan Publishing Co., 1986:25–52 160. Burnstine MA, Fruch BR, Elner VM: Angiosarcoma metastatic to the orbit. Arch Ophthalmol 114:93–96, 1996 161. Shields JA, Shields CL, Brotman HK, et al: Cancer metastatic to the orbit: the 2000 Robert M. Curts Lecture. Ophthal Plast Reconstr Surg 17:346–354, 2001 162. Char DH, Miller T, Kroll S: Orbital metastases: Diagnosis and course. Br J Ophthalmol 81:386, 1997 163. Shields JA, Bakewell B, Augsburger JJ, et al: Classification and incidence of space-occupying lesions of the orbit. Arch Ophthalmol 102:1601–1611, 1984 164. Henderson JW, Campbell RJ, Farrow GM, et al: Orbital Tumors. New York: Raven Press, 1994:43–52 165. Gunalp I, Gunduz K: Biopsy-proven orbital lesions in Turkey. A survey of 1092 cases
over 30 years. Orbit 13:67–69, 1994 166. Seregard S, Sahlin S: Panaorama of orbital space-occupying lesions. The 24-year
experience of a referral center. Acta Ophthalmol Scand 77:91–98, 1999 167. Jakobiec FA, Bilyk JR, Font RL: Lacrimal gland tumors. In Spencer WH (ed): Ophthalmic Pathology. An Atlas and Textbook, 4th edition. Philadelphia: WB Saunders Company, 1996:2485–2525 168. Grossniklaus HE, Abbuhl MF, McLean IW: Immunohistologic properties of benign and malignant mixed tumor of the
lacrimal gland. Am J Ophthalmol 110:540–549, 1990 169. Shields JA, Shields CL: Malignant transformation of presumed pleomorphic adenoma of lacrimal gland
after 60 years. Arch Ophthamol 105:1403, 1987 170. Tellad MV, McLean I, Specht CS, et al: Adenoid cystic carcinomas of the lacrimal gland in childhood and adolescence. Ophthalmology 104:1622–1625, 1997 171. Wright JE, Rose GE, Garner A: Primary malignant neoplasms of the lacrimal gland. Br J Ophthalmol 76:401–407, 1992 172. Gamel JW, Font RL: Adenoid cyst carcinoma of the lacrimal gland: the clinical significance
of a basaloid histologic pattern. Hum Pathol 13:219–225, 1982 173. Eviatar JA, Hornblass A: Mucoepidermoid carcinoma of the lacrimal gland: 25 cases and a review and
update of the literature. Ophthal Plast Reconstr Surg 9:170–181, 1993 174. Levin LA, Popham J, To K, et al: Mucoepidermoid carcinoma of the lacrimal gland: Report of a case with oncocytic
features arising in a patient with chronic dacryops. Ophthalmology 98:1551, 1991 175. Bullock JD, Fleishman JA, Rosset JS: Lacrimal duct cysts. Ophthalmology 93:1355–1360, 1986 176. Monica M, Long DA, Karcioglu ZA: Bilateral dacryops. Ann Ophthalmol 20:259–263, 1988 177. Margo CE, Karcioglu ZA, Naugle TC: Ectopic lacrimal gland tissue of the orbit and sclerosing dacryoadenitis. Ophthalmic Surg 16:178, 1985 178. Harris GJ: Orbital vascular malformations: A consensus statement on terminology and
its clinical implications. Am J Ophthalmol 127:453–455, 1999 179. Haik BG, Karcioglu ZA, Gordon RA, Pechos B: Capillary Hemangioma. [Major Review]. Survey Ophthalmol 38:399–426, 1994 180. Chang E, Boyd A, Nelson CC, et al: Successful treatment of infantile hemangiomas with interferon-alpha–2B. J Pediatr Hematol Oncol 19:237–244, 1997 181. Burstein FD, Simms C, Cohen SR, et al: Intralesinal laser therapy of extensive hemangiomas in 100 consecutive
