1. Lewis R, Nussbaum R, Stambolian, D: Mapping X-linked ophthalmic diseases.
IV. Provisional assignment of the locus for X-lined congenital cataracts
and microcornea (the Nance-Horan syndrome) to Xp22.2-p22.3. Ophthalmology,
97:110, 1990.
2. Mondino G, Cohn H: Corectopia with nystagmus and corneal changes. Acta
Ophthalmol (Copenhag), 59:85, 1981.
3. Ghose S, Mehta U: Microcornea with corectopia and macular hypoplasia
in a family. Jpn Jf Ophthalmol 28:126, 1984.
4. Frydman M, Steinberger J, Shabta F et al: Interstitial deletion 2q14q21.
Am J Med Genet 34:476, 1989.
5. Judisch F, Martin-Casals A, Hanson J et al: Oculodentodigital dysplasia.
Four new reports and a literature review. Arch Ophthalmol 97:878, 1979.
6. Streeten B, Karpic A, Spritzer K: Posterior keratoconus associated
with systemic abnormalities. Arch Ophthalmol 101:616, 1983.
7. Brooks J, Coccaro P, Zarbin M: The Rieger anomaly concomitant with
multiple dental, craniofacial and somatic midline anomalies and short
stature. Oral Surg Oral Medl Oral Pathol 68:717, 1989.
8. May M, Beauchamp G: Collagen maturation defects in Ehlers-Danlos syndrome.
J Pediatr Ophthalmol Strabismus 24:780, 1987.
9. Frenkel L, Keys M, Hefferfen S et al: Unusual eye abnormalities associated
with congenital cytomegalovirus. Pediatrics 66:763, 1980.
10. Cibis G.Waeltermann J, Harris D: Peters' anomaly in association with
ring 21 chromosomal abnormality. Am J Ophthalmol 100:733, 1985.
11. Kivlin J, Fineman R, Crandall A et al: Peters' anomaly as a consequence
of genetic and non-genetic syndromes. Arch Ophthalmol 104:61, 1986.
12. Holbach L, Font R, Shivitz I et al: Bilateral keloid-like myofibrobastic
proliferations in the cornea in children. Ophthalmology 97:1188, 1990.
13. Bahn D, Falls H, Yarley G et al: Classification of corneal endothelial
disorders based on neural crest origin. Ophthalmology 91:558, 1984.
14. Pe'er J, BenEzra D, Sela M et al: Cryptophthalmos syndrome. Clinical
and histopathologic findings. Ophthalmic Paediatr Genet 8:177, 1097.
15. Fraser G: Our genetic load: A review of some aspects of genetic variation.
Ann Hum Genet 25:387, 1962.
16. Thomas IT, Frias JL, Felix V et al:, Isolated and syndromic cryptophthalmos.
Am J Med Gen 25:85, 1986.
17. Berg C, Geipel A, Germer U et al: Prenatal detection of Fraser syndrome
without cryptophthalmos: Case report and review of the literature. Ultrasound
Obstet Gynecol 18:76, 20010.
18. Gattuso J, Patton MA, Baraitser M: The clinical spectrum of the Fraser
syndrome: Report of three new cases and review. J Med Genet 24:549, 1987.
19. Grayson M: Diseases of the Cornea, pp 27–43. St. Louis, Mosby, 1883.
20. Salmon J, Wallis C, Murray A: Variable expressivity of autosomal dominant
microcornea with cataract. Arch Ophthalmol 106:505, 1988.
21. Fukuchi T, Ueda J, Hara H et al: Glaucoma with microcornea; morphometry
and differential diagnosis. Nippon Ganka Gakkai Zasshi 102:746, 1998.
22. Schuller M, Barnett M, Strassburger K et al: Oculodentodigital dysplasia.
Oral Surg Oral Med Oral Pathol 61:418, 1986.
23. Cameron JA: Corneal abnormalities in Ehlers-Danlos syndrome type VI.
Cornea 12:54, 1993.
24. Seemanova E, Lesny I: X-linked microcephaly, microophthalmia, microcornea,
congenital cataract, hypogenitalism, mental deficiency, growth retardation,
spasticity. Possible new syndrome. Am J Med Genet 66:178, 1996.
25. Saatci A, Soylev M, Kavukcu S et al: Bilateral megalocornea with unilateral
lens subluxation. Ophthalmic Paediatr Genet 18:35, 1997.
26. Meire F, Delleman J: Biometry in X-lined megalocornea: Pathognomonic
findings. Br J Ophthalmol 78:781, 1994.
27. Skuta G, Sugar J, Ericson E: Corneal endothelial cell measurements
in megalocornea. Arch Ophthalmol 101:51, 1983.
28. Eriksson AW, Lehmann W, Forsius J: Congenital cornea plana in Finland.
Clini Genet 4:301, 1973.
29. Forsius H, Damsten M, Eriksson AW et al: Autosomal recessive cornea
plana. A clinical and genetic study of 78 cases in Finland. Acta Ophthalmol
Scand 76:96, 1998.
30. Tahvanainen E, Forsius H, Karila E et al: Cornea plana congenita gene
assigned to the long arm of chromosome 12 by linkage analysis. Genomics
26:290, 1995.
31. Tahvanainen E, Villanueva AS, Forsius H et al: Dominantly and recessively
inherited cornea plana congenita map to the same small region of chromosome
12. Genome Res 6:249, 1996.
32. Forsius H, Demsten M, Eriksson AW et al: Autosomal recessive cornea
plana. A clinical and genetic study of 78 cases in Finland. Acta Ophthalmol
Scand 76:196, 1998.
33. Pellegata NS, Dieguez-Lucena JL, Joensuu T et al: Mutations in KERA,
encoding keratocan, cause cornea plana. Nature Genet 25:91, 2000.
34. Lehmann OJ, El-ashry MF, Ebenezer ND et al: A novel keratocan mutation
causing autosomal recessive cornea plana. Invest Ophthalmol Vis Sc 42:3118,
2001.
35. Fishman A, Ackerman J, Kanarek I et al: Cornea plana. A case report.
Ann Ophthalmol 14:47, 1982.
36. Vesaluoma MH, Sankila EM, Gallar J et al: Autosomal recessive cornea
plana: In vivo corneal morphology and corneal sensitivity. Invest Ophthalmol
Vis Sci 41:2120, 2000.
