ANTERIOR SEGMENT
Conjunctival Sickle Sign
Abnormalities of the bulbar conjunctival blood vessels provide direct evidence of the vaso-occlusive process and were one of the earliest reported ocular changes.38–43 These abnormalities are believed to be the result of flow obstruction or impedance by sickled cells. The severity of the conjunctival changes ranges from linear dilatations to isolated groups of truncated, comma-shaped segments. These changes have been correlated with the ISC count, Hb S concentration, and the intraerythrocytic hemoglobin concentration (Fig. 1).44–47 Although they are known as the conjunctival sickle sign, these vascular abnormalities are not completely pathognomonic of sickle cell disease: in rare cases they are seen in patients with AIDS, chronic myelogenous leukemia, and other vaso-occlusive diseases.47–49
|
Iris Atrophy and Neovascularization
Occlusions of the iris vessels can result in atrophy, and patients may present with asymptomatic white patches of the iris.50,51 The area of atrophy may be extensive (Fig. 2) and may be associated with pupillary irregularity. Iris neovascularization may develop in eyes with chronic retinal detachment or major arteriole occlusions and can in rare cases cause a secondary neovascular glaucoma.52
|
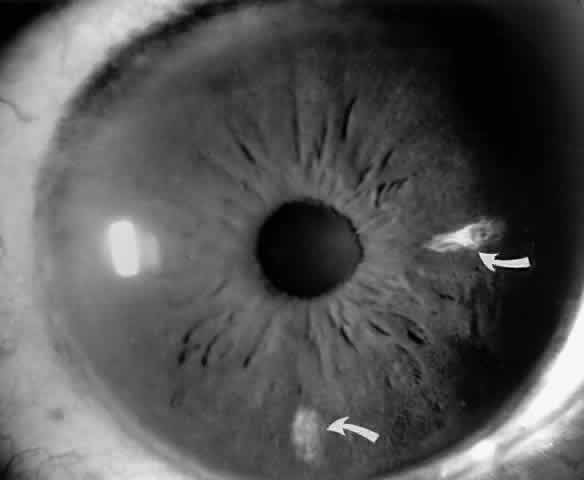
Hyphema
Sickle cell patients, including patients with sickle cell trait, are susceptible to developing central retinal artery occlusions and optic atrophy secondary to elevated intraocular pressure.53,54 Therefore, the potential for permanent ocular damage is an important consideration in sickle cell patients with even minimal hyphema from surgery or anterior segment trauma.55–57 The environment in the anterior chamber promotes Hb S polymerization and secondary impairment of outflow from blockage of the trabecular meshwork by sickled cells.58–63
It is warranted to order a sickle screen for every black American patient with hyphema and for every patient with hyphema associated with elevated intraocular pressure.64 If Hb S is present, the intraocular pressure should be closely monitored and should not be allowed to remain higher than 25 mmHg for more than 24 hours.56,57 Medical management should be restricted, if possible, to topical β-blockers and possibly oxygen (100%) inhalation or oxygen goggles, which deliver oxygen through the cornea to the aqueous humor.65 A judicious trial of 25 mg of oral methazolamide twice daily may be included. Methazolamide has been found to reduce intraocular pressure without altering renal bicarbonate secretion and causing systemic acidosis. Preferably, other carbonic anhydrase inhibitors and osmotic agents should be avoided because they may induce hemoconcentration and acidosis, causing further Hb S polymerization. If the intraocular pressure cannot be controlled by medical means, surgical intervention with anterior chamber lavage, repeated as necessary, is indicated.66
POSTERIOR SEGMENT
Disc Sign
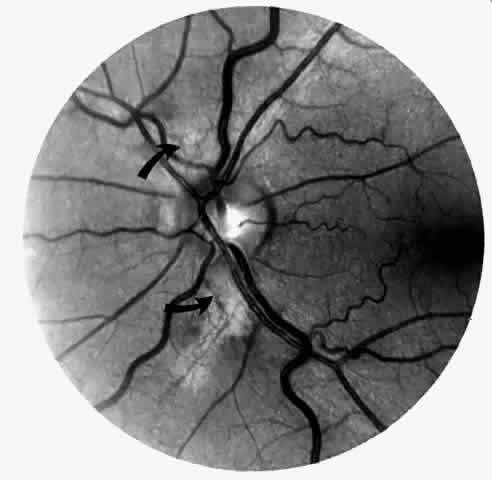
Transient dark red spots (similar to conjunctival commas), representing plugs of sickled erythrocytes within superficial capillaries, may be seen on the surface of the optic disc (Fig. 3 and Color Plate 1A). These disc changes are not associated with any functional or anatomic abnormalities. They are found in 11% of all patients with sickle cell disease, but appear to be more common in patients with homozygous sickle cell anemia, occurring in 29% of these patients.67 The disc sign correlates with the presence of conjunctival commas and ISCs.
|
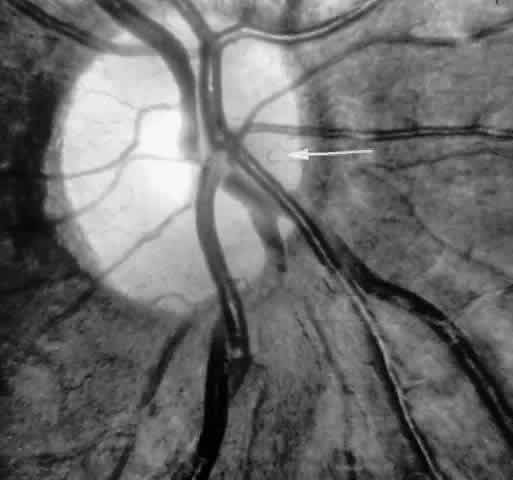
|
Unlike some retinal vaso-occlusive diseases, sickle cell retinopathy is rarely associated with optic disc neovascularization.68 In our extensive experience with sickle cell disease, we have seen only one such case. The low incidence of optic disc neovascularization may be due to the peripheral location of the ischemia and to the localized changes, much of the retina not being significantly affected by the ischemia.69 Peripheral retinal scatter photocoagulation is effective in stimulating regression of optic disc neovascularization.
