| Several developmental variations of the peripheral retina, defined as localized
topographic and structural deviations related to ocular development, are
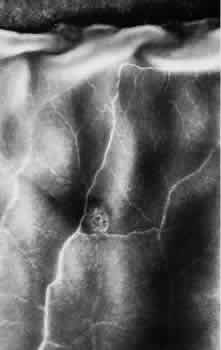
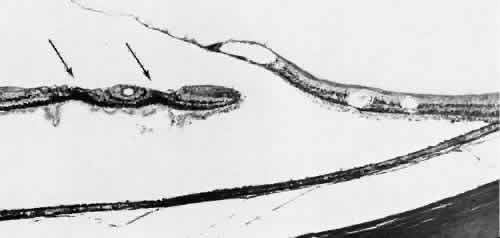
superimposed on the normal topography, structure, and relationships. MERIDIONAL FOLD A meridional fold is a radially oriented, ridgelike elevation on the peripheral
retina that projects into the vitreous, is aligned with a dentate
process or with the middle of an ora bay, originates at theora serrata, and
extends posteriorly for 0.6 to 6 mm.The surface of the fold
is slightly irregular; the thickened retina contains irregular cystoid
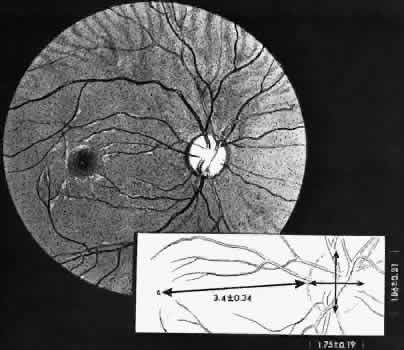
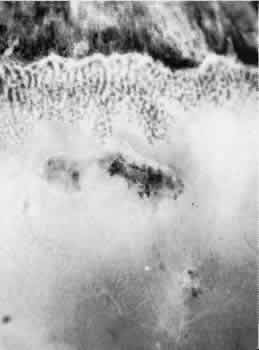
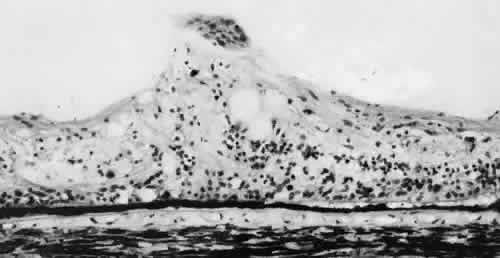
degeneration (Fig. 8).  Fig. 8. Meridional fold in a young patient. Retina is thickened along course of
fold, which shows microcystoid change near the surface and a cap of dense-staining
glial cells along its surface. Middle and outer layers of
the retina are largely unremarkable. Pigment epithelium shows focal
redundancy anteriorly. (Hematoxylin-eosin; × 150.) Fig. 8. Meridional fold in a young patient. Retina is thickened along course of
fold, which shows microcystoid change near the surface and a cap of dense-staining
glial cells along its surface. Middle and outer layers of
the retina are largely unremarkable. Pigment epithelium shows focal
redundancy anteriorly. (Hematoxylin-eosin; × 150.)
|
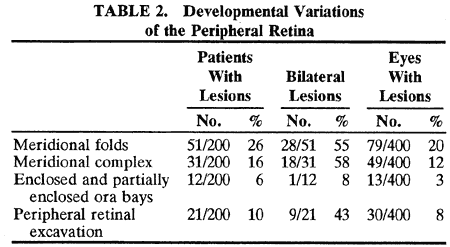
Meridional folds are present in 26% of the population and are bilateral
in 55% of the affected patients; thus, they are present in 20% of all
eyes (Table 2). Meridional folds are multiple in 27% of affected eyes; they are most
common in the superior nasal quadrant.5,6
MERIDIONAL COMPLEX A meridional complex is the occurrence of a dentate process and a ciliary
process within the same meridian. When this abnormal alignment occurs, the
den-tate process is exceptionally large, usually combined with
a meridional fold, usually continuous with an enlarged ciliary process, and
often associated with peripheral retinal excavation in the corresponding
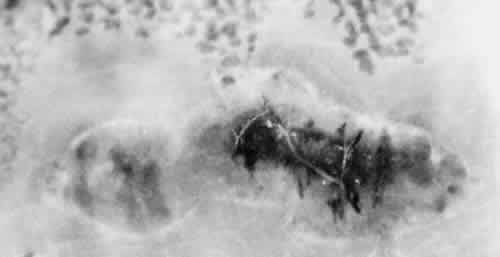
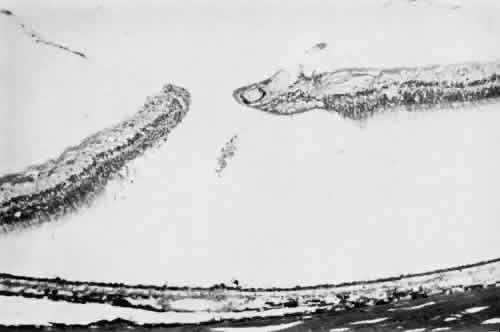
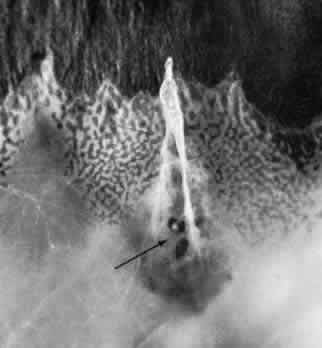
meridian (Fig. 9). The large dentate process and meridional fold are composed of excessive, disorganized, somewhat degenerated retinal tissue (Fig. 10). 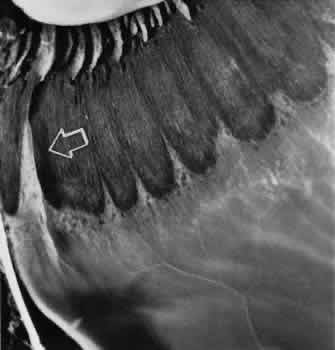 Fig. 9. Meridional complex (arrow). Note its basic constituent, an atypical dentate process, which aligns