37. Cavara V: Keratoconus and keratoglobus. A contribution to the nosological
interpretation of keratoglobus. Br J Ophthalmol 34:621, 1950.
38. Pouliquen Y, Dhermy P, Espinasse MA et al: Keratoglobus.J Fr Ophtalmol 8:43, 198539. Cameron JA: Keratoglobus. Cornea 12:124, 1993. 39.
40. Jacobs D, Green W, Maumenee A: Acquired keratoglobus. Am J Ophthalmol
77:393, 1974.
41. Karabatsas CH, Cook SD: Topographic analysis in pellucid marginal
corneal degeneration and keratoglobus. Eye 10:451, 1996.
42. Sega, P, Insull W, Chambless L et al:, The association with corneal
arcus and antelasma. The Lipid Research Clinics Program Prevalence Study.
Circulation 73:1108, 1986.
43. Chambless L, Fuchs F, Linn S et al: The association of corneal arcus
with coronary heart disease and cardiovascular disease mortality in the
Lipid Research Clinics Mortality Follow-up Study. Am J Public Health 80:1200,
1990.
44. Datubo-Brown D: Keloids. A review of the literature. Br J Plast Surg
43:70, 1990.
45. Holbach L, Font R, Shivitz I et al: Bilateral keloid-like myofibroblastic
proliferations of the cornea in children. Ophthalmology 97:1188, 1990.
46. Cibis G, Tripathi R, Tripathi B et al: Corneal keloid in Lowe's syndrome.
Arch Ophthalmol 100:1795, 1982.
47. Meija L, Acosta C, Santamaria J: Clinical, surgical and histopathologic
characteristics of corneal keloid. Cornea 20:421, 2001.
48. Stone D, Kenyon K, Green W et al: Congenital central corneal leukoma
(Peters' anomaly). Am J Ophthalmol 81:173, 1976.
49. Fogle J, Green W, Kenyon K et al: Peripheral Peters anomaly: A histopathologic
case report. Pediatr Ophthalmol Strabismus 15:71, 1978.
50. Heckenlively J, Kielar R: Congenital perforated cornea in Peters'
anomaly. Am J Ophthalmol 88:63, 1979.
51. Varley M, Grossniklaus H, Lass J: Corneal perforation at birth secondary
to Peters' anomaly. Am J Ophthalmol 104:303, 1987.
52. Brownstein S, Kirkham T, Kalousek D: Bilateral renal agensis with
multiple congenital ocular anomalies. Am J Ophthalmol 82: 770, 1976.
53. Ginsberg J, Buchino J, Menefee M et al: Multiple congenital ocular
anomalies with bilateral agenesis of the urinary tract. Ann Ophthalmol
11:1021, 1979.
54. Abu-Judeh HH, Methratta S: Intestinal malrotation: Another systemic
anomaly associated with Peters' syndrome. J Pediatr Ophthalmol Strabismus
37:313, 2000.
55. Green J, Johnson G: Congenital cataract with micro-cornea and Peters'
anomaly as expression of one autosomal dominant gene. Ophthalmic Paediatr
Genet 7: 187, 1986.
56. Polack F, Graue E: Scanning electron microscopy of congenital corneal
leukomas (Peters' anomaly). Am J Ophthalmol 88:169, 1979.
57. Kuper C, Kuwabara K, Stark W: The histopathology of Peters' anomaly.
Am J Ophthalmol 80:653, 1975.
58. Lee C, Yue B, Robin J: Immunohistochemical studies of Peters' anomaly.
Ophthalmology 96:958, 1989.
59. Churchill A, Booth A, Anwar R et al: PAX 6 is normal in most cases
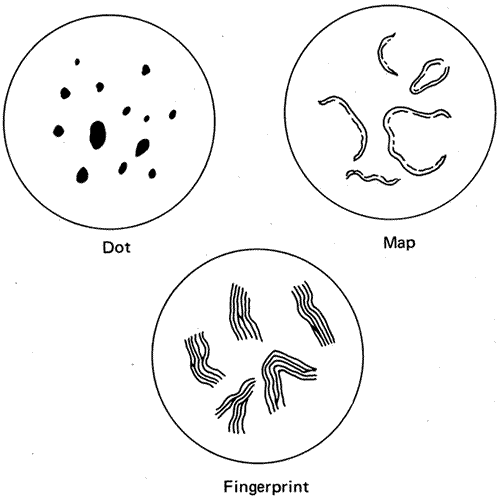
of Peters' anomaly. Eye 12:299, 1998.
60. Ozeki H, Shirai S, Nozaki M et al: Ocular and systemic features of
Peters' anomaly. Graefes Arch ClinExp Ophthalmol 238:833, 2000.
61. Streeten B, Karpik A, Spitzer K: Posterior keratoconus associated
with systemic abnormalities. Arch Ophthalmol 101:616, 1983.
62. Krachmer J, Rodrigues M: Posterior keratoconus. Arch Ophthalmol 96:1867,
1978.
63. Rao S, Padmanabhan P: Posterior keratoconus. An expanded classification
scheme based on corneal topography. Ophthalmology 105:1206, 1998.
64. Alward WL: Axenfeld-Rieger syndrome in the age of molecular genetics.
Am J Opthalmol 130:107, 2000.
65. Riise R, Storhaug K, Brondum-Nielsen K: Rieger's syndrome is associated
with PAX6 deletion. Acta Ophthalmol Scand 79:201, 2001.
66. Shields MB, Buckley E, Klintworth GK: Axenfeld-Rieger syndrome. A
spectrum of developmental disorders. Surv Ophthalmol 29:387, 1985.
67. Burian H, Braley A, Braley AL: Visibility of the ring of Schwalbe
and the trabecular zone: An interpretation of the posterior corneal embryotoxon
and the so-called congenital hyalin membranes on the posterior corneal
surface. Arch Ophthalmol 53:767, 1955.
68. Ozeki H, Shirai S, Majima A et al: Clinical evaluation of posterior
embryotoxon in one institution. Jpn J Ophthalmol 41:422, 1997.
69. Loewenstein A: “Knobs” at the periphery of Descemet's
membrane. Br J Ophthalmol 34:246, 1950.
70. Shields M, Buckley E, Klintworth G et al: Axenfeld-Rieger syndrome.
A spectrum of developmental disorders. Surv Ophthalmol 29:387, 1985.