pediatric patients. Ann Plast Surg 44:188–194, 2000 182. Kushner BJ: Intralesional corticosteroid injection for infantile adnexal hemangioma. Am J Ophthalmol 93:496–506, 1982 183. Karcioglu ZA, Nasr MN, Haik BG: Orbital hemangiopericytoma: Clinical and morphologic features. Am J Ophthalmol 124:661–672, 1997 184. Rice CD, Kersten RC, Mrak RE: An orbital hemangiopericytoma recurrent after 33 years. Arch Ophthalmol 107:552, 1989 185. Harris GJ, Jakobiec FA: Cavernous hemangioma of the orbit: A clinicopathologic analysis of sixty-six
cases. In Jakobiec FA (ed): Ocular and Adnexal Tumors, Birmingham AL:. Aesculapius 1978:741 186. Sullivan TJ, Aylward GW, Wright JE, et al: Bilateral multiple cavernous hemangiomas of the orbit. Br J Ophthalmol 76:627–629, 1992 187. Henderson JW, Campbell RJ, Farrow GM, et al: Orbital Tumors. New York: Raven Press, 1994:89–118 188. Tunc M, Sadri E, Char DH: Orbital lymphangioma: an analysis of 26 cases. Br J Ophthalmol 83:760–780, 1999 189. Rootman J, Hay E, Graeb D, et al: Orbital-adnexal lymphangiomas: A spectrum of hemodynamically isolated
vascular hamartomas. Ophthalmology 93:1558, 1986 190. Harris GJ, Sakel PJ, Bonavolonta G, et al: An analysis of thirty cases of orbital lymphangioma: Pathophysiologic considerations
and management recommendations. Ophthalmology 97:1583, 1990 191. Shields JA, Dolinskas C, Augsburger JJ, et al: Demonstration of orbital varix with computed tomography and valsalva maneuver. Am J Ophthalmol 97:108–110, 1984 192. Keltner J, Satterfield, D, Dublin AB, et al: Dural and carotid cavernous sinus fistulas. Ophthalmology 94:1585–1600, 1987 193. Wright JE, Sullivan TJ, Garner A: Orbital venous anomalies. Ophthalmology 104:905–913, 1997 194. Gunduz K, Shields JA, Shields CL, et al: Cutaneous angiosarcoma with eyelid involvement. Am J Ophthalmol 125:870–871, 1998 195. Hufnagel T, Ma L, Kuo TT: Orbital angiosarcoma with subconjunctival presentation. Ophthalmology 94:72–77, 1987 196. Shields JA, Shields CL, Eagle RC Jr , et al: Intravascular papillary endothelial hyperplasia with presumed bilateral
orbital varices. Arch Ophthalmol 117:1247–1249, 1999 197. Jolly SS, Brownstein S, Jordan DR: Leiomyoma of the anterior orbit and eyelids. Can J Ophthalmol 30:366–370, 1995 198. Kaulkarni V, Rajshekhar V, Chandi SM: Orbital apex leiomyoma with intracranial extension. Surg Neurol 54:327–330, 2000 199. Smith DL, Kincaid MC, Nicolitz E: Angiolymphoid hyperplasia with eosinophilia (Kumura's disease) of
the orbit. Arch Ophthalmol 106:793–795, 1988 200. Karcioglu ZA, Yulug A, Haik BG: Tumors of the optic nerve. In, Margo C, et al (eds): Diagnostic Problems in Clinical Ophthalmology. Philadelphia: Saunders, 1994:105–106 201. Alvord EJ, Lofton S: Gliomas of the optic nerve or chiasm. Outcome by patient's age, tumor
site and treatment. J Neurosurg 68:85–98, 1988 202. Dutton JJ: Gliomas of the anterior visual pathway. Surv Ophthalmol 38:427–452, 1994 203. Brodovsky S, Hove MW, Pinkerton RM, et al: An enhancing optic nerve lesions: malignant glioma of adulthood. Can J Ophthalmol 32:409–413, 1997 204. Chateil JF, Sousotte C, Pedespan JM, et al: MRI and clinical differences between optic pathway tumours in children
with and without neurofibromatosis. Br J Radiology 74:24–31, 2001 205. Deliganis AV, Geyer JR, Berger MS: Prognostic significance of type I neurofibromatosis (von Recklinghausen
Disease) in childhood optic glioma. Neurosurgery 38:1114–1118, 1996 206. Lee AG, Dutton J: A practice pathway for the management of gliomas of the anterior visual