Vascular Tortuosity
Dilation and tortuosity of the retinal veins was one of the first recognized abnormalities of sickle cell eye disease. Although it is not pathognomonic of sickle cell disease, it reportedly occurs in up to 47% of patients with homozygous sickle cell anemia and 32% of patients with SC disease (Fig. 4).70 The significance of this venous tortuosity is unknown, and the incidence does not appear to be related to age.71
|
Angioid Streaks
Angioid streaks occur in association with sickle cell disease, with an overall incidence of less than 6%.72–75 The changes are more common in patients with homozygous sickle cell anemia and are age-dependent, occurring in 2% of sickle cell anemia patients less than 40 years of age versus 22% in those who are more than 40 years of age (Fig. 5).76
|
Unlike the angioid streaks seen in patients with pseudoxanthoma elasticum, choroidal neovascularization and disciform disease are uncommon in association with sickle cell disease. Elastic tissue degeneration, as is seen in pseudoxanthoma elasticum, has not been demonstrated in the skin biopsy specimens of sickle hemoglobinopathy patients with angioid streaks.73,75 Initially, the etiology of angioid streaks in sickle cell disease was hypothesized to be secondary to iron deposition due to chronic hemolysis, causing brittleness of Bruch's membrane. Histopathologic examination of angioid streaks in a patient with homozygous sickle cell anemia, however, revealed heavy calcification of Bruch's membrane without evidence of iron or hemosiderin.77
Epiretinal Membranes
Epiretinal membranes may produce visual loss in patients with sickle cell disease. Macular epiretinal membranes are seen more frequently in eyes with retinal neovascularization, retinal tears, and vitreous hemorrhage, as well as in eyes that have had laser treatment or surgery of the retina or vitreous.78 Progressive visual loss from macular distortion has been reported in up to 32% of these patients over a 2.5-year period.79 Peripheral neovascularization may stimulate formation of epiretinal membranes by transudation of plasma and erythrocytes into the vitreous, disrupting the vitreous cortex and inducing posterior vitreous detachment. Successful treatment of the neovascular tissue reduces the risk of epiretinal membrane development by approximately 30%.79 Although spontaneous separation of epiretinal membranes following treatment of peripheral neovascularization has been observed, surgical removal may be considered when patients exhibit moderate to severe visual loss (Fig. 6).80,81
|
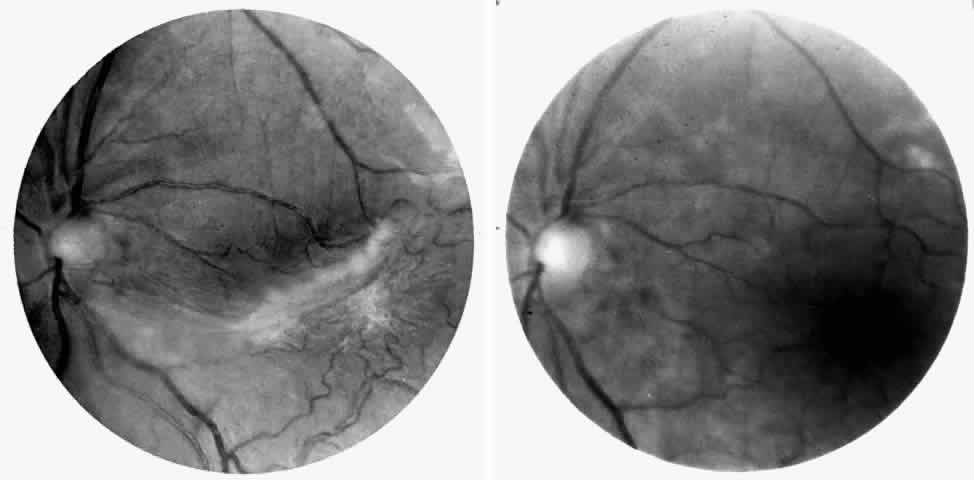
Traction across the macula from peripheral neovascularization is thought to contribute to the formation of macular holes in sickle cell retinopathy.82
Retinal Artery Occlusions
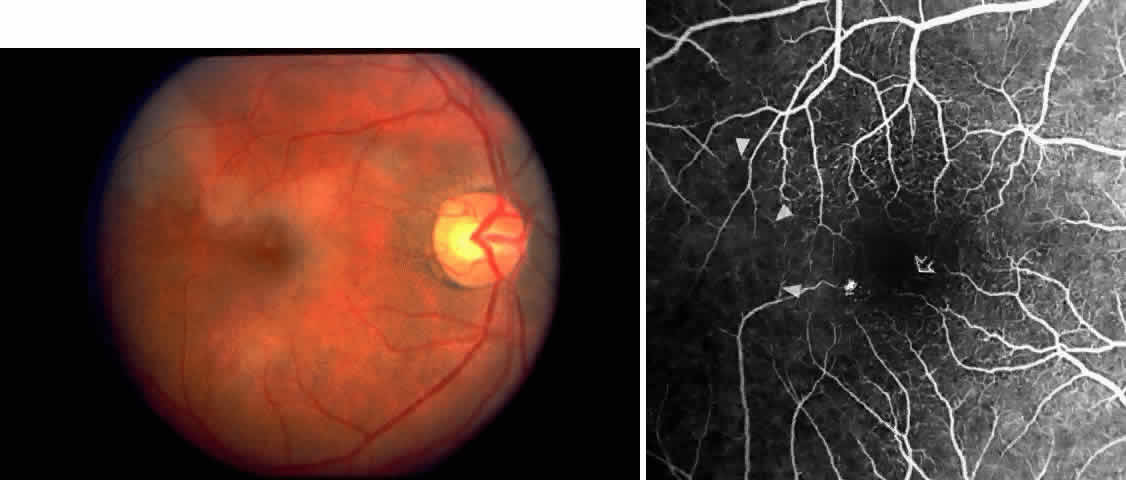
Occlusions of the central retinal artery and major arteriolar branches are probably most frequent in young patients with homozygous sickle cell anemia; however, they may also occur with other sickling genotypes (Fig. 7).39,83,84 They may cause permanent or transient visual loss and can occur simultaneously in both eyes.85–87 Arterial occlusion has also been reported to occur as a complication of retrobulbar anesthesia and following compression of the eye during photocoagulation (Fig. 8).88
|
|
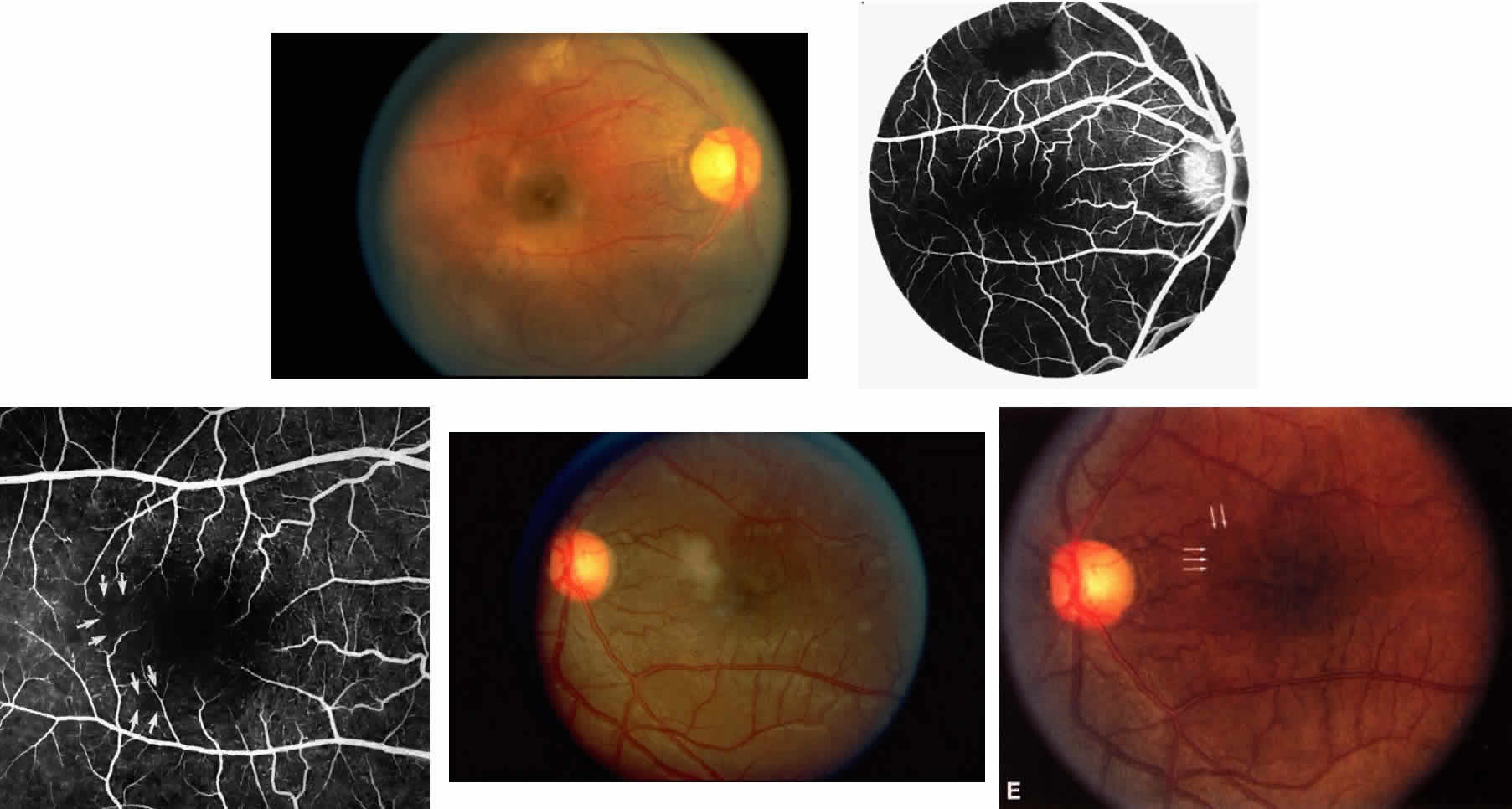
Retinal artery occlusion has also been reported in patients with sickle cell trait secondary to airplane travel,70 with elevated intraocular pressure following blunt trauma,53,54 with extreme dehydration,89 and in association with tuberculosis and systemic lupus erythematosus.90
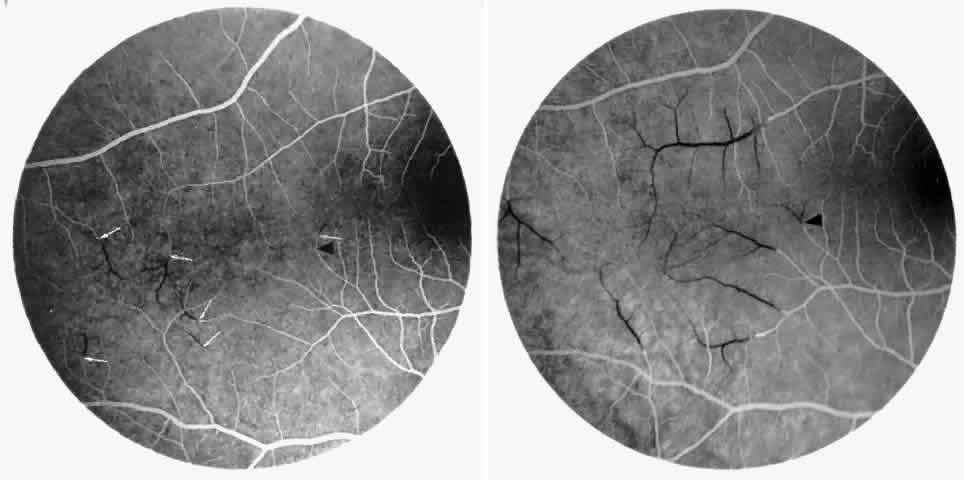