71. Shields M: Axenfeld-Rieger syndrome: A theory of mechanism and distinctions
from the iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc 81:736,
1983.
72. Chisholm I, Chudley A: Autosomal dominant iridogoniodysgenesis with
associated somatic anomalies: Four-generation family with Rieger's syndrome.
Br J Ophthalmol 67:529, 1983.
73. Schanzlin D, Robin J, Erickson G et al:, Histopathologic and ultrastructural
analysis of congenital corneal staphyloma. Am J Ophthalmol 95:506, 1983.
74. Leff S, Shields JAugsburger J et al: Congenital corneal staphyloma:
Clinical, radiological and pathologic correlation. Br J Ophthalmol 70:427,
1986.
75. Pallikaris I, Kymionis G, Astyrakakis N: Corneal ectasia induced by
laser in situ keratomileusis. J Cataract Refract Surg 27:1796, 2001.
76. Elliott J, Feman S, O'Day D et al: Hereditary sclerocornea. Arch Ophthalmol
103:676, 1985.
77. Waring GO3rd , Rodrigues MM: Ultrastructure and successful keratoplasty
of sclerocornea in Mietens' syndrome. Am J Opthalmol 90:469, 1980.
78. Kim T, Cohen EJ, Schnall BM et al: Ultrasound biomicroscopy and histopathology
of sclerocornea. Cornea 17:443, 1998.
79. Kasner L, Mietz H, Green WR: Agenesis of Bowman's layer. A histopathological
study of four cases. Cornea 12:163, 1993.
80. Moriarty AP, Kerr-Muir MG: Sclerocornea and interstitial deletion
of the short arm of chromosome 6—(46XY del[6 [p22, p24). J Pediatr
Ophthalmol Strabismus 29:177, 1992.
81. Lindor NM, Michels VV, Hoppe DA et al: Xp22.3 microdeletion syndrome
with microphthalmia, sclerocornea, linear skin defects, and congenital
heart defects. Am J Med Genet 44:61, 1992.
82. Cox TC, Cox LL, Ballabio A: A very high density microsatellite map
(1 STR/41 kb) of 1.7 Mb on Xp22 spanning the microphthalmia with linear
skin defects (MLS) syndrome critical region. 7Eur J Hum Genet 6:406, 1998.
83. Henkind P, Marinoff G, Manas A et al: Bilateral corneal dermoids.
Am J Ophthalmol 76:972, 1973.
84. Mann I: Developmental Anomalies of the Eye, p 357. Philadelphia, Lippincott, 1957.
85. Sugar H: The oculoauriculo-vertebral syndrome of Goldenhar. Am J Ophthalmol
62:678, 1966.
86. Mandelcorn M, Merin S, Cardarelli J: Goldenhar's syndrome and phocomelia:
Case report and etiologic considerations. Am J Ophthalmol 72:618, 1966.
87. Margolis S, Aleksic S, Charles N: Retinal and optic nerve findings
in Goldenhar-Gorlin syndrome. Ophthalmology 91:1327, 1984.
88. Jones B: Thygeson's superficial punctate keratitis. Trans Ophthalmol
Soc UK 89:22, 1963.
89. Goldberg DB, Scanlon D, Brown S: Management of Thygeson's superficial
punctate keratitis. Am J Ophthalmol 89:22, 1980.
90. Goto E, Shimmura S, Shimazai, J et al: Treatment of superior limbic keratoconjunctivitis by application of autologous
serum.Cornea 20:807, 2001.
91. Theodore F, Ferry A: Superior limbic keratoconjunctivitis: Clinicopathologic
correlations. Arch Ophthalmol 84:481, 1970.
92. Vastine D: Viral Diseases: Adenovirus and miscellaneous viral infections.
In Smolin G, Thoft R (eds): Cornea, p 216. Boston, Little, Brown and Company,
1987.
93. Hammer L, Perry H, Donnenfeld E et al: Symblepharon formation in epidemic
keratoconjunctivitis. Cornea 4:338, 1990
94. Laibson PR., Dhiri S, Oconer J: Corneal infiltrates in epidemic keratoconjunctivitis.
Response to double-blind corticosteroid therapy. Arch Ophthalmol 84:36,
1970.
95. T'ang F, Chang H, Wank K: Studies on the etiology of trachoma with
special reference to isolation of viris in the chick embryo. Clini Med
J 75:429, 1957.
96. Dawson C, Juster R, Marx R et al: Limbal disease in trachoma and other
chlamydial infections: Risk factors for corneal neovascularization. Eye
3:204, 1989.
97. Hidayat A, Risco JM: Amyloidosis of corneal stroma in patients with
trachoma. A clinicopathologic study of 62 cases. Ophthalmology 96:203,
1989.
98. Ffytche T: Leprosy: Hansen's disease. In Gold D, Weingeist T (eds):
The Eye in Systemic Disease Philadelphia, Lippincott, 1990.
99. Alien J, Byers J: The pathology of leprosy. I. Cornea.Arch Ophthalmol 64:216, 1960.
100. Foulks G, Sanfilippo F, Locascio J: Histocompatibility testing for
keratoplasty in high-risk patients. Ophthalmology 90:230, 1893.
101. Passo M, Rosenbaum J: Ocular syphilis in patients with human immunodeficiency
virus infection. Am J Ophthalmol 97:281, 1988.
102. Duke-Elder S: Diseases of the outer eye. In Duke-Elder (ed): System of Ophthalmology, pp 236–242. St. Louis, Mosby, 1965. 103. Tamesis R, Foster C: Ocular syphilis.Ophthalmology 97:1281, 1990.
104. Waring GO3rd , Font R, Rodrigues MM et al: Alterations of Descemet's
membrane in interstitial keratitis. Am J Ophthalmol. 81:773, 1976.
105. Scattergood K, Breen E, Hirst L: Scrolls of Descemet's membrane in
healed syphilitic interstitial keratitis. Ophthalmology 90:1518, 1983.
106. Steere A, Malawista S, Snydman D et al: Lyme arthritis: An epidemic
of oligoarticular arthritis in children and adults in three Connecticut
communities. Arthritis Rheum 20: 7, 1977.
107. Winterkorn J: Lyme disease: Neurologic and ophthalmic manifestations.
Surv Ophthalmol 35:191, 1990.