pathway: an update and an evidence-based approach. Neuroophthalmology 22:139–155, 1999 207. Levin LA, Jakobiec FA: Optic nerve tumors of childhood: A decision analytical approach to their
diagnosis. Int Ophthalmol Clin 32:223–240, 1992 208. Mulvihill JJ, Parry MD, Sherman JL, et al: NIH Conference. Neurofibromatosis 1 (Recklinghausen disease) and
neurofibromatosis 2 (bilateral acoustic neurofibromatosis. An update. Ann Intern Med 113:39–52, 1990 209. Pearson-Webb MA, Kaiser-Kupfer MI, Eldridge R: Eye findings in bilateral acoustic (central) neurofibromatosis: Association
with presenile lens opacities and cataracts but absence
of Lisch nodules (letter). N Engl J Med 35:1553–1554, 1986 210. Cummings TJ, Provenzale JM, Hunter SB, et al: Gliomas of the optic nerve: histological, immunohistochemical (MIB–1 and
p53), and MRI analysis. Acta Neuropathologica 99:563–570, 2000 211. Bilgic S, Erbengi A, Tinaztepe B, et al: Optic glioma of childhood: Clinical, histopathological, and histochemical
observations. Br J Ophthalmol 73:832–837, 1989 212. Imes RK, Hoyt WF: Magnetic resonance imaging signs of optic nerve gliomas in neurofibromatosis. Am J Ophthalmol 111:729–734, 1991 213. Stern J, Jakobiec FA, Housepian EM: The architecture of optic nerve gliomas with and without neurofibromatosis. Arch Ophthalmol 98:505–511, 1980 214. Cooling RJ, Wright JE: Arachnoid hyperplasia in optic nerve glioma: Confusion with orbital meningioma. Br J Ophthalmol 63:596–599, 1979 215. Fuss M, Hug EB, Schaefer RA, et al: Proton radiation therapy (PRT) for pediatric optic pathway gliomas: comparison
with 3D planned conventional photons and standard photon
technique. Int J Rad Oncol Biol Physics 45:1117–1126, 1999 216. Muci-Mendoza R, Arevalo JF, Ramella M, et al: Optociliary veins in optic nerve sheath meningioma. Indocynanine green
videoangiography findings. Ophthalmology 106:311–318, 1999 217. Cunliffe IA, Moffat DA, Hardy DG, et al: Bilateral optic nerve sheath meningiomas in a patient with neurofibromatosis
type 2. Br J Ophthalmol 76:310–312, 1992 218. Mafee MF, Goodwin J, Dorodi S: Optic nerve sheath miningiomas. Role of MR imaging. Radiol Clin North Am 37:37–58, 1999 219. Turbin RE, Thompson CR, Kennderdell JS, et al: A long-term visual outcome comparison in patients with optic nerve
sheath meningioma managed with observation, surgery, radiotherapy, or
surgery and radiotherapy. Ophthalology 109:890–900, 2002 220. Moyer PD, Golnik KC, Breneman J: Treatment of optic nerve sheath meningioma with three-dimensional
conformal radiation. Am J Ophthalmol 129:694–696, 2000 221. Rootman J, Goldberg C, Robertson W: Primary orbital Schwannomas. Br J Ophthalmol 66:194–204, 1982 222. Carroll GS, Haik BG, Fleming JC, et al: Peripheral nerve tumors of the orbit. Rad Clin North Am 37:195–202, 1999 223. Rose GE, Wright JE: Isolated peripheral nerve sheath tumours of the orbit. Eye 5:668–673, 1991 224. Abet T, Kawamura N, Homma H, et al: MRI of orbital schwannomas. Head Neck Radiol 42:466–468, 2000 225. Schatz H: Benign orbital neurilemmoma: sarcomatous transformation in von Recklinghausen's
disease. Arch Ophthalmol 86:268–273, 1971 226. Harkin JC, Reed RJ: Tumors of the peripheral nervous system. Atlas of tumor pathology. Washington, DC, 1968:29–59 227. Lam DSC, Ng JSK, To TAF, et al: Cystic schwannoma of the orbit. Eye 11:798–800, 1997 228. Jakobiec FA, Font RL, Zimmerman LE: Malignant peripheral nerve sheath tumors of the orbit: a clinical pathological