Macular Small Vessel Occlusions
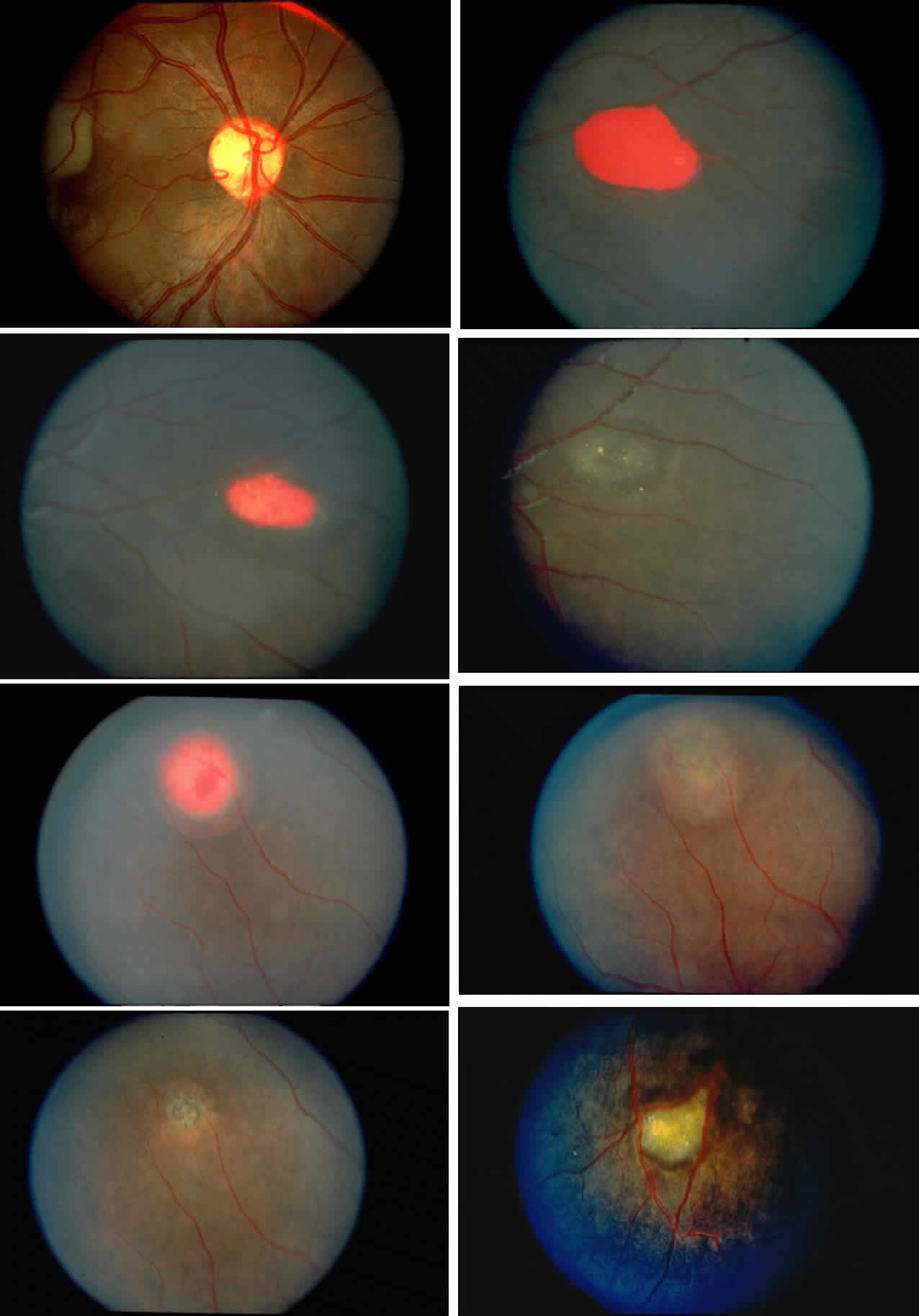
Occlusions of the fine vasculature of the macular and perimacular area have been reported in 10% to 40% of patients with sickle cell disease.18,83,91–99 In the acute phase, the occluded vessel will have a dark red appearance and may appear as a dark line on fluorescein angiography (Fig. 9). Nerve fiber layer infarcts (cotton-wool spots) are seen (see Fig. 8D and E;Fig. 10).100
|
|
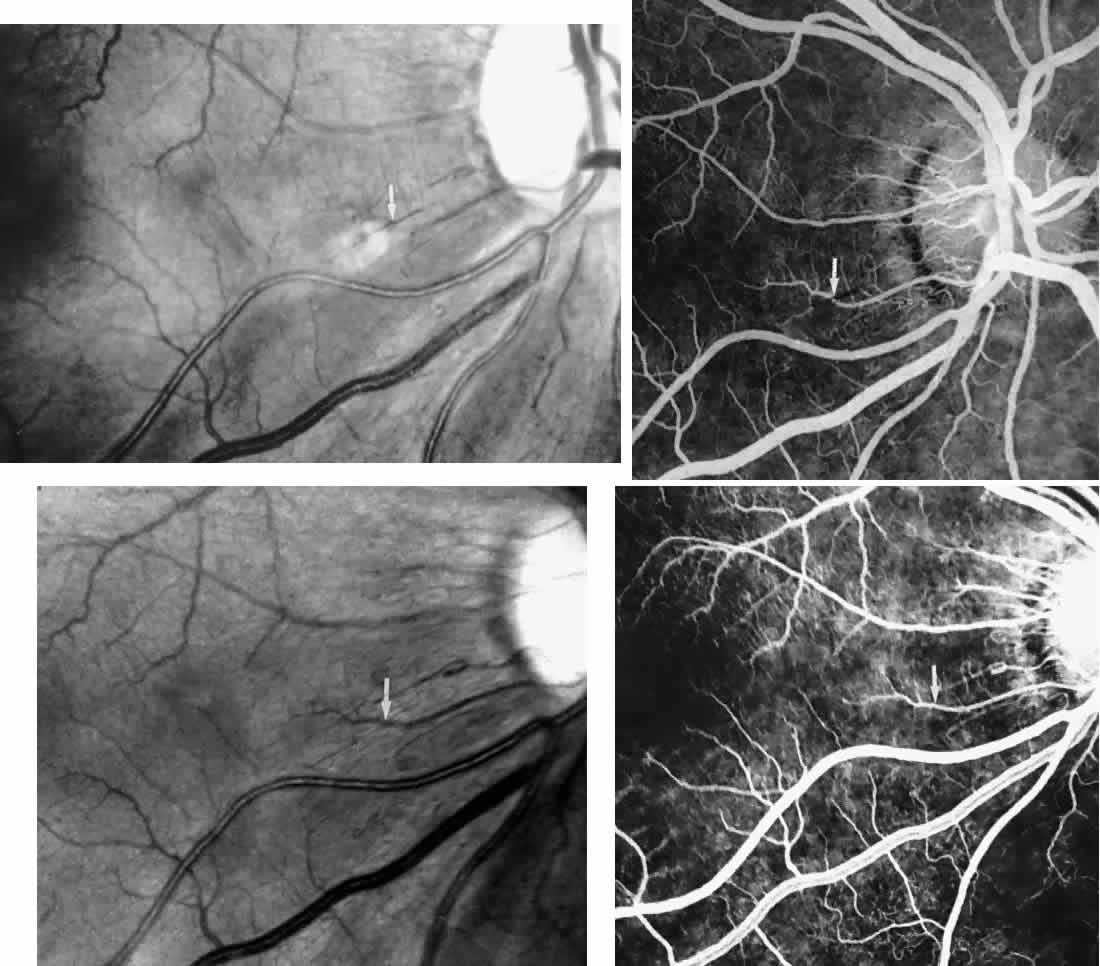
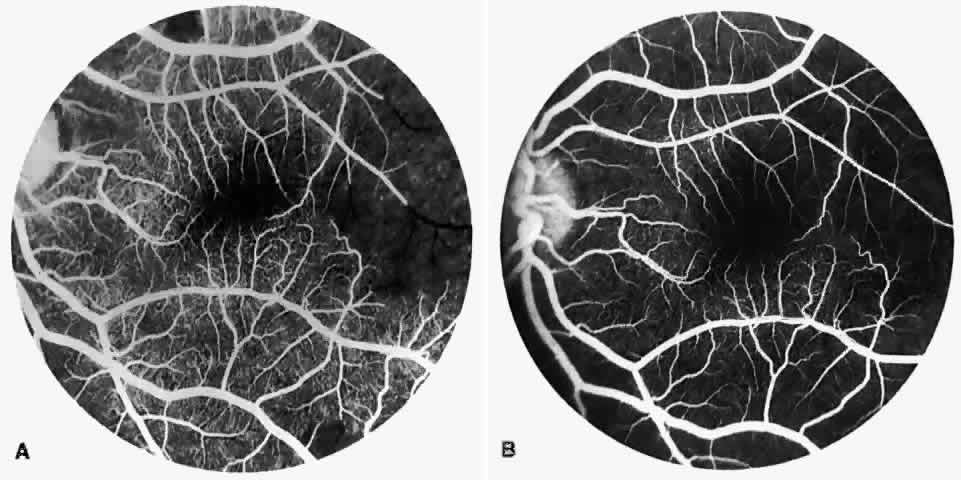
Other macular and perimacular changes include microaneurysm-like dots, dark and enlarged segments of arterioles, hairpin-shaped venular loops, pathologic avascular zones, and widening and irregularities of the foveal avascular zone (Figs. 11 and 12). In the Jamaican cohort study evaluating children with homozygous sickle cell anemia and SC disease between the ages of 5.0 and 7.5 years of age, no pathologic avascular zones could be identified despite a high incidence of peripheral vascular closure.31 In evaluating patients with homozygous sickle cell anemia, no relationship between ISC counts and macular abnormalities or visual acuity could be found.101 Using fluorescein angiography, investigators have found the foveal avascular zone to be significantly larger in eyes with clinical evidence of sickle cell maculopathy as compared with normal eyes and eyes without clinical evidence of sickle cell maculopathy.102–104
|
|
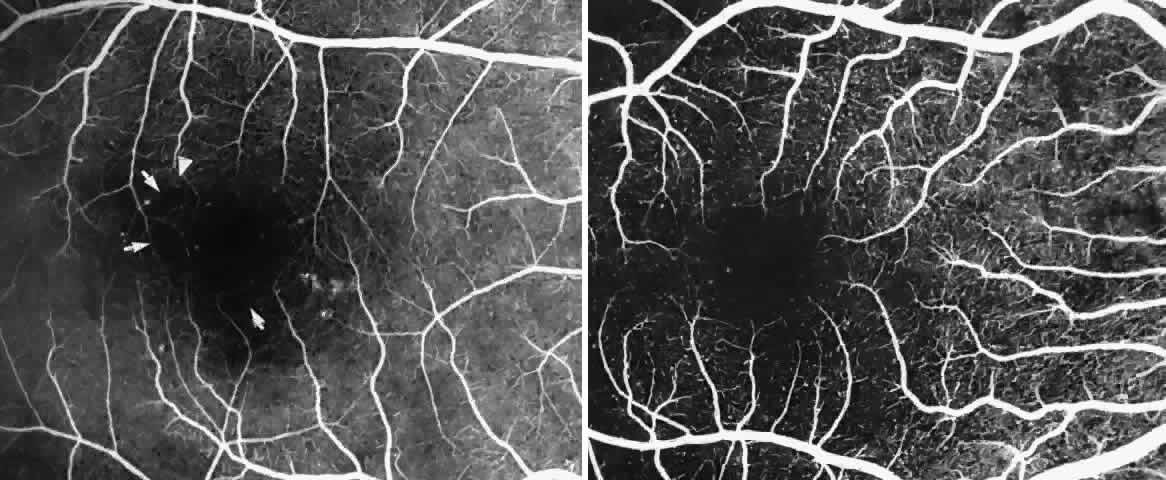
Careful examination by fluorescein angiography, looking for areas of capillary dropout and other capillary abnormalities, is often necessary to identify the macular changes. These changes may be transient, and the macula may appear normal on subsequent fluorescein angiograms (Fig. 13). Although fluorescein angiography may or may not demonstrate reperfusion of a previously occluded capillary bed, a loss of the inner retinal layers results in an ophthalmoscopic focal concavity with an abnormal reflex (retinal depression sign) (see Fig. 8E).105,106 These changes are usually permanent. The retinal depression sign is not pathognomonic of sickle cell disease and may be seen with other arteriolar occlusive diseases, such as embolic retinopathy, vasculitis, and hypertension.
|
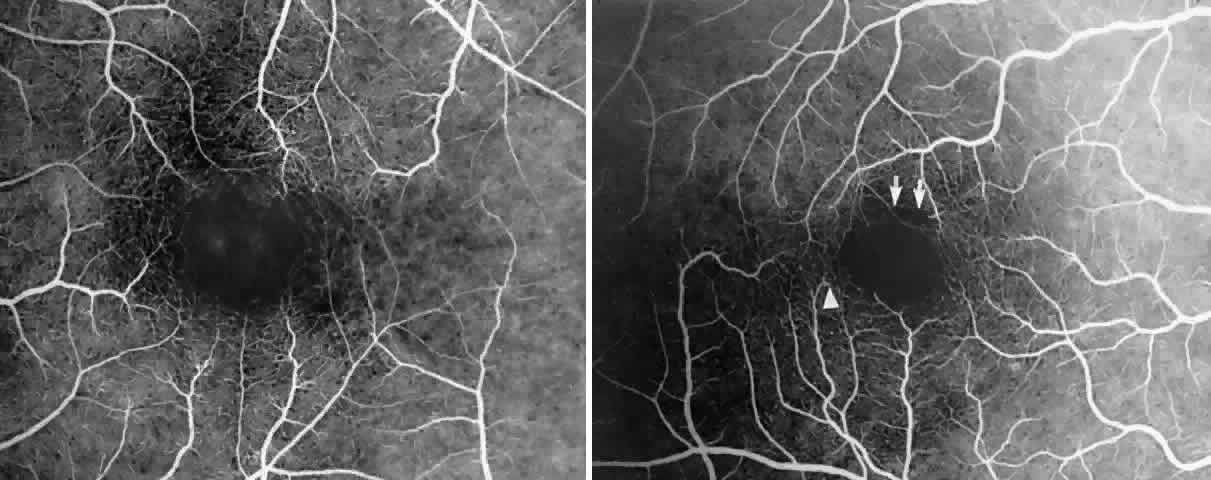
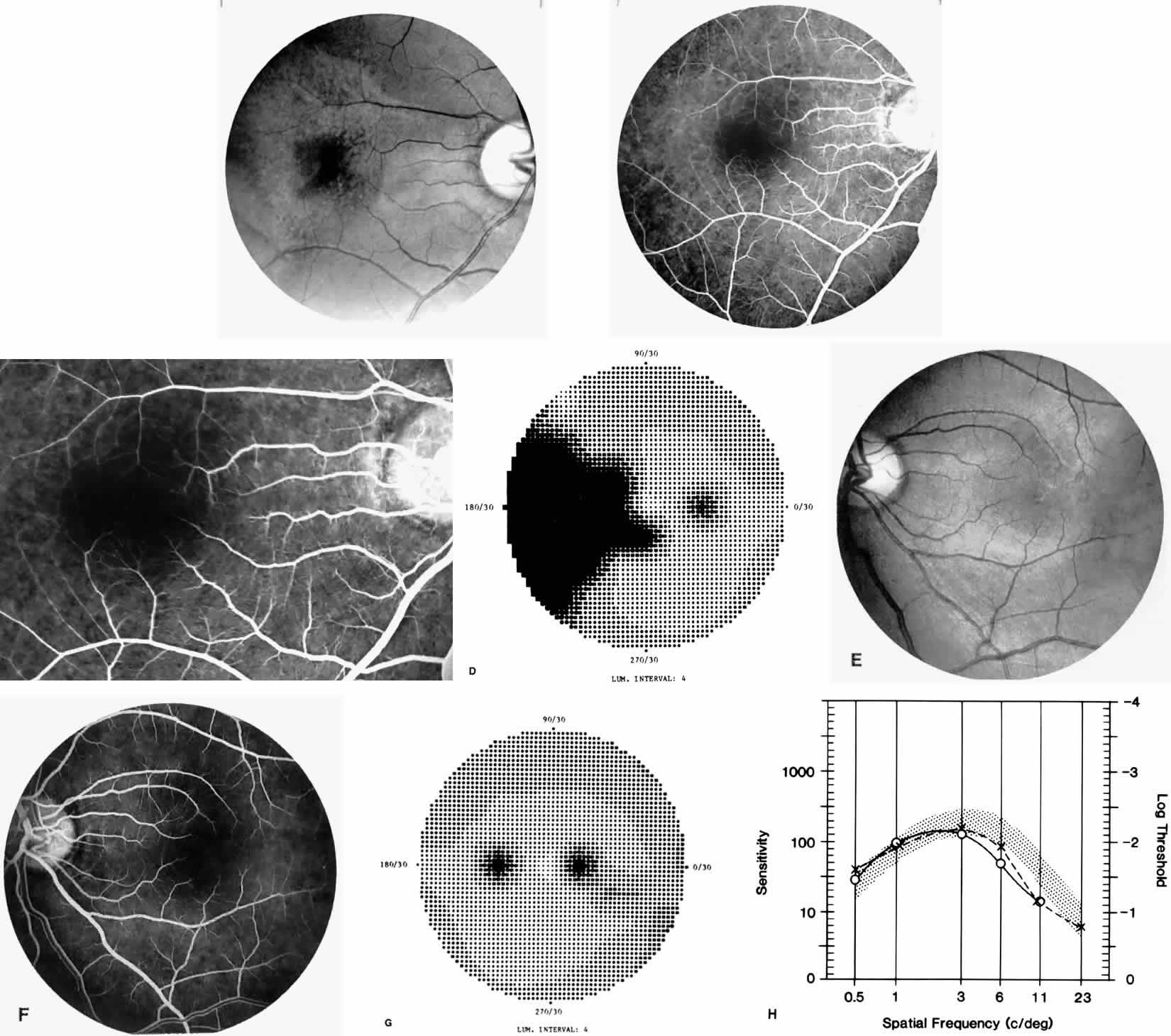
Macular Function Testing in Sickle
Cell MaculopathyThe visual acuity in patients with sickle cell disease is often normal, despite the presence of an enlarged foveal avascular zone or other evidence of sickle cell maculopathy (Fig. 14). In addition, patients with sickle cell maculopathy have a remarkable absence of visual complaints. Although 55% of patients with homozygous sickle cell anemia had abnormal contrast sensitivity, no significant relationship was demonstrated between contrast sensitivity and macular vascular abnormalities.101 Automated visual field analysis has demonstrated significantly larger scotomas in patients with abnormally enlarged foveal avascular zone.102 Color vision testing has revealed a greater incidence of blue-yellow defects in patients with sickle cell retinopathy; however, no significant correlation has been demonstrated between color vision defects and the presence of sickle cell maculopathy.98,107
|
Retinal Venous Occlusions
Retinal venous occlusions are surprisingly uncommon in sickle cell disease.108 We have seen a patient with homozygous sickle cell anemia and a branch vein occlusion, but this patient had hypertension. We have not seen central vein occlusion. An underlying systemic disease associated with a higher incidence of venous occlusions (e.g., hypertension) should be suspected when a venous occlusion occurs in a patient with sickle cell disease. The anemia and low blood pressure present in sickle cell patients may actually protect against venous occlusions.