108. Suresh P, Campbell I, Herzig S et al: Myobacterium keratitis following
hyperopic laser in situ keratomileusis. Can J Ophthalmol 36:194, 2001.
109. Lennarson P, Barney N: Interstitial keratitis as presenting ophthalmic
sign of sarcoidosis in a child. J Pediatr Ophthalmol Strabismus 32:194,
1995.
110. Basanez M, Boussinesq M: Population biology of human onchocerciasis.
Philos Trans R Soc Lond B Biol Sci 354:809, 1999.
111. Hubbard A, Centifanto-Fitzgerald Y: Variability among HSV-1 strains and its importance in disease. In Ninth International Herpes Virus Workshop. 1984. Seattle, 1984. 112. Collin B, Abelson M: Herpes simplex virus in humanArch Ophthalmol 94: 1726, 1976.
113. Green WR, Zimmerman L: Granulomatous reactions to Descemet's membrane.
Am J Ophthalmol 64:555, 1967.
114. Naumann G, Gass J, Font R: Histopathology of herpes zoster ophthalmicus.
Am J Opthalmol 65:533, 1968.
115. Allen N, Cox C, Cobo M et al: Use of immunosuppressive agents in
the treatment of severe ocular and vascular manifestations of Cogan's
syndrome. Am J Med 88:296, 1990.
116. Cobo L, Haynes B: Early corneal findings in Cogan's syndrome. Ophthalmology
91:903, 1984.
117. Blaustein B, Gurwood A: Recurrent phlyctenular keratoconjunctivitis:
A forme fruse manifestation of rosacea. Optometry 72:179, 2001.
118. Jones D: A plan for antimicrobial therapy. Trans Am Acad Ophthalmol
Otolaryngol 79:95, 1975.
119. Musch D, Sugar A, Meyer R: Demographic and predisposing factors in
corneal ulceration. Arch Ophthalmol 101:1545, 1983.
120. Pavan-Langston D: Herpetic infections in the cornea. In Smolin G (ed): Cornea, pp 240–254. Boston, Little, Brown and Company, 1987.
121. Font R: Chronic ulcerative keratitis caused by herpes simplex. Arch
Ophthalmol 90:382, 1973
122. Ishibashi Y, Kaufman HE: Corneal biopsy in the diagnosis of keratomycosis.
Am J Ophthalmol 101:288, 1986.
123. Stehr-Green J, Bailey T, Visvesvara : The epidemiology of Acanthamoeba
keratitis in the United States. Am J Ophthalmol 107:331, 1989.
124. Moore M, McCulley J, Kaufman HE et al: Radial keratoneuritis as a
presenting sign of Acanthamoeba keratitis. Ophthalmology. 93:1310, 1986.
125. Wilhelmus K, Osato M, Font R: Rapid diagnosis of Acanthamoeba keratitis
using Calcofluor white. Arch Ophthalmol 104:1309, 1986.
126. Kuwabara T, Ciccarelli E: Meesmann's corneal dystrophy. A pathological
study. Arch Ophthalmol 71:676, 1964.
127. Nakanishi I, Brown SI: Ultrastructure of the epithelial dystrophy
of Meesmann. Arch Ophthalmol 93:259, 1975
128. Irvine AD, Corden LD, Swensson O et al: Mutations in cornea-specific
keratin K3 or K12 genes cause Meesmann's corneal dystrophy. Nat Genet
16:184, 1997.
129. Corden L, Swensson O, Swensson B et al: Molecular genetics of Meesmann's
corneal dystrophy. Ancestral and novel mutations in keratin 12 (K12) and
complete sequence of the human KRT12 gene. Exper Eye Res , 7041, 2000.
130. Rodrigues MM, Fine B, Laibson PR et al: Disorders of the corneal
epithelium. A clinicopathologic study of dot, geographic and finger print
patterns. Arch Ophthalmol 92:475, 1974.
131. Cogan D, Donaldson D, Kuwabara T et al: Microcystic dystrophy of
the corneal epithelium. Trans Am Ophthalmol Soc 62:213, 1964.
132. Lisch W, Steuhl KP, Lisch C et al: A new band shaped and whorled
microcystic dystrophy of the corneal epithelium. Am J Ophthalmol 114:35,
1992.
133. Cibis G, Tripathi R, Harris D: Mucolipidosis I. Birth Defects 18:359,
1982.
134. Lisch W, Buttner A, Oeffner F et al: Lisch corneal dystrophy is genetically distinct form Meesmann corneal dystrophy
and maps to xp22.3. Am J Ophthalmol 130:461, 2000.
135. Grayson M, Wilbrandt H:, Dystrophy of the anterior limiting membrane
of the cornea (Reis-Bucklers type). Am J Ophthalmol 61:345, 1966.
136. Kuchle M, Green WH, Volcker H: Reevaluation of corneal dystrophies
of Bowman's layer and anterior stroma (Reis-Bucklers and Thiel-Behnke
types): A light and electron microscopic study of eight corneas and a
review of the literature. Cornea 14:333, 1995.
137. Perry H, Fine B, Caldwell D: Reis-Bucklers' dystrophy. Arch Ophthalmol
97:644, 1979.
138. Munier F, Korvatska E, Djemai A et al: Kerato-epithelin mutations
in four 5q31-linked corneal dystrophies. Nat Genet 15:247, 1997.
139. Yee R, Sullivan L, La H et al: Linkage mapping of Thiel-Behnke corneal
dystrophy (CDB2) to chromosome 10q23-q24. Genomics 46:152, 1997.
140. Streeten BW, Qi Y, Klintworth GK et al: Immunolocalization of beta
ig-h3 protein in 5q31-linked corneal dystrophies and normal corneas. Arch
Ophthalmol 117:67, 1999.
141. Kocak-Altintas A, Kocak-Midillioglu I, Alakreu A et al: BIGH3 gene
analysis in the differential diagnosis of corneal dystrophies. Cornea
20:64, 2001.
142. Stone E, Mathers W, Rosenwasser G et al: Three autosomal dominant
corneal dystrophies map to chromosome 5q. Nat Genet 6:47, 1994.
143. Rodrigues MM, McGavic J: Recurrent corneal granular dystrophy: A
clinicopathologic study. Trans Am Ophthalmol Soc 73:306, 1975.
144. Stuart J, Mund M, Iwamoto T et al: Recurrent granular corneal dystrophy.
Am J Ophthalmol 79:18, 1975.