study of eight cases. Trans Am Ophthalmol Soc 83:332–366, 1985 229. Lewis RA, Gerson LP, Axelson KA, et al: Von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology 91:929–935, 1984 230. Henderson JW, Campbell RJ, Farrow GM, et al: Orbital Tumors. New York: Raven Press, 1994:221–37 231. Jackson IT, Laws ERJr , Martin RD: The surgical management of orbital neurofibromatosis. Plast Reconst Surg 71:751–758, 1983 232. Bednar MM, Trainer TD, Aitken, PA, et al: Orbital paraganglioma: case report and review of the literature. Br J Ophthalmol 76:183–185, 1992 233. Gunalp I, Gunduz K, Duruk K, Kanpolat Y: Neurogenic tumors of the orbit. Jpn J Ophthalmol 38:185–190, 1994 234. Myssiorek D: Head and neck paragangliomas: an overview. Otolaryngol Clin North Am 34:829–836, 2001 235. Castillo BVJr , Kaufman L: Pediatric tumors of the eye and orbit. Pediatr Clin North Am 50:149–172, 2003 236. He Y, Song G, Ding Y: Histopathologic classification of 3,476 orbital diseases. Zhonghua Yan Ke Za Zhi 38:396–398, 2002 237. Shields JA, Shields CL: Rhabdomyosarcoma: review for the ophthalmologist. Surv Ophthalmol 48:39–57, 2003 238. Knowles DM II , Jakobiec FA, Potter GD, et al: Ophthalmic striated muscle neoplasms. Surv Ophthalmol 21:219–261, 1976 239. Margo CE, Karcioglu ZA: Diagnostic electronmicroscopy In Karcioglu ZA (ed): Laboratory
Diagnosis in Ophthalmology. New York: McMillian, 1987:120–130 240. Buwalda J, Schouwenburg PF, Blank LE, et al: A novel local treatment strategy for advanced stage head and neck rhabdomyosarcomas
in children: results of the AMORE protocol. Eur J Cancer 39:1594–602, 2003 241. Seregard S: Management of alveolar rhabdomyosarcoma of the orbit. Acta Ophthalmol Scand 80:660–664, 2002 242. Luckenbach M: Idiopathic inflammatory pseudotumor of childhood. Park City, UT:. Hogan-Theobald Society Meeting, 1984 243. Shields CL, Shields JA, Honavar SG, Demirci H: Primary ophthalmic rhabdomyosarcoma in 33 patients. Trans Am Ophthalmol Soc 99:133–142, 2001 244. Fetkenhour DR, Shields CL, Chao , et al: Orbital cavitary rhabdomyosarcoma masquerading as lymphangioma. Arch Ophthalmol 11:1208–1210, 2001 245. Seedat RY, Hamilton PD, de Jager LP, et al: Orbital rhabdomyosarcoma presenting as an apparent orbital subperiosteal
abscess. Int J Pediatr Otorhinolaryngol 52:177–181, 2000 246. Gordon LK: Diagnostic dilemmas in orbital inflammatory disease. Ocul Immunol Inflamm 11:3–15, 2003 247. Hou LC, Murphy MA, Tung GA: Primary orbital leiomysarcoma: a case report with MRI findings. Am J Ophthalmol 135:408–410, 2003 248. Klippenstein KA, Wesley RE, Glick AD: Orbital leiomyosarcoma after retinoblastoma. Ophthalmic Surg Lasers 30:579–583, 1999 249. Weichens B, Werner JA, Luttges J, et al: Primary orbital leimyoma and leiomyosarcoma. Ophthalmologica 213:159–164, 1999 250. Karcioglu ZA, Carroll G, Nasr A, Haik BG: Fibrous lesions of the orbit and periorbital tissues. Amsterdam: XXVIII
International Congress of Ophthalmology (ICO), 1998:8 251. Dalley RW: Fibrous histiocytoma and fibrous tissue tumors of the orbit. Radiol Clin North Am 1999;37:185–194 252. Jakobiec FA, Font RL: Orbit. In Spencer WH, Font RL, Green WR, et al (eds): Ophthalmic Pathology. An Atlas and Textbook. Vol. 3. Philadelphia: W.B. Saunders Co., 1986:2568–2580 253. Caballero LR, Rodriguez AC, Sopelana AB: Angiomatoid fibrous histiocytoma of the orbit. Am J Ophthalmol 92:13–15, 1981 254. Lui D, McCann P, Kini RK, et al: Malignant fibrous histiocytoma of the orbit in a 3-year-old