Choroidal Vascular Occlusions
Choroidal vascular occlusions may occur focally at the level of the choroidal precapillary arteriole or capillary bed (Elschnig's spots) or from posterior ciliary artery occlusion. Although focal precapillary arteriole occlusions have not been specifically identified with sickle cell disease, clinical and histopathologic evidence of spontaneous posterior ciliary artery occlusions have been reported in sickle cell disease.109,110 The findings are similar to those described following compression of the eye during general anesthesia and after heavy peripheral photocoagulation.111,112 In the acute phase, the occlusions appear as white, circumscribed, triangular patches at the level of the retinal pigment epithelium and outer retina. Over the following weeks, the white lesions fade and retinal pigment epithelial mottling develops (Fig. 15). Since patients with acute ciliary artery occlusions may be asymptomatic and the diagnosis is often based solely on the appearance of peripheral pigment mottling, the frequency of this complication remains uncertain.
|
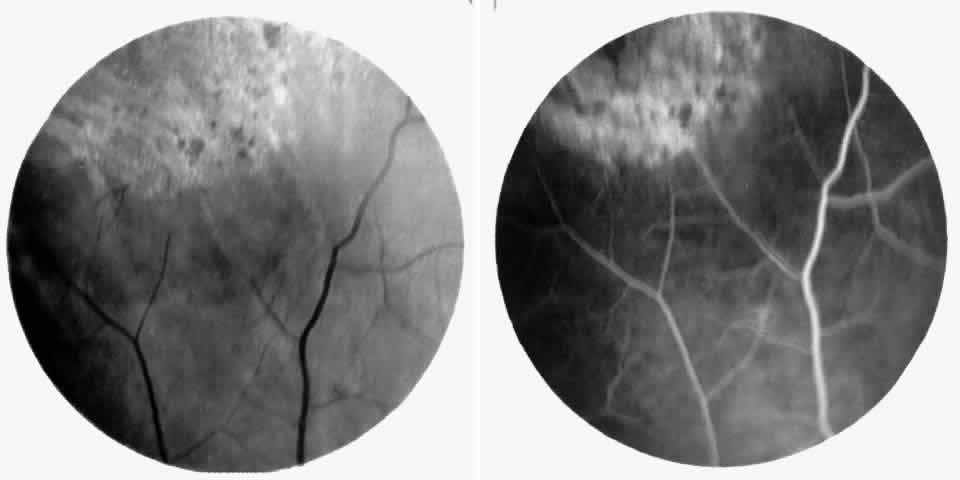
Focal areas of atrophy of the choriocapillaris have been seen histopathologically on postmortem examination of patients with homozygous sickle cell anemia. Mild sclerosis of the choriocapillaris, focal peripheral photoreceptor loss, and areas of choroidal neovascularization also have been noted.110 It has been suggested that choroidal ischemia plays a role in the development of angioid streaks in sickle cell disease. The one histopathologic report of angioid streaks indicated that the basement membrane of the underlying choriocapillaris endothelial cells was slightly thickened and that sickled erythrocytes were found within patent lumina of the choriocapillaris.77 No specific evidence exists to support a relationship between choroidal ischemia and angioid streaks.
Retinal Hemorrhages, Iridescent Spots, and Black Sunbursts
Retinal hemorrhages (“salmon patches”), found most commonly in the equatorial periphery, can be observed after an abrupt occlusion and rupture of an intermediate-sized retinal arteriole (Fig. 16).113 Because the hemorrhages typically appear adjacent or distal to an intraluminal obstruction, it is likely that ischemic necrosis causes a weakening of the vessel wall and that reperfusion of the vessel causes a rupture of the damaged vessel wall, resulting in a hemorrhage (Fig. 17).100 Acutely, these hemorrhages are bright red, but after several days, the partially degenerated blood acquires a characteristic orange-red color (hence the name salmon patch). In most cases, these hemorrhages are asymptomatic. The majority of these hemorrhages remain confined to the sensory retina; however, blood may leak through the internal limiting membrane into the vitreous or dissect deeper into the subretinal space (Fig. 18).114 Resolution occurs over days to weeks and may result in a focal area of atrophic split retina (a “schisis” cavity), a pigmented retinal scar, or a grayish-white vitreous deposit, depending on the location of the hemorrhage (Color Plate 1B through G).115 The blood is slowly cleared by macrophages.
|
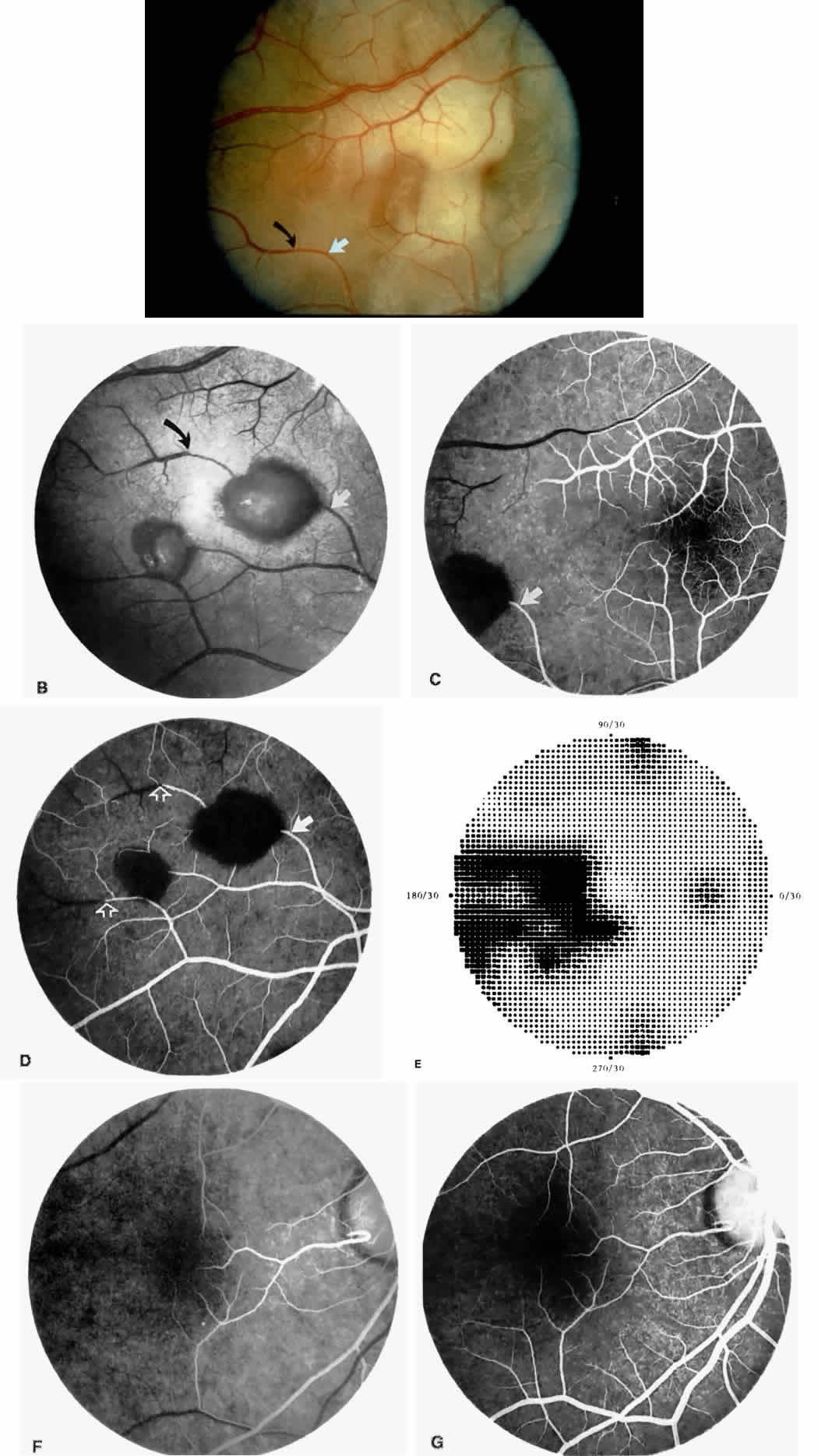
|
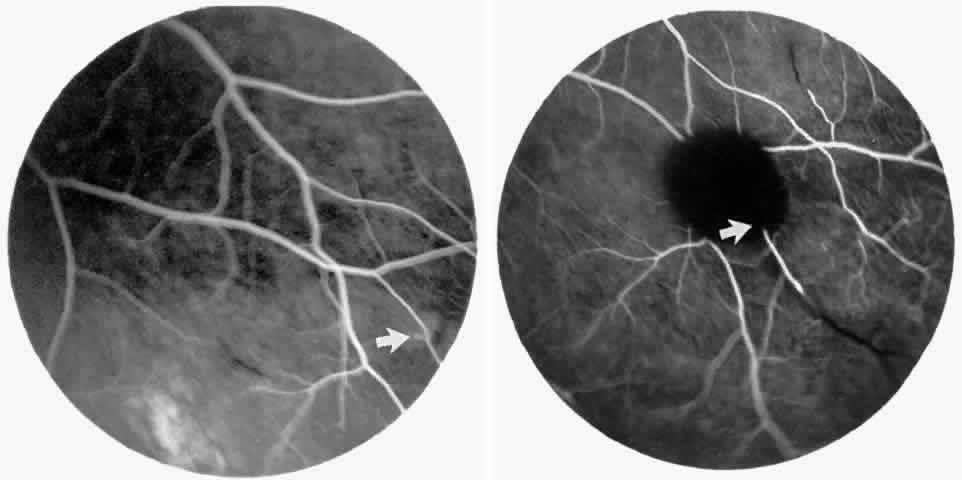
|
Intraretinal blood breakdown products, either extracellular or within macrophages, may appear as refractile copper-colored granules (“iridescent spots”") (Color Plate 1H). Macular iridescent schisis lesions have not been described clinically, but they have been observed on histologic examination.114
The occluded vessels may reopen, and the capillary network in the area of a schisis cavity may appear normal; however, more commonly, the vessels will remain closed (Fig. 19). In rare cases, an area of retinal neovascularization may be found within a schisis cavity (Fig. 20).