145. Herman S, Hughes W: Recurrence of hereditary corneal dystrophy following
keratoplasty. Am J Ophthalmol 75:689, 1973.
146. Lanier J, Fine M, Togni B: Lattice corneal dystrophy. Arch Ophthalmol
94:921, 1976.
147. Klintworth GK: Proteins in ocular disease. In GarnerA, Klintworth GK (eds): Pathobiology of Ocular Disease. A Dynamic Approach, pp 999–1000. New York, Marcel Dekker, 1982.
148. Klintworth GK: Advances in the molecular genetics of corneal dystrophies.
Am J Ophthalmol 128:747, 1999.
149. Haddad AM, Font R, Fine B: Unusual superficial variant of granular
dystrophy of the cornea. Am J Ophthalmol 83:213, 1977
150. Jones S, Zimmerman L: Histopathologic differentiation of granular,
macular and lattice dystrophies of the cornea. Am J Ophthalmol 51:394,
1961.
151. Garner A: Histochemistry of corneal granular dystrophy.Br J Ophthalm 53:799, 1969.
152. Brownstein S, Fine B, Sherman M et al: Granular dystrophy of the
cornea: Light and electron microscopic confirmation of recurrence in a
graft. Am J Ophthalmol 77:701, 1974
153. Iwamoto T, Stuart J, Srinivasan B et al: Ultrastructural variation in granular dystrophy of the cornea.Graefes Archiv Clin Exp Ophthalmol 194:1, 1975.
154. Klintworth GK, Vogel F: Macular corneal dystrophy: An inherited acid
mucopolysaccharide storage disease of the corneal fibroblast. Am J Pathol
45:565, 1981.
155. Thonar E, Meyer R, Dennis R et al: Absence of normal keratin sulfate
in blood of patients with macular corneal dystrophy. Am J Ophthalmol 102:561,
1986.
156. Donnenfeld E, Cohen EJ, Ingraham H et al: Corneal thinning in macular
corneal dystrophy. Am J Ophthalmol 101:112, 1986.
157. Dubord P, Krachmer JH: Diagnosis of early lattice corneal dystrophy.
Arch Ophthalmol 100:788, 1982.
158. Stansbury F: Lattice type of hereditary corneal degeneration: A report
of five cases, including one of a child of two years. Arch Ophthalmol
40:189, 1948.
159. Meretoja J: Familial systemic paramyloidosis with lattice dystrophy
of the cornea, progressive cranial neuropathy, skin changes and various
internal symptoms. I. A previously unrecognized heritable syndrome. Ann
Clin Res 1:314, 1969.
160. Meretoja J: Comparative histopathological and clinical findings in
eyes with lattice corneal dystrophy of two different types. Ophthalmologica
165:15, 1972.
161. Kivela T, Tarkkanen A, Frangione B et al: Ocular amyloid deposition
in familial amyloidosis, Finnish: an analysis of native and variant gelsolin
in Meretoja's syndrome. Invest Ophthalmol Vis Sci 35:3759, 1994.
162. Seitz, Weidle EG, Naumann G: Unilateral type II (Hida) lattice stromal
corneal dystrophy. Klin MonatsblAugenheilk 203:279, 1993.
163. Yanoff M, Fine B, Colosi N et al: Lattice corneal dystrophy.Arch Ophthalmol 95:651, 1977.
164. Freddo T, Polack FM, Leibowitz H: Ultrastructural changes in posterior
layers of the cornea in Schnyder's crystalline dystrophy. Cornea 8:170,
1989.
165. Burns R, Connor W, Gipson I: Cholesterol turnover in hereditary corneal
dystrophy of Schnyder. Trans Am Ophthalmol Soc 76:184, 1978.
166. Shearman A, Hudson T, Andersen J et al: The gene for Schnyder's crystalline
corneal dystrophy maps to human chromosome 1p34. 1-p36. Hum Mol Genet
5:1667, 1996.
167. Kiskadon B, Campbell R, Waller R et al: Fleck dystrophy of the cornea:
Case report. Ann Ophthalmol 12:700, 1980.
168. Nicholson D, Green WR, Cross H et al: A clinical and histopathologic
study of Francois-Neetens speckled corneal dystrophy. Am J Ophthalmol
83:554, 1977.
169. Witschel H, Fine B, Grutzner P et al: Congenitial hereditary stromal
dystrophy of the cornea. Arch Ophthalmol 96:1043, 1978.
170. Krachmer JH, Feder FS, Belin MW: Keratoconus and related noninflammatory
corneal thinning disorders. Surv Ophthalmol 1984. 28: p. 293.
171. Rabinowitz W, Maumenee I, Lundergan M et al: Molecular genetic analysis
in autosomal dominant keratoconus. Cornea 11:302, 1992.
172. Morrison D, Rosser E, Clouue C: Keratoconus associated with a chromosome
7,11 translocation. Eye 15:556, 2001.
173. Kenyon KR: Ocular manifestations and pathology of systemic mucopolysaccharidoses.
Birth Defects 12:133, 1976.
174. Gahl W, Bashan N, Tietze F et al:, Cystine transport is defective
in isolated leukocyte lysosomes from patients with cystinosis. Science
217:1263, 1982.
175. Group TCCR: Linkage of the gene for cystinosis to markers on the
short arm of chromosome 17. Nat Genet 10:246, 1995.
176. Sanderson P, Kuwabara T, Stark W: Cystinosis: A clinical, histopathologic
and ultrastructural study. Arch Ophthalmol 19:270, 1974.
177. Kaiser-Kupfer M, Chan C, Rodrigues MM et al: Nephropathic cystinosis:
Immunohistochemical and histologic studies of cornea, conjunctiva and
iris. Curr Eye Res 6:617, 1987.
178. Frxier P, Wong V: Cystinosis. Arch Ophthalmol 80:87, 1968.
179. Li Q, Ashraf M, Shen D et al: The role of apoptosis in the pathogenesis
of Fuchs' endothelial dystrophy of the cornea. Arch Ophthalmol 119: 1597,
2001.
180. Lipman R, Rubenstein J, Torczynski E: Keratoconus and Fuchs' corneal
dystrophy in a patient and her family. Arch Ophthalmol 108:993, 1990.