girl. Arch Ophthalmol 105:895–896, 1987 255. Font RL, Hidayat AA: Fibrous histiocytoma of the orbit. A clinicopathologic study of 150 cases. Hum Pathol 13:199–209, 1982 256. Ros PR, Kursunoglu S, Batlle JF, et al: Malignant fibrous histiocytoma of the orbit. J Clin Neuro-ophthalmol 5:116–119, 1985 257. Masson JK, Soule EH: Desmoid tumors of the head and neck. Am J Surg 112:6115–6122, 1966 258. Perry RH, Ramani PS, McAllister V, et al: Nodular fasciitis causing unilateral proptosis. Br J Ophthalmol 59:404–408, 1975 259. Font RL, Zimmerman LE: Nodular fasciitis of the eye and adnexa. A report of ten cases. Arch Ophthalmol 75:475–481, 1966 260. Ferlito A, Recher G, Tomazzolli L: Radiation-induced fibrosarcoma of the mandible following treatment
for bilateral retinoblastoma. J Laryngol Otol 93:1015–1020, 1979 261. Karcioglu ZA, Al-Rasheed W, Gray A: Second neoplasms in retinoblastoma. Middle East J Ophthalmol 5:99–104, 1997 262. Polito E, Tosi M, Toti P, et al: Orbital solitary fibrous tumor with aggressive behavior. Three cases and
the review of the literature. Grafes Arch Exp Ophthalmol 240:570–574, 2002 263. Kim HY, Lee SY, Kang SJ, et al: Solitary fibrous tumor of the orbit: a poorly recognized orbital lesion. Acta Ophthalmol Scand 77:704–708, 1999 264. Lanuza A, Lazaro R, Salvador M, et al: Solitary fibrous tumour of the orbit. Report of a new case. Int Ophthalmol 22:265–268, 1998 265. Caroll G, Haik BG, Karcioglu ZA: Orbital giant cell fibroblastoma. Orbit 18:25–32, 1999 266. Van Furth R, Cohn ZA, Hirsch JG, et al: The mononuclear phagocyte system: a new classification of macrophages, monocytes
and their precursor cells. Bull World Health Org 46:845–855, 1972 267. Chu AC: The confusion state of the histicytoses. Br J Dermatol 143:475–476, 2000 268. Karcioglu ZA, Sharara N, Boles, TL, Nasr AM: Orbital xanthogranuloma, clinical and morphologic features in 8 patients. Ophthal Plast Reconstr Surg 19:372–381, 2003 269. Malone M: The histiocytosis of childhood. Histopathology 19:105–119, 1991 270. Dahlmann AH, Simmons I, Picton S, et al: Single-Langerhans cell histiocytosis of the orbit. Eye 14:109–110, 2000 271. Veyssier-Belot C, Cacoub P, Caparros-Lefebvre D, et al: Erdheim-Chester disease. Clinical and radiologic characteristics
of 59 cases. Medicine (Baltimore) 75:157–169, 1996 272. Chetritt J, Paradis V, Dargere D, et al: Chester-Erdheim disease: a neoplastic disorder. Hum Pathol 30:1093–1096, 1999 273. Alper MG, Zimmerman LE, la Piana FG: Orbital manifestations of Erdheim-Chester disease. Trans Am Ophthalmol Soc 81:64–85, 1983 274. Shields JA, Karcioglu ZA, Shields CL, et al: Orbital and eyelid involvement with Erdheim-Chester disease. A report
of two cases. Arch Ophthalmol 109:850–854, 1991 275. Miszkiel KA, Sohaib SAA, Rose GE: Radiological and clinicopathological feaures of orbital XG. Br J Ophthalmol 84:251–258, 2000 276. Karcioglu ZA: Bone and cartilage tumors. In Karcioglu ZA (ed): Orbital Tumors. New York: Springer-Verlag, 2004:(in press) 277. Earwaker J: Perinasal sinus osteomas: a review of 46 cases. Skel Radiol 22:417–423, 1993 278. Tucker MA, D'Angio GJ, Boice JD Jr. , et al: Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med 317:588–593, 1987 279. Shields JA: Lymphoid tumors and leukemias. In Shields JA (ed): Diagnosis and Management of Orbital Tumors, Philadelpha: WB Saunders, 1989:316 280. Jakobiec FA, Font RL: Orbit: lymphoid tumors. In Spencer WH, Font RL, Green WR, et al (eds): Ophthalmic Pathology: An Atlas and Textbook. Vol 3, ed. Philadelphia: WB Saunders Co., 1986:2663–2711 281. Sharara N, Holden JT, Wojno TH, et al: Ocular adnexal lymphoid proliferations. Clinical, histological, flow cytometry