|
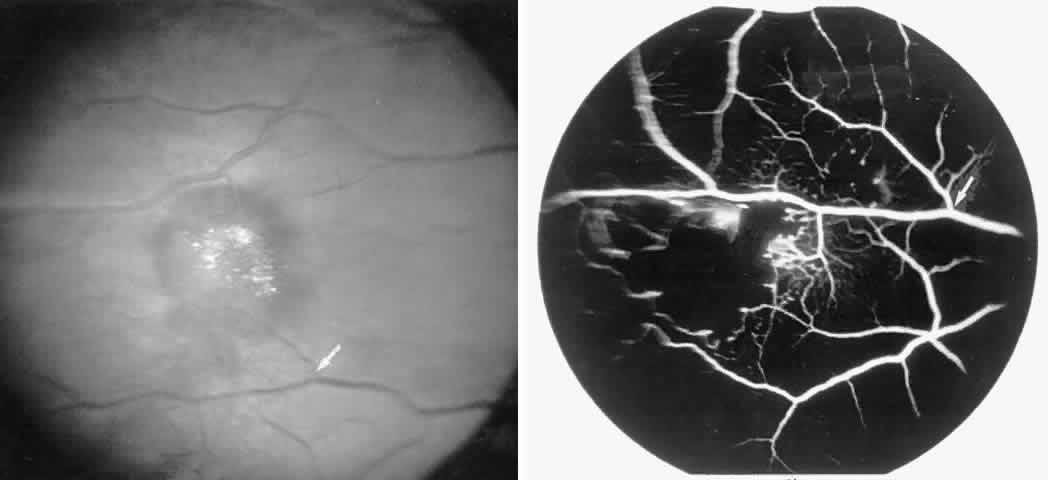
|
Black pigmented spiculate or stellate chorioretinal lesions (“black sunbursts”) are typically found around or anterior to the equator and adjacent to an arteriole.70 Occasionally, a pigmented lesion may be seen trailing from an arteriole or as a cuff of pigment overlying the vessel (Color Plate 2A).83 Additionally, the overlying arteriole may be occluded. Refractile deposits are often seen interspersed with the pigment. Black sunbursts are believed to be due to deep retinal blood stimulating pigment epithelial migration, hyperplasia, and hypertrophy.116,117 Histopathologic findings support this hypothesis,114 and the development of black sunbursts has been documented in an area of previous intraretinal and subretinal hemorrhage (see Color Plate 1E, F, and G).114,115 An alternative explanation for black sunbursts is the occurrence of choroidal ischemia and aborted choroidal neovascularization.22,118 A spontaneous chorioretinal neovascular membrane was shown to occur within a black sunburst in a 14-year-old girl with homozygous sickle cell anemia.119
|
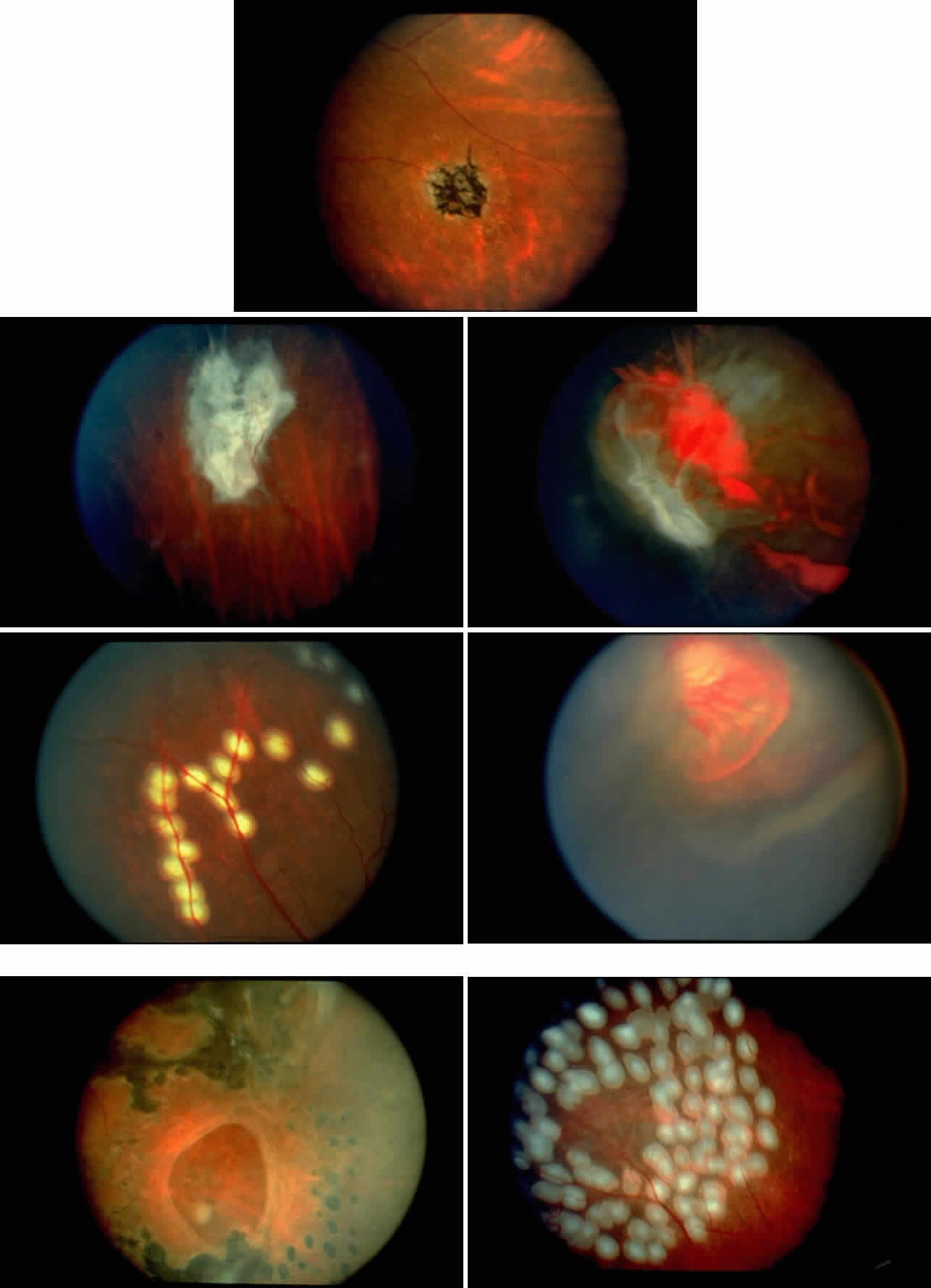
Dark- and White-Without-Pressure Fundus Lesions
Flat, geographic dark brown areas have been identified in the posterior pole or midperiphery of the retina in patients with sickle cell disease without any signs of a previous hemorrhage or definite evidence of retinal or choroidal vascular occlusion. These dark-without-pressure lesions are transient, changing shape or disappearing over weeks to months.120,121 Areas of white-without-pressure, which are possibly secondary to condensation of the overlying basal vitreous, also have been described.83,122 These two distinct fundus lesions have been noted in black American patients with sickle hemoglobinopathies, but their relationship to sickle hemoglobin, if any, remains uncertain.
Peripheral Retinal Manifestations
The retinal capillary network in the retinal periphery thins to a single layer approximately 1 mm from the ora serrata. A similar thinning of the retinal capillary network occurs around the foveal avascular zone.123–125 These two areas appear to be the most susceptible to vascular occlusions from sickle cell retinopathy. Repeated vascular closures and reopenings of the retinal periphery results in a dynamic remodeling and a centripetal recession of the vascularized border toward the posterior pole.100,126 Peripheral vascular occlusions are seen more frequently on the temporal side, tend to be more rapidly progressive in children and adolescents than in adults, and are significantly more common in homozygous sickle cell anemia than in SC disease.31,32
Redirection of blood flow results in the formation of arteriolar-venular anastomosis at the border of the perfused and nonperfused peripheral retina. This process of vascular closures and arteriolar-venular anastomosis formation is similar to that seen in the spleen, brain, and kidney.127,128 Perhaps unique to the retina, however, is the subsequent development of neovascularization near areas of arteriolar-venular anastomoses.129
The sequential changes of the peripheral retina form the basis of Goldberg's five-stage pathologic classification:
Stage I: Peripheral arteriolar occlusions
Stage II: Peripheral arteriolar-venular anastomoses
Stage III: Preretinal neovascularization
Stage IV: Vitreous hemorrhage
Stage V: Retinal detachment (Table 3)130,131
TABLE 3. Pathogenetic Classification of Proliferative Sickle Retinopathy
| Stage | Peripheral Retinal Findings |
| I | Peripheral arteriolar occlusions |
| II | Peripheral arteriolar-venular anastomosis |
| III | Neovascular and fibrous proliferation |
| IV | Vitreous hemorrhage |
| V | Retinal detachment |
Fluorescein angiography remains the best method of identifying these peripheral retinal changes and documenting the presence of neovascularization.19
Examination of the Jamaican Sickle Cohort children with homozygous sickle cell anemia and SC disease, comparing their peripheral retinas with those of age- and sex-matched normal (Hb AA) controls, has revealed that the peripheral retina demonstrates a characteristic change that may allow identification of an increased risk of progressing to neovascularization. A classification scheme has been proposed that divides the peripheral retinal manifestations into qualitatively normal and qualitatively abnormal (Table 4). In the new classification scheme, if there is a continuous arteriolar-venular anastomosis at the margin of the perfused and nonperfused peripheral retina, it is considered qualitatively normal. An abnormal peripheral retinal vascular pattern has capillary stumps extending into nonperfused retina or an irregular capillary border.
TABLE 4. New Classification of Sickle Retinopathy
| Peripheral Retinal Vascular Pattern | |
| Type I | Qualitatively normal; continuous arteriolar-venular loops with thinned capillary bed |
| Type II | Qualitatively abnormal |
| a. | Capillary stumps extending into nonperfused retina |
| b. | Irregular capillary border without arteriolar-venular loops or capillary stumps |
| Type III | Indeterminant |
Although Talbot and co-workers found that vascular occlusions occurred earlier in homozygous sickle cell disease (Hb SS) than in heterozygous sickle cell disease (Hb SC), a significantly larger proportion of subjects with SC disease versus sickle cell anemia had an abnormal peripheral vascular pattern.31–33 It appears that once an abnormal peripheral retinal vascular pattern appears, it remains qualitatively abnormal despite further loss of the capillary bed. An abnormal border probably occurs as a radical alteration of retinal perfusion and correlates with the subsequent development of neovascularization. A normal border, even if undergoing a posterior regression, results from a gradual modification of the capillary bed and indicates a low risk for PSR.34 However, caution must be exercised, and the entire extent of the peripheral retina must be examined: even though a part of the peripheral retina is qualitatively normal, there may be still be neovascularization in other areas.