181. Rodrigues MM, Krachmer JH, Hacket J et al: Fuchs' corneal dystrophy.
A clinicopathologic study of the variation in corneal edema. Ophthalmology
93:789, 1986.
182. Eagle RC Jr, Laibson PR, Arentsen J: Epithelial abnormalities in
chronic corneal edema: A histopathologic study. Trans Am Ophthalmol Soc
87:107, 1990.
183. Krachmer JH: Posterior polymorphous dystrophy: A disease characterized
by epithelial-like cells which influence management and prognosis. Trans
Am Ophthalmol Soc 83:413, 1985.
184. Laganowski H, Sherrard E, Uir M et al: Distinguishing features of
the iridocorneal endothelial syndrome and posterior polymorphous dystrophy:
Value of endothelial specular microscopy. Br J Ophthalmol 175:212, 1991.
185. Judisch GF, Maumenee I: Clinical differentiation of recessive congenital
hereditary endothelial dystrophy and dominant hereditary endothelial dystrophy.
Am J Ophthalmol 85:606, 1978.
186. Hand C, Harmon D, Kennedy S et al: Localization of the gene for autosomal
recessive congenital hereditary endothelial dystrophy (CHED2) to chromosome
20 by homozygosity mapping. Genomics 61:1, 1999.
187. Callaghan M, Hand C, Kennedy S et al: Homozygosity mapping and linkage
analysis demonstrate that autosomal recessive congenital hereditary endothelial
dystrophy (CHED) and autosomal dominant CHED are genetically distinct.
Br J Ophthalmol 83:115.
188. Fraunfelder F, Wright P, Tripathi R: Corneal mucus plaques. Am J
Ophthalmol 83:191, 1977.
189. Nelson J, Havener V, Cameron J: Cellulose acetate impressions of
the ocular surface. Dry eye states. Arch Ophthalmol 101:1869, 1983.
190. Dodds H Laibson PR: Filamentary keratitis following cataract extraction.
Arch Ophthalmol 88:609, 1972.
191. Fox R, Howell F, Bone R: Primary Sjogren's syndrome: Clinical and
immunologic features. Semin Arthritis Rheum 14:77, 1984.
192. Daniels T: Salivary histopathology in the diagnosis of Sjogren's
syndrome. Scand J Rheumatol 61:36, 1986.
193. Turner K, Pflugfelder S, Ji Z et al: Interleukin-6 levels in the
conjunctival epithelium of patients with dry eye disease treated with
cyclosporine ophthalmic emulsion. Cornea 19:492, 2000.
194. Sall K, Stevenson O, Mundorf T et al: Two multicenter randomized
studies of the efficacy and safety of cyclosporine ophthamic emulsion
in moderate to sever dry eye disease. CsA Phase 3 Study Group. Ophthalmology
107:631, 2000.
195. Brown N, Bron A: Recurrent erosion of the cornea.Br J Ophthalmol , 1976. 60:p. 84.
196. Franceschetti A: Hereditare rezidivierende erosion der Hornhaut.
Z Augenheilkd 66:309, 1928.
197. Kenyon KR: Recurrent corneal erosion: Pathogenesis and therapy. Int
Ophthalmol Clin 19:169, 1979.
198. Judge D, Payant J, Frase S et al: Anterior stromal micropuncture
electron microscopic changes in the rabbit cornea. Cornea 9:152, 1990.
199. Goldman J, Dohlman C, Dravit B: The basement membrane of the human
cornea in recurrent epithelial erosion syndrome. Trans Am Acad Ophthalmol
Otolaryngol 73:471, 1969.
200. Tripathi R, Bron : Ultrastructural study of non-traumatic corneal
erosion. Br J Ophthalmol 56:73, 1972.
201. Smith R, Farrell T, Bailey T: Keratomalacia. Surv Ophthalmo 20:213,
1975.
202. Sommer A: Nutritional Blindness: Xerophthalmia and Keratomalacia,
Vol 145. New York, Oxford University Press, 1982.
203. Sommer A, Sugana T, Djunaedi E et al: Vitamin A-responsive panocular
xerophthalmia in a healthy adult. Arch Ophthalmol 96:1630, 1978.
204. Sommer A, Green WR, Kenyon KR: Clinical-histopathogic correlation
in xerophthalmic ulceration and necrosis. Arch Ophthalmol 100:953, 1982.
205. Sommer A, Green WR, Kenyon KR: Bitot's spots responsive and nonresponsive
to vitamin A: Clinicopathologic correlations. Arch Ophthalmol 99:2014,
1981.
206. Cogan D, Kuwabara T: Arcus senilis: Its pathology and histochemistry.
Arch Ophthalmol 61:353, 1959.
207. Hill J, Maske : Pathogenesis of pterygium. Eye. 3:218, 1989.
208. Ioachim-Velogianni E, Tsironi E, Agnantis E: HLA-DR antigen expression
in pterygium epithelial cells and lymphocyte subpopulations: An immunohistochemistry
study. Ger J Ophthalmol 4:123, 1995.
209. Guyer D, Barraquer J, McDonnell P et al: Terrien's marginal degeneration: Clinicopathologic case reports.Graefes Arch Clini Exp Ophthalmol 225:19, 1987.
210. Pouliquen Y, Dhermy P, Renard G et al: Terrien's disease: Clinical
and ultrastructural studies, five case reports. Eye 3:791, 1989.
211. Soong H, Fitzgerald J, Boruchoff S et al: Corneal hydrops in Terrien's
marginal degeneration. Ophthalmology 93:340, 1986.
212. Brown SI: Mooren's ulcers. Histopathology and proteolytic enzymes
of adjacent conjunctiva. Br J Ophthalmol 59:670, 1975.
213. Young R, Watson P: Light and electron microscopy of corneal melting
syndrome (Mooren's ulcer). Br J Ophthalmol. 66:341, 1982.
214. Zhao J, Jin X: Immunological analysis and treatment of Mooren's ulcer
with cyclosporin A applied topically. Cornea 12:481, 1993.
215. Foulks G, Hatchell D, Proia A et al: Histopathology of silicone oil
keratopathy in humans. Cornea 10:29, 1991.
216. Binder P, Deg J, Kohl F: Calcific band keratopathy after intraocular
chondroitin sulfate. Arch Ophthalmol 105:1243, 1987.