and molecular analysis of 43 cases. Ophthalmology 110:1245–1254, 2003 282. Freedman AS, Nadler LM: NonHodgkin's lymphomas. In Bast RC, Kufe DW, Pollock RE, et al (eds): Cancer Medicine 5th ed. BC Decker Inc., 2000:Section 36, Ch 130 283. Chan JKC: Tumors of the lymphoreticular system, including skin and thymus. In, Fletcher CDM (ed): Diagnostic histopathology of tumors, 2nd ed. London: Churchill Livingston, 2000:1099–1316 284. Harris NI, Jaffe ES, Stein H, et al: A revised European-American classification of lymphoid neoplasms: a
proposal from the International Lymphoma Study Group. Blood 84:1361–1392, 1994 285. Armitage JO, Weisenburger DD: New approach to classifying nonHodgkin's lymphomas: Clinical features
of the major histologic subtypes. J Clin Oncol 16:178–195, 1998 286. Coupland SE, Krause L, Des Velecluse HJ, et al: Lymphoproliferative lesions of the ocular adnexa: analysis of 112 cases. Ophthalmology 105:1430–1441, 1998 287. Jenkins C, Rose GE, Bunce C, et al: Histological features of ocular adnexal lymphoma (REAL classification) and
their association with patient morbidity and survival. Br J Ophthalmol 84:907–913, 2000 288. Jakobiec FA, Lefkowich J, Knowles DM: B- and T-lymphocytes in ocular disease. Ophthalmology 91:635, 1984 289. Coupland SE, Hummel M, Stein H: Ocular adnexal lymphomas, five case presentations and a review of the literature. Surv Ophthalmol 47:470–490, 2002 290. Au Eong, Choo CT: Burkitt lymphoma manifesting as acute proptosis. Am J Ophthalmol 123:856–858, 1997 291. Uceda-Montanes A, Blanco G, Saornil MA, et al: Extramedullary plasmocytoma of the orbit. Acta Ophthalmol Scand 78:601–603, 2000 292. Rodman HI, Font RL: Orbital involvement in multiple myeloma: Review of the literature and report
of three cases. Arch Ophthalmol 87:30, 1972 293. Kennedy RE: An evaluation of 820 orbital cases. Trans Am Ophthalmol Soc 82:134, 1984 294. Rootman J: Disease of the orbit. A multidisciplinary approach. Philadelphia: JB Lippincott Co., 1988:405–427 295. Jensen OA: Metastatic tumors of the eye and orbit: a histopathologic analysis of a
Danish series. Acta Pathol Microbiol Scand 212:201, 1970 296. Font RL, Ferry AP: Carcinoma metastatic to the eye and orbit. III: A clinicopathologic study
of 28 cases metastatic to the orbit. Cancer 38:1326, 1976 297. Shields CL, Shields JA, Peggs M: Tumors metastatic to the orbit. Ophthalmic Plast Reconstr Surg 4:73, 1988 298. Henderson JW, Campbell RJ, Farrow GM, Garrity JA: Orbital tumors, 3rd ed. New York: Raven Press, 1993;361–375 299. Shields JA, Shields CL, Brotman HK, et al: Cancer metastatic to the orbit: the 2000 Robert M. Curts Lecture. Ophthal Plast Reconstr Surg 17:346–354, 2001 300. Goldberg RA, Rootman J, Cline RA: Tumors metastatic to the orbit: A changing picture. Surv Ophthalmol 35:1, 1990 301. Katz SE, Rootman J, Goldberg RA: Secondary and metastatic tumors of the orbit. In Tasman W, Jaeger EA (eds): Duane