STAGE I: PERIPHERAL ARTERIOLAR OCCLUSIONS. This stage may be further subdivided into three grades: grade I, narrowing of the peripheral arterioles with tortuosity and abnormal looping of the peripheral venules; grade II, tortuosity, dilation, and microaneurysmal formation in the capillary network; and grade III, occlusion of the peripheral capillaries and arterioles.83
Histologic and trypsin digest studies support the theory of a sudden occlusion of the precapillary arteriolar circulation followed by degeneration of the occluded vessels and the distal nonperfused retina. The presence of focal areas of small vessel degeneration and vascular beading (but not typical retinal microaneurysms) also have been confirmed.132
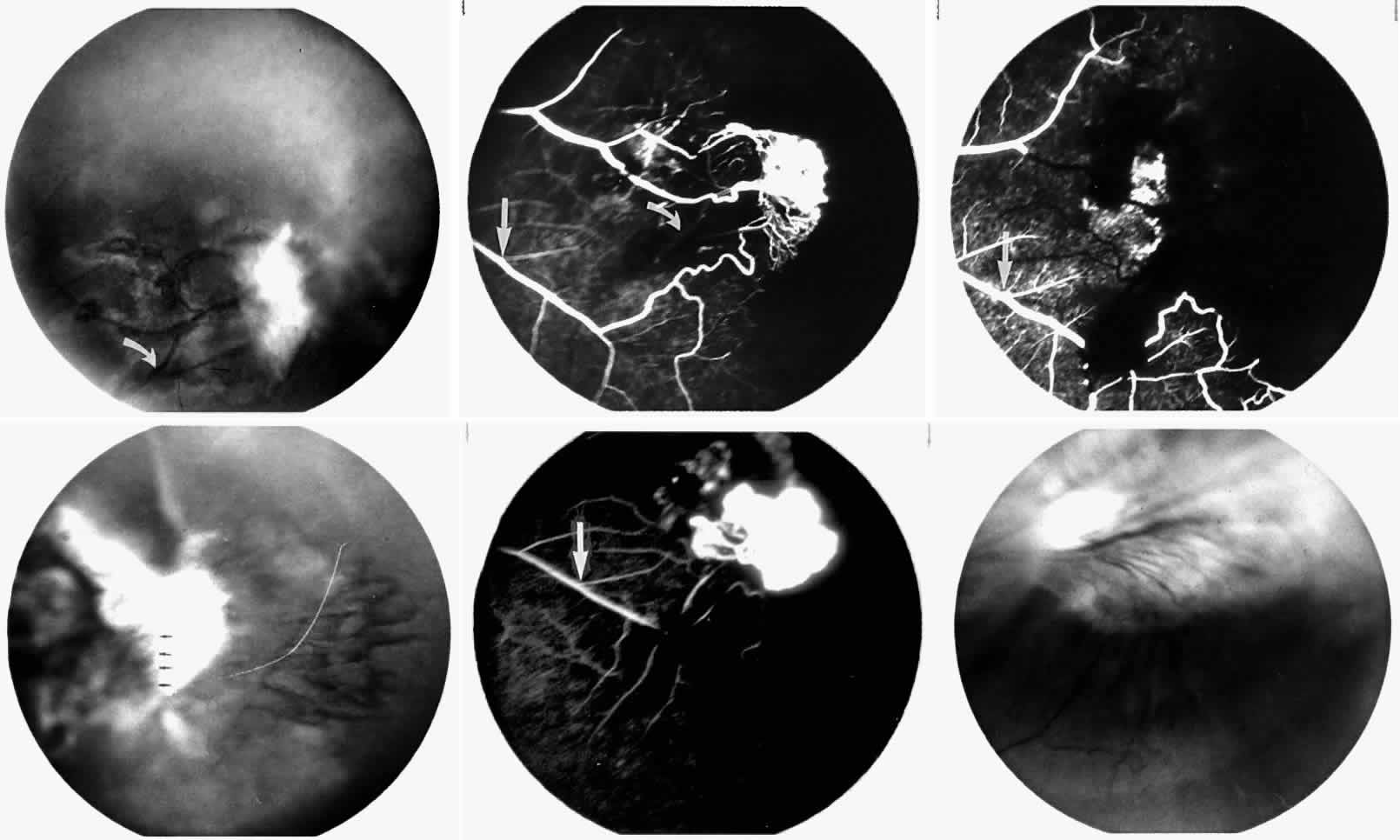
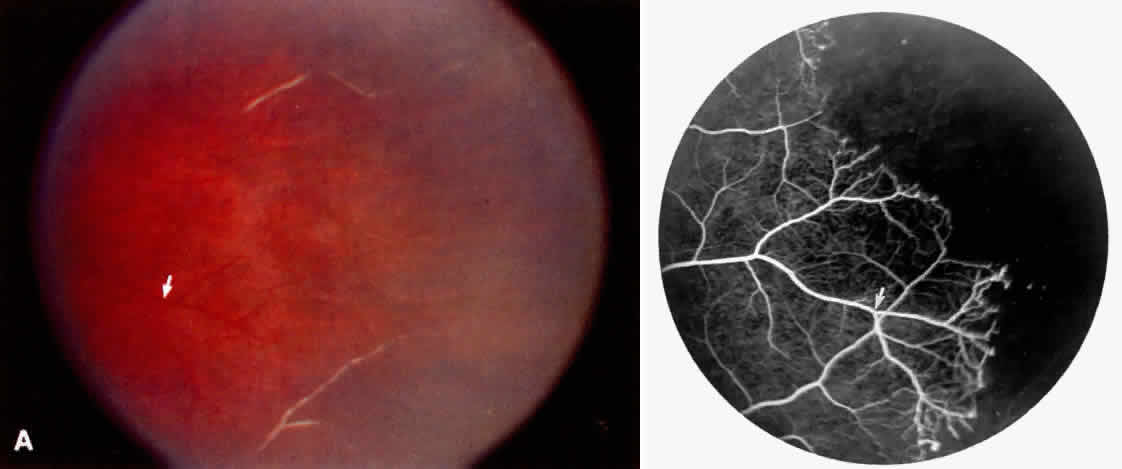
The occluded arterioles may be invisible or may have a “silver-wire” or chalk-white appearance, as first described by Goodman and colleagues39 (Fig. 21). Fluorescein angiography may demonstrate an abrupt complete occlusion at the interface between peripheral nonperfused and posterior perfused retina. Frequently, this occlusion will take place just distal to a branching vessel, giving the appearance of a freshly pruned rose bush. The nonperfused anterior peripheral retina will have a grayish brown appearance and on fluorescein angiography will appear blurred without clearly defined fundus markings.
|
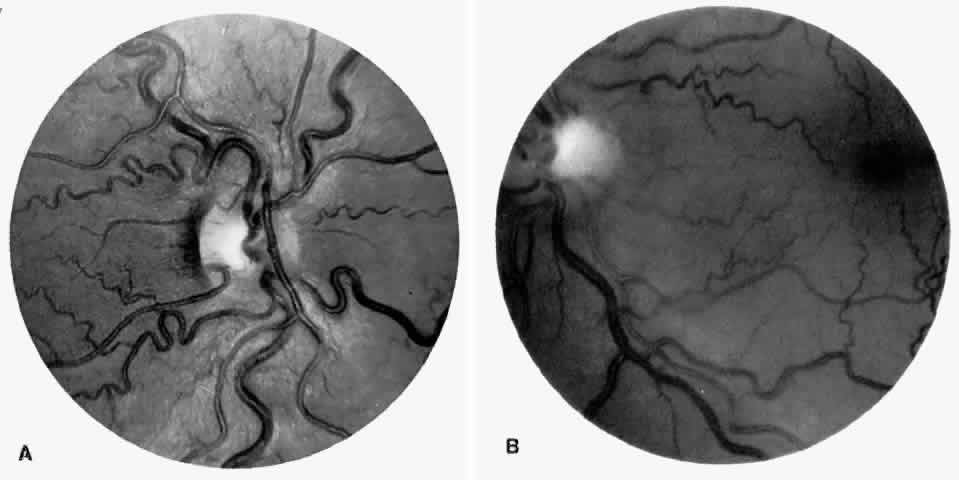
STAGE II: PERIPHERAL ARTERIOLAR-VENULAR ANASTOMOSES. Following occlusion of the terminal arterioles, anastomotic channels form to channel the blood from the occluded arteriole to the nearest venules. These anastomoses form at the interface between the perfused and nonperfused retina. Most likely, they are dilated preexisting capillaries rather than new vessels, since they do not leak on fluorescein angiography. The redirection of blood flow is probably due to hydrostatic forces (Figs. 22 and 23).