217. Cursino J, Fine B: A histologic study of calcific and non-calcific
band keratopathies. Am J Ophthalmol 82:395, 1976.
218. Tabbara K: Climatic droplet keratopathy. Int Ophthalmol Clin 26:63,
1986.
219. Matta C, Tabbara K, Cameron JA et al: Climatic droplet keratopathy
with corneal amyloidosis. Ophthalmology 98:192, 1991.
220. Garner A, Morgan G, Tripathi R: Climatic droplet keratopathy. II.
Pathologic findings. Arch Ophthalmol 75:799, 1973.
221. Brownstein S, Rodrigues MM, Fine B et al: The elastotic nature of
hyaline corneal deposits. Am J Ophthalmol 75:799, 1973.
222. Garner A, Fraunfelder F, Barras T et al: Spheroidal degeneration
of cornea and conjunctiva. Br J Ophthalmol 60:473, 1976.
223. Ferry A: A “new” iron line of the superficial cornea:
Occurrence in patients with filtering blebs. Arch Ophthalmol 79:142, 1968.
224. Rodman R, Burnstine M, Esmaeli B et al: Wilson's disease: Presymptomatic
patients with Kayser-Fleischer rings. Ophthalmic Genet 18:79, 1997.
225. Ferry A, Zimmerman L: Black cornea: A complication of topical use
of epinephrine. Am J Ophthalmol 58:205, 1964.
226. Calkins L: Corneal epithelial changes occurring during chloroquine
(Aralyn) therapy. Arch Ophthalmol 60:981, 1958.
227. Slowik C, Samodi S, von Gruben C et al: Detection of morphological
corneal changes caused by chloroquine thereapy using confocal in vivo
microscopy. Ophthalmologe 94:549, 1997.
228. D'Amico DJ, Kenyon KR, Ruskin JN: Amiodarone keratopathy: Drug-induced
lipid storage disease. Arch Ophthalmol 99:257, 1981.
229. Ciancaglini M, Carpineto P, Zuppardi E, Nubile M et al: In vivo confocal
microscopy of patients with amiodarone-induced keratopathy. Cornea 20:368,
2001.
230. Mantyjarvi M, Tuppurainen K, Ikaheimo K: Ocular side effects of amiodarone.
Surv Ophthalmol 42:360, 1998.
231. Dolan BJ, Flach AJ, Peterson JS: Amiodarone keratopathy and lens
opacities. J Am Optom Assoc 56:468, 1985.
232. Pollak PT: Clinical organ toxicity of antiarrhythmic compounds: Ocular
and pulmonary manifestations. Am J Cardiol 84:37R, 1999.
233. Macaluso DC, Shults WT, Fraunfelder FT: Features of amiodarone-induced
optic neuropathy. Am J Opthalmol 127:610, 1999.
234. Fine B, Yanoff M: Ocular Histology: A Text and Atlas, pp 159–165, Vol 168, New York, Harper & Row, 1972.
235. Hamanaka T: Scleral spur and ciliary muscle in man and monkey. Jpn
J Ophthalmol 33:221, 1989.
236. Dutton J, Anderson R, Schelper R et al: Orbital malignant melanoma
and oculodermal melanocytosis: Report of two cases and review of the literature.
Ophthalmology 91:497, 1984.
237. Sugar H, Ilgren E, Adams C: Nevus of Ota associated with meningeal
melanosis and intracranial melanoma. Case report. J Neurosurg 58:280,
1983.
238. Mansour AM, Barber JC, Reinecke RD et al: Ocular choristomas. Surv
Ophthalmol 33:330, 1989.
239. Hered R, Hiles D: Epibulbar osseous choristoma and ectopic lacrimal
gland underlying a dermolipoma. J Pediatr Ophthalmol Strabismus 24:255,
1987.
240. Conway V, Brownstein S, Chisholm I: Lacrimal gland choristoma of
the ciliary body. Ophthalmology 92:449, 1985.
241. Pittke E, Marquardt R, Mohr W: Cartilage choristoma of the eye. Arch
Ophthalmol 101:1569, 1983.
242. Shields JA, Shields C, Eagle RC Jr et al: Ocular manifestations of
the organoid nevus syndrome. Ophthalmology 104:549, 1997.
243. Khalil M: Subhyaloid hemorrhage in osteogenesis imperfecta tarda.
Can J Ophthalmol 18:251, 1983.
244. Howard F: Lax ligament syndrome in children associated with blue
sclera and bat ears. Br J Gen Pract 40:233, 1990.
245. Zlotogora J, BenEzra D, Cohen T et al: Syndrome brittle cornea, blue
sclera, and joint hyperextensibility. Am J Med Genet 36:269, 1990.
246. Pouliquen Y, Dhermy P, Espinasse MA et al: Keratoglobe. J Fr Ophtalmol
8:43, 1985.
247. Zimmerman L, Fischer R, Winterhalter K et al: Comparative studies
of collagens in normal and keratoconus corneas. Exp Eye Res 46:431, 1988.
248. Sussman M, Kelly T, Rosenbaum K et al: Abnormality of cartilage collagen
in a patient with unclassified chondrodystrophy. J Orthop Res 2:339, 1984.
249. Roxburgh S: Atypical retinitis pigmentosa with hypophosphatasia.
Trans Ophthalmol Soc UK 103:513, 1983.
250. Chan C, Green WR, de la Cruz Z et al: Ocular findings in osteogenesis
imperfecta congenita. Arch Ophthalmol 100:1459, 1982.
251. Mori S, Komatsu H, Watari H: Spontaneous posterior bulbar perforation
of congenital scleral coloboma and its surgical treatment: A case report.
Ophthalmic Surg 16:433, 1985.
252. Gass J: Uveal effusion syndrome: A new hypothesis concerning pathogenesis
and technique of surgical treatment. Trans Am Ophthalmol Soc 81:246, 1983.
253. Brockhurst R: Nanophthalmos with uveal effusion. Trans Am Ophthalmol
Soc 72:371, 1974.
254. Hotz F, Boehmer H, Mechtersheimer G et al: Uveal non-Hodgkin's lymphoma
with epibulbar extension simulating choroidal effusion syndrome. Retina
19:343, 1999.
255. Donoso L, Shields JA, Nagy R: Epibulbar lesions simulating extraocular
extension of uveal melanomas. Ann Ophthalmol 14:120, 1982.