Ophthalmology CD-ROM Edition. Hagerstown, MD:. Lippincott, Williams & Wilkins, 2001:Clinical Volume 2, Chapter 46 302. Healy JF: Computed tomographic evaluation of metastases to the orbit. Ann Ophthalmol 15:1026, 1983 303. Peyster RG, Shapiro MD, Haik BG: Orbital metastasis: Role of magnetic resonance imaging and computed tomography. Radiol Clin North Am 25:647, 1987 304. Dresner SD, Kennerdell JS, Dekker A: Fine needle aspiration biopsy of metastatic orbital tumors. Surv Ophthalmol 27:397, 1983 305. Kennerdell JS, Slamovits TL, Dekker A, et al: Orbital fine needle aspiration biopsy. Am J Ophthalmol 99:547, 1985 306. Das DK, Das J, Bhatt NC, et al: Orbital lesions. Diagnosis by fine needle aspiration cytology. Acta Cytol 38:158–164, 1994 307. Krohel GB, Tobin DR, Chavis RM: Inaccuracy of fine needle aspiration biopsy. Ophthalmology 92:666, 1985 308. Liu D: Complications of fine needle aspiration biopsy of the orbit. Ophthalmology 92:1768, 1985 309. Shetlar DJ, Font RL, Ordonez N, et al: A clinicopathologic study of three carcinoid tumors metastatic to the orbit. Ophthalmology 97:257, 1990 310. Fan JT, Buettner H, Bartley GB, Bolling JP: Clinical features and treatment of seven patients with carcinoid tumor
metastatic to the eye and orbit.Am J Ophthalmol 119:211, 1995 311. Amemiya T, Hayashida H, Dake Y: Metastatic orbital tumors in Japan: a review of the literature. Ophthalmic Epidemiol 9:35–47, 2002 312. Evans AR: Congenital neuroblastoma. J Clin Pathol 18:54–62, 1965 313. Reynolds P, Smith R, Frenkel B: The diagnostic dilemma of small, round cell neoplasm. Cancer 48:2088, 1981 314. Henderson JW, Farrow GM: Orbital tumors (2nd ed.), New York: Brian C. Decker (Thieme-Stratton), 1980:425–450 315. Crotty TB, Scheithauer BW, Young WF, et al: Papillary craniopharyngioma: a clinical study of 48 cases. J Neurosurg 83:206–214, 1995 316. Wilson WB: Meningiomas of the anterior visual system. Surv Ophthalmol 26:109, 1981 317. Carrau RL, Segas J, Nuss DW, et al: Squamous cell carcinoma of the sinonasal tract invading the orbit. Laryngoscope 109:230–235, 1999 318. Karcioglu ZA, et al: Proptosis pseudocyst formation from inverted papilloma. Ann Ophthalmol 14:443–448, 1982 319. Hurwitz JJ: Lacrimal sac tumours. Chapter 28, In Hurwitz JJ (ed): The Lacrimal System. Philadelphia: Lippincott-Raven, 1996:195 320. Shields JA, Shields CL: Orbital malignant melanoma: 2002 Sean B. Murphy lecture. Ophthalmol Plas Reconstr Surg 19:262–269, 2003 321. Finger PT, Harbour JW, Karcioglu ZA: Risk factors for metastasis in retinoblastoma. Surv Ophthalmol 47:1–16, 2002 322. Liesegang GK: Amyloid infiltration of the levator palpebral superioris muscle. Ann Ophthalmol 15:610–613, 1983 323. Gorevic PD, Rodrigues MM: Ocular amyloidosis. Am J Ophthalmol 113:529–532, 1994 324. Sevino PJ, Schatz NJ, Rodrigues MM: Orbital amyloidosis. Can J Ophthalmol 11:252–255, 1976 325. Wheeler GE, Eiferman RA: Immunohistochemical identification of the AA protein in lattice dystrophy. Exp Eye Research 36:181, 1983 326. Breathnach SN: Amyloid and amyloidosis. J Am Acad Derm 18:1–16, 1988 327. Okamoto K, Ito J, Emura T, et al: Focal orbital amyloidosis presenting as rectus muscle enlargement: CT and
MR findings. AJNR Am J Neuroradiol 19:1799–1801, 1988 328. Murdoch IE, Sullivan TJ, Moseley I, et al: Primary localized amyloidosis of the orbit. Br J Ophthalmol 80:1083–1086, 1996 329. Massry GG, Harrison W, Hornblass A: Clinical and computed tomographic characteristics of amyloid tumors of