|
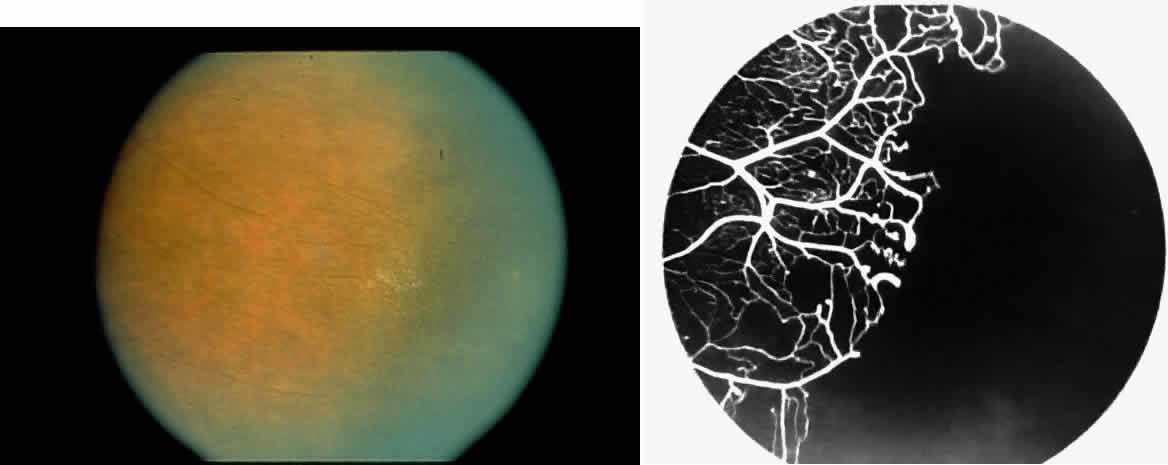
|
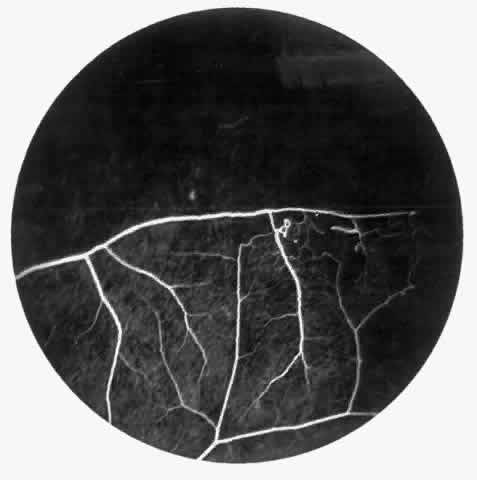
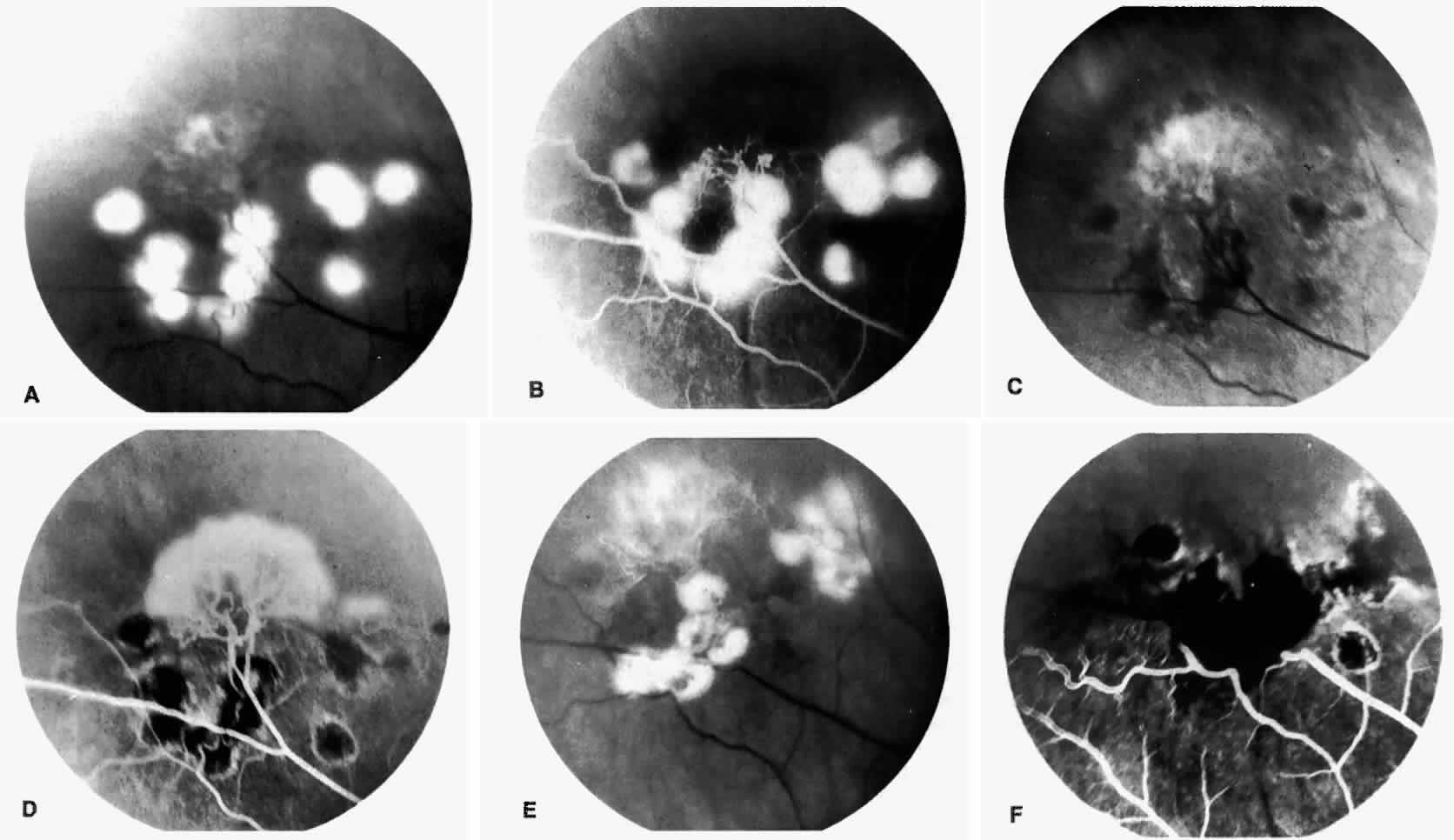
STAGE III: PRERETINAL NEOVASCULARIZATION (PROLIFERATIVE SICKLE RETINOPATHY). “Sea fan”-shaped neovascularization typically develops on the venular side of an arteriolar-venular anastomosis, mimicking the normal development of retinal capillaries (Fig. 24).125 A lowered oxygen tension and angiogenic factors released on the venular side may be the stimulus for neovascular growth.125,126 In most instances, the direction of growth is toward the ora serrata, from the perfused retina toward the nonperfused retina. Presumably, this represents an abortive attempt to revascularize the nonperfused retina, initiated by vasoproliferative factors.
|
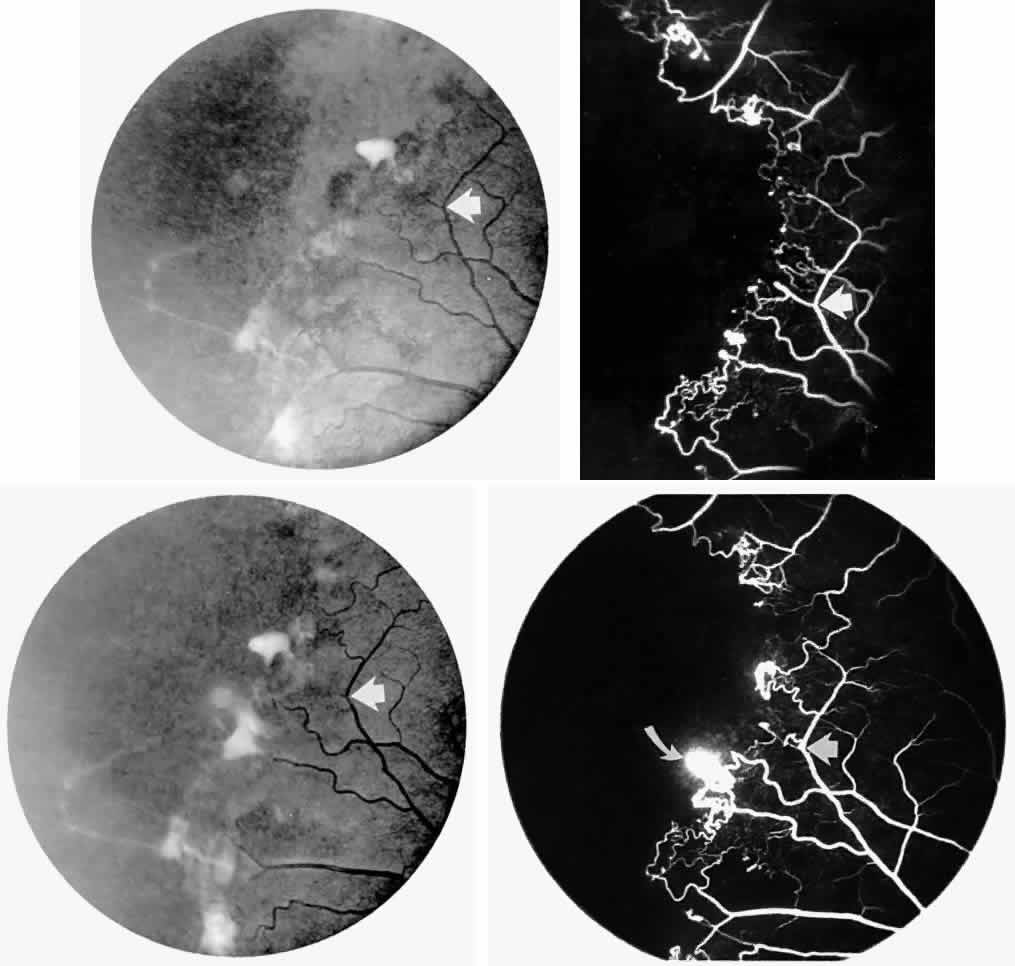
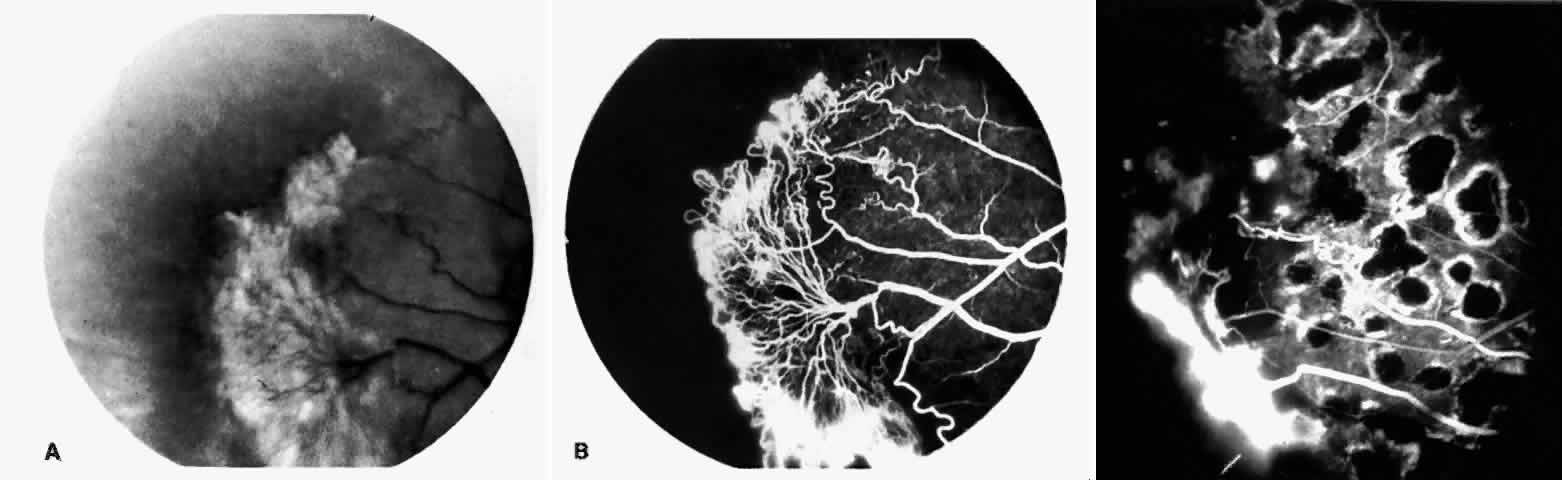
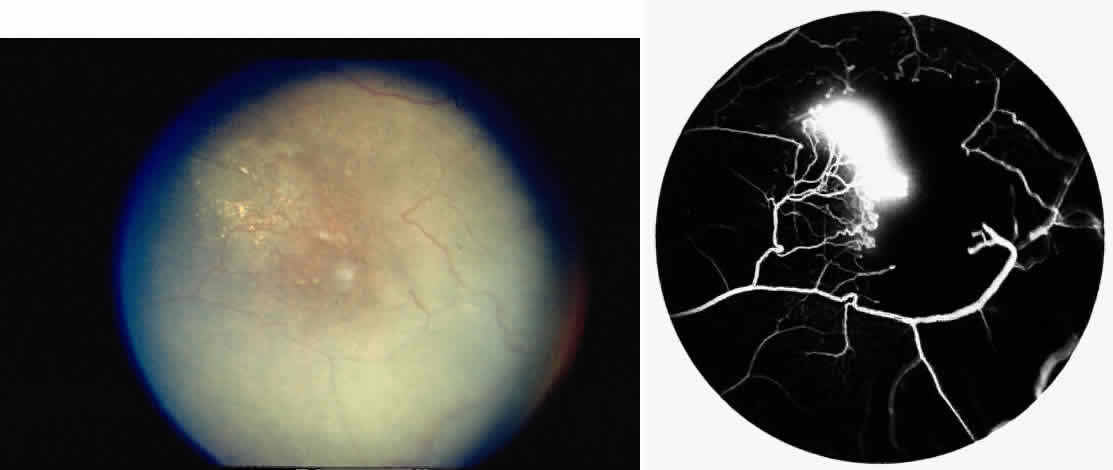
The characteristic neovascular lesions of PSR are called sea fans because they resemble the marine invertebrate Gorgonia flabellum.70 They tend to occur more commonly in the temporal periphery, but they have been reported to occur in the temporal macula in the presence of extensive nonperfusion.130,133 Initially they grow on the surface of the retina, but they often become elevated into the vitreous and adhere to a partially detached posterior hyaloid.114 It may be difficult to visualize small sea fans ophthalmoscopically; however, fluorescein angiography clearly demonstrates leakage of dye into the vitreous (Fig. 25). The feeding arteriole is usually more tortuous than the draining venule (Fig. 26). Early on, the neovascular lesion is fed by a single arteriole and drained by a single venule, but with time, additional arterioles and venules become arborized within the lesion (Fig. 27).129 Growth of the sea fan often occurs circumferentially, rather than radiallyÜmh- 1Ý, toward the ora serrata. Progressive circumferential growth may lead to neovascular lesions extending around the entire periphery. As it matures, a white fibroglial mantle often covers the neovascular tissue (Color Plate 2B).