256. Manschot W: Senile scleral plaques and senile scleromalacia. Br J
Ophthalmol 62:376, 1978.
257. Carroll C, Peyman G, Raichand M: Surgical management of senile scleromalacia.
Ophthalmic Surg 11:719, 1980.
258. Klintworth GK: Radiographic abnormalities in eyes with retinoblastoma
and other disorders. Br J Ophthalmol 62:365, 1978.
259. Friedman E, Ivry M, Ebert E et al: Increased scleral rigidity and
age-related macular degeneration. Ophthalmology 96:104, 1989.
260. Norn T: Topography of scleral emissaries and sclera-perforating blood
vessels. Acta Ophthalmol (Copenh) 63:320, 1985.
261. Pollak M, Chou Y, Cerda J et al: Homozygosity mapping of the gene
for alkaptonuria to chromosome 3q2. Nat Genet 5:201, 1993.
262. Daicker B, Riede U: Histological and ultrastructural findings in
alkaptonuric ocular ochronosis. Ophthalmologica 69:277, 1974.
263. Kampik A, Sani J, Green WR: Ocular ochronosis. Clinicopathological,
histochemical, and ultrastructural studies. Arch Ophthalmol 98:1441, 1980.
264. Gaines J: The pathology of alkaptonuric chronosis. Hum Pathol 20:40,
1989.
265. Phinney R, Mondino B, Abrahim A: Corneal icterus resulting from stroma
bilirubin deposition. Ophthalmology 96:1212, 1989.
266. Lascari A: Carotenemia. A review. Clin Pediatr 20:25, 1981.
267. Congdon P, Kelleher J, Edwards P et al: Benign carotenaemia in children.
Arch Dis Child 56:292, 1981.
268. Angeloni V, Salasche S, Ortiz R: Nail, skin, and scleral pigmentation
induced by minocycline. Cutis 40:229, 1987.
269. Henderson R, Lander R: Scleral discoloration associated with long-term
prednisone administration. Cutis 34:76, 1984.
270. Watson P, Young R: Changes at the periphery of a lesion in necrotizing
scleritis: Anterior segment fluorescein angiography correlated with electron
microsocpy. Br J Ophthalmol 69:656, 1985.
271. Flach A, Lavoie P: Episcleritis, conjunctivitis, and keratitis as
ocular manifestations of Lyme disease. Ophthalmology 97:973, 1990.
272. Zaidman GW: Episcleritis and symblepharon associated with Lyme keratitis.
Am J Ophthalmol 109:487, 1990.
273. McCluskey P, Wakefield D, Penny R: Scleritis and the spectrum of
external inflammatory eye disease. Aust N Z J Ophthalmol 13:159, 1985.
274. Kleiner R, Raber I, Passero F: Scleritis, pericarditis, and aortic
insufficiency in a patient with rheumatoid arthritis. Ophthalmology 91:941,
1984.
275. Robertson D, Winkelman R: Ophthalmic features of necrobiotic xantogranuloma
with paraproteinemia. Am J Ophthalmol 97:173, 1984.
276. Lyne A, Pitkeathley D: Episcleritis and scleritis: Association with
connective tissue disease. Arch Ophthalmol 80:171, 1968.
277. McGavin D, Williamson J, Forrester J: Episcleritis and scleritis:
A study of their clinical manifestations and association with rheumatoid
arthritis. Br J Ophthalmol 60:192, 1976.
278. Wilhelmus K, Yokoyama C: Syphilitic episcleritis and scleritis. Am
J Ophthalmol 104:595, 1987.
279. Verhoeff F, King M: Scleromalacia perforans: A report of a case in
which the eye was examined microscopically. Arch Ophthalmol 20:1013, 1938.
280. Ashton N, Hobbs H: Effect of cortisone on rheumatoid nodules of the
sclera (scleromalacia perforans). Br J Ophthalmol 36:373, 1976.
281. Ferry A: Histopathology of rheumatoid episcleral nodules: An extraarticular
manifestation of rheumatoid arthritis. Arch Ophthalmol 82:77, 1969.
282. Fraunfelder F, Watson P: Evaluation of eyes enucleated for scleritis.
Br J Ophthalmol 60:227, 1976.
283. Lebowitz M, Jakobeic F, Donnenfeld E et al: Bilateral epibulbar rheumatoid
nodulosis. A new ocular entity. Ophthalmology 95:256, 1988.
284. Lindenmuth K, Sugar A, Kincaid M et al: Invasive squamous cell carcinoma
of the conjunctiva presenting as necrotizing scleritis and scleral perforation
and uveal prolapse. Surv Ophthalmol 33:50, 1988.
285. Kim R, Seiff S, Howes E et al: Necrotizing scleritis secondary to
squamous cell carcinoma in acquired immunodeficiency syndrome. Am J Ophthalmol
109:231, 1990.
286. Kaufman L, Folk E, Miller M et al: Necrotizing scleritis following
strabismus surgery for thyroid ophthalmopathy. J Pediatr Ophthalmol Strabismus
26:236, 1989.
287. Young R, Watson P: Microscopical studies of necrotizing scleritis.
II. Collagen degradation in the scleral stroma. Br J Ophthalmol 68:781,
1984.
288. Rao N, Marak G, Hidayat A: Necrotizing scleritis. A clinicopathologic
study of 41 cases. Ophthalmology 92:1542, 1985.
289. Young R, Watson P: Microscopical studies of necrotizing scleritis.
I. Cellular aspects. Br J Ophthalmol 68:770, 1984.
290. Mamalis N, Johnson M, Haines J et al: Corneal-scleral melt in association
with cataract surgrey and intraocular lenses: A report of four cases.
J Cataract Refract Surg 16:108, 1990.
291. Finger P, Perry H, Packer S et al: Posterior scleritis, as an intraocular
tumor. Br J Ophthalmol 74:21, 1990.
292. Benson W: Posterior scleritis. Surv Ophthalmol 32:297, 1988.
293. Singh G, Guthoff R, Foster C: Observations on long-term follow-up
of posterior sceleritis. Am J Ophthalmol 101:570, 1986.
294. Bernauer N, Buchi E, Daicker B: Immunopathological findings in posterior
scleritis. Int Ophthalmol 18:229, 1994.
295. Shields C, Shields JA, Varenhorst M: Transscleral leiomyoma. Ophthalmology
98:84, 1991.
|