the lacrimal gland. Ophthalmology 103:1233–1236, 1996 330. Ustun MO, Ekinci N, Payzin B: Extramedullary plasmocytoma of the parotid gland. Report of a case with
extensive amyloid deposition masking the cytology and histopathologic
picture. Acta Cytol 45:449–453, 2001 331. Jabs DA, Miller NA, Neuman SA, et al: Optic neuropathy in systemic Lupus erythematosus. Arch Ophthalmol 104:564, 1986 332. Grimson BS, Simons KB: Orbital inflammation, myositis, and systemic lupus erythematosis. Arch Ophthalmol 101:736–738, 1983 333. Ibanez HE, Bardenstein DS, Korman NJ, et al: Exuberant conjunctival pseudopolyposis in a patient with dermatomyositis. Ann Ophthalmol 25:326–327, 1993 334. Leib ML, Odel JG, Cooney MJ: Orbital polymyositis and giant cell myocarditis. Ophthalmology 101:950–954, 1994 335. Selva-O'Callaghan A, Mijares-Boeckh-Behrens T, Solans-Laque R, et al: Dermatomyositis and Graves' disease. Clin Exp Rheumatol 19:595–596, 2001 336. Watson PG, Hazelman BL: The sclera and systemic disorders. Philadelphia: WB Saunders, 1976 337. Foster CS, Sainz de la Masa M: The sclera. New York: Springer-Verlag, 1994 338. Horan EC: Ophthalmic manifestations of progressive systemic sclerosis. Br J Ophthalmol 53:388–392, 1969 339. Manschot WA: Generalized scleroderma with ocular symptoms. Ophthalmologica 149:131–137, 1965 340. Anzai H, Tajima S: Systemic scleroderma associated with Graves' disease. J Dermatol 23:896–898, 1996 341. Nicholson D, White S, Lipson A, et al: Progressive systemic sclerosis and Graves' disease. Report of three
cases. Arch Intern Med 146:2350–2352, 1986 342. Koike R, Yamada M, Matsunaga T, et al: Polyarteritis nodosa (PN) complicated with unilateral exophthalmos. Intern Med 32:232–236, 1993 343. Ishida K, Yokota T, Wada Y, et al: Unilateral facial swelling and exophthalmos in a patient with polyarteritis
nodosa. Intern Med 31:500–503, 1992 344. Prasad TS, Suresh PS, Rodrigues G, et al: Orbital involvement with angiolymphois hyperplasia eosinophilia—a
benign condition for the practicing ophthalmologist to be aware of. Eye 14:390–392, 2000 345. Buggage RR, Spraul CW, Wojno TH, et al: Kimura disease of the orbit and ocular adnexa. Surv Ophthalmol 44:79–91, 1999 346. Cook HT, Stafford ND: Angiolymphoid hyperplasia with eosinophilia involving the lacrimal gland: case
report. Br J Ophthalmol 72:710–712, 1988 347. Francis IC, Kappagoda MB, Smith J, et al: Kimura's disease of the orbit. Ophthal Plast Reconstr Surg 4:235–239, 1988 348. Al-Malki AF, Hussain H, Al-Ma'ani JR, et al: Extraocular myositis in Behcet's Syndrome. VI International Symposium
on Ocular Inflammation. Istanbul, Turkey, June 2000 349. Sale S, Patterson R: Recurrent Churg-Strauss vasculitis. With exophthalmos, hearing loss, nasal
obstruction, amyloid deposits, hyperimmunoglobulinemia E, and
circulating immune complexes. Arch Intern Med 141:1363–1365, 1981 350. Iliff NT: The ophthalmic implications of the correction of late enophthalmos following
severe midfacial trauma. Trans Am Ophthalmol Soc 89:455–548, 1991 351. Cline RA, Rootman J: Enophthalmos. A clinical review. Ophthalmology 91:229, 1984 352. Nasr AM, Ayyash I, Karcioglu ZA: Unilateral enophthalmos secondary to acquired hemilipodystrophy. Am J Ophthalmol 124:572–575, 1997 |