|
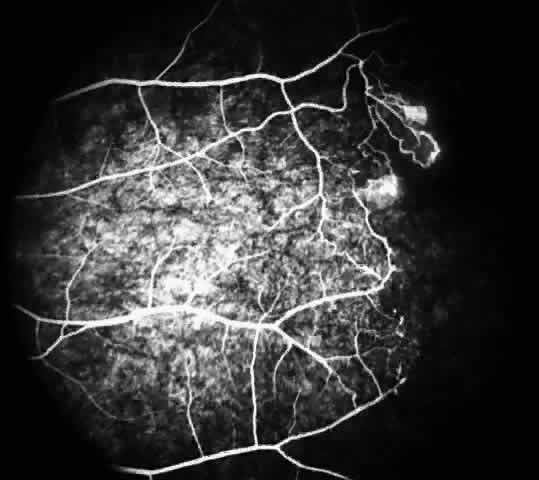
|
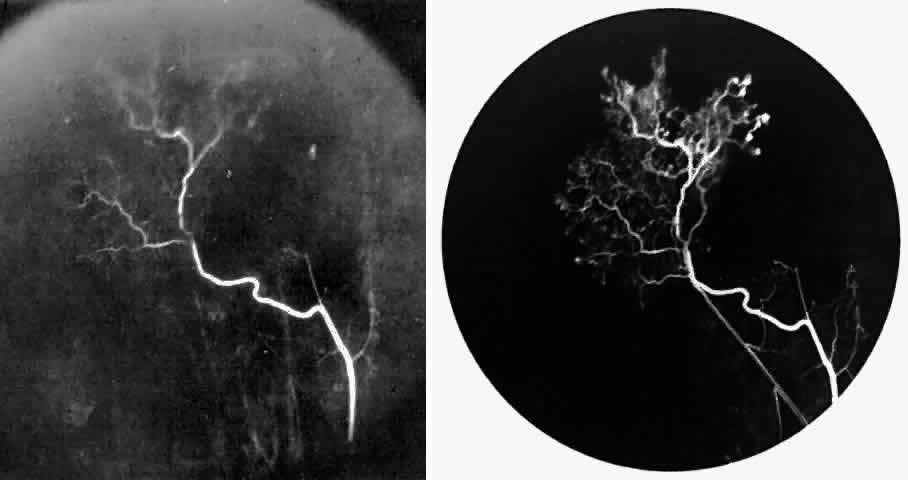
|
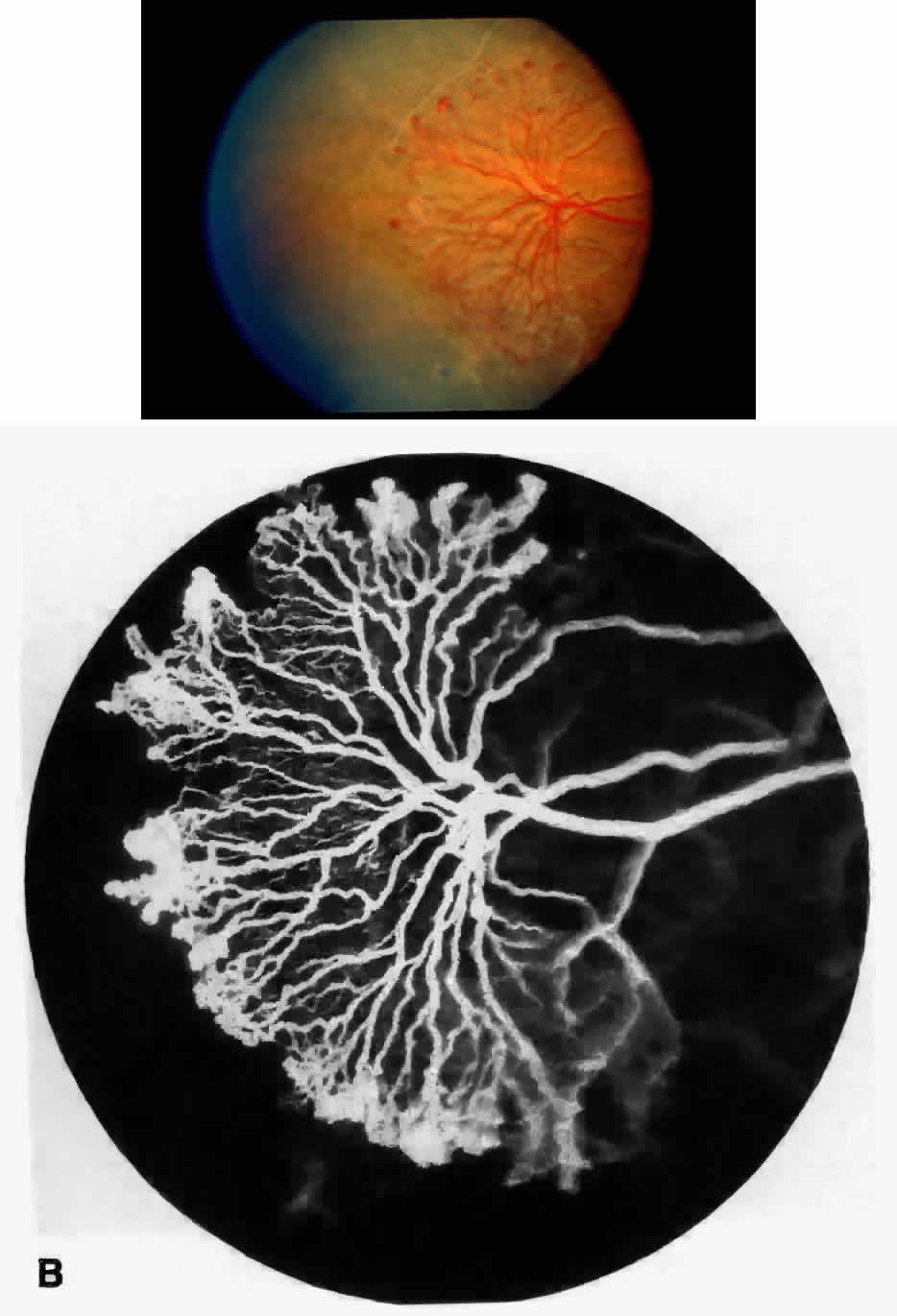
PSR is associated with the severe vision-threatening sequelae of sickle cell disease: vitreous hemorrhage (stage IV) and retinal detachment (stage V). These stages are believed to result from transudation of blood components into the vitreous through the incompetent neovascular tissue (Fig. 28). Vitreous fluorophotometry has quantified the leakage from the peripheral neovascularization.134 This leads to premature syneresis and collapse of the vitreous, inducing tractional forces on the retina that lead to vitreous hemorrhage, retinal tears, and tractional and rhegmatogenous retinal detachment. In rare cases, an exudative detachment may occur.
|

Spontaneous nonperfusion or autoinfarction, accompanied by regression of the neovascular lesion, occurs in 20% to 60% of eyes with PSR.135,136 The peak incidence of autoinfarction is 2 years after the development of PSR. It appears that autoinfarction occurs primarily as a result of (1) occlusion of the feeding arteriole due to traction on the neovascular lesion by contracting vitreous, or (2) occlusion by sickled RBCs. The latter probably is more common in homozygous sickle cell anemia, which is more commonly associated with autoinfarction and complete vascular occlusion.
Sea fans develop an average of 14 months after diagnosis of stage II, with an approximate incidence of 14% per year in patients with SC disease.129 In a series of selected patients with different hemoglobinopathies in Jamaica, the incidence of PSR was reported as follows: 2.6% with sickle cell anemia83; 14.0% with Hb S-β-thalassemia91; and 32.8% with SC disease (see Table 1).92 It is a common misconception that patients with homozygous sickle cell anemia have a very low risk of developing retinal neovascularization. Although the risk is recognizably lower, it must be remembered that the development of neovascularization is not only genotype-dependent but also age-dependent, increasing with age in both genotypes with the highest risk period of 20 to 34 years of age in SC disease and 40 to 50 years of age in homozygous sickle cell anemia.26,137 The incidence of PSR in patients with homozygous sickle cell anemia is 14% for patients older than 40 years and up to 29% in patients older than 50 years.25,76 In a series of 786 patients with homozygous sickle cell anemia, the prevalence of PSR was demonstrated to increase gradually, peaking in patients aged 30 years and older. The first cases were observed in patients in their late teens, the incidence rate increasing in patients who were 25 years and older.29 In the same study, a series of 533 patients with SC disease demonstrated that the prevalence of PSR reached a maximum in men in their late 20s and in women older than 40 years, whereas the incidence rates of PSR peaked in men in their early 20s and in women in their late 20s. The prevalence of PSR has been reported to be as high as 68% in SC disease patients 45 years of age and older.26 An ongoing cohort study in Jamaica will undoubtedly provide information on the evolution of PSR in patients with the various genotypes.
As mentioned previously, the otherwise benign condition of sickle cell trait can be associated in rare cases with ocular complications, including neovascularization. Nagpal and associates138 reported that their patients with sickle cell trait who had neovascularization also had associated underlying systemic diseases, including diabetes, hypertension, tuberculosis, syphilis, and sarcoidosis. The neovascularization in these cases was similar to that seen in sickle cell disease. They proposed that any patient with sickle cell trait and PSR be examined thoroughly for other underlying diseases. An additional consideration, and a frequent source of misdiagnosis, is the failure to perform a hemoglobin electrophoresis to clarify a positive sickle cell test. A sickle test can detect the presence of Hb S, but it cannot clarify whether a patient has sickle cell trait or one of the other genotypes associated with sickle cell eye disease. Sickle cell trait may be confused with Hb S-β+ -thalassemia unless a quantitative hemoglobin electrophoresis is done. This may account for some apparent cases of PSR with sickle cell trait. It is still unclear whether sickle cell trait alone can produce PSR.
Once PSR is established, the rates of progression vary greatly among patients. PSR does progress more rapidly in young patients with SC disease and homozygous sickle cell anemia.135
STAGE IV: VITREOUS HEMORRHAGE. Vitreous hemorrhage often complicates PSR. In a selected series of patients with untreated SC disease, vitreous hemorrhage was found in 28% at diagnosis and in 44% after 31 months.130 In the presence of neovascularization, the three risk factors for the development of vitreous hemorrhage include SC disease, more than 60° of perfused sea fans, and the presence of old blood in the eye.139 In a long-term follow-up of an untreated control group participating in a randomized clinical trial of feeder vessel photocoagulation for PSR, vitreous hemorrhage occurred in 45% of eyes and was recurrent in two thirds of these eyes.140
Transudation of plasma results in vitreous syneresis and fibrosis and induces collapse of the formed vitreous, which causes traction on the adherent neovascular tissue. The fragile elevated vessels in the neovascular membranes are prone to rupture, resulting in hemorrhage.141 The hemorrhage is frequently localized in the periphery near the sea fan, but diffuse hemorrhage does occur and may obscure fundus details (Color Plate 2C).
STAGE V: RETINAL DETACHMENT. Vitreous traction on the retina may cause tractional retinal detachments or retinal breaks that can result in localized or total rhegmatogenous retinal detachments.131,142 The retinal breaks are usually found adjacent to neovascular tissue and may be difficult to detect because of overlying hemorrhage. Exudative retinal detachments may occur in rare cases and reportedly have resolved after photocoagulation of the neovascularization.143