with and extends to an enlarged ciliary process. Complex also has a
meridional fold which extends along the dentate process and posteriorly
into the peripheral retina. Fig. 9. Meridional complex (arrow). Note its basic constituent, an atypical dentate process, which aligns
with and extends to an enlarged ciliary process. Complex also has a
meridional fold which extends along the dentate process and posteriorly
into the peripheral retina.
|
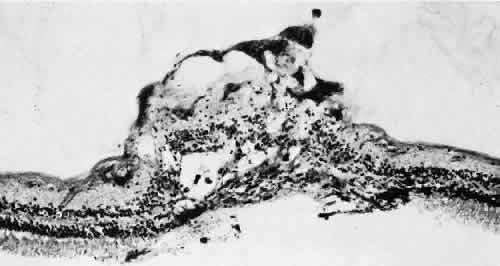
 Fig. 10. Microsection of meridional complex through atypical dentate process and
its meridional fold. Anteriorly (on the left) the complex shows marked
redundancy of pigmented epithelium in its outer aspect and a dense glial
plaque on its inner aspect. Posteriorly (on the right) there is microcystoid
change, nonspecific degeneration, and dense-staining glial
cells along its surface. (Hematoxylin-eosin; × 63.) Fig. 10. Microsection of meridional complex through atypical dentate process and
its meridional fold. Anteriorly (on the left) the complex shows marked
redundancy of pigmented epithelium in its outer aspect and a dense glial
plaque on its inner aspect. Posteriorly (on the right) there is microcystoid
change, nonspecific degeneration, and dense-staining glial
cells along its surface. (Hematoxylin-eosin; × 63.)
|
Meridional complexes are present in 16% of the population, are bilateral
in 58% of affected patients, and thus are present in 12% of all eyes (see Table 2). The complexes are multiple in 45% of the affected eyes; they are most
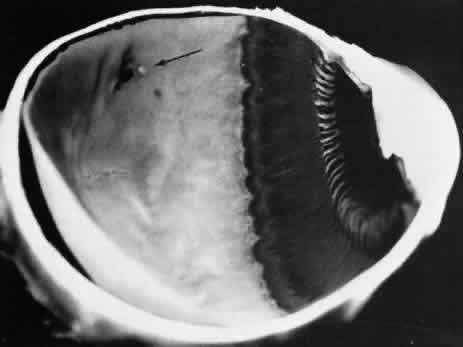
common in the superior nasal quadrant. ENCLOSED ORA BAY Enclosed and partially enclosed ora bays are relatively uncommon developmental
variations. These are oval islands of pars plana epithelium located
immediately posterior to the ora serrata and com-pletely or almost
completely circumscribed by the peripheral retina (Figs. 11 and 12). The enclosed ora bay is composed of a thin layer of nonpigmented pars
plana epithelium surrounded by neurosensory retina. Enclosed and partially
enclosed ora bays are evident in 6% of patients, are bilateral
in 8% of affected individuals, and are present in 3% of all eyes
(see Table 2). These lesions are equally prevalent nasally and temporally near the
horizontal meridian.6,7  Fig. 11. Partially enclosed ora bay in a 20-year-old woman. Posteriorly, the ora
bay extends 1.8 mm behind general line of ora serrata, and the retina
shows a large area of typical cystoid degeneration. Anteriorly, the ora
bay is embraced by two long dentate processes that converge toward, but
do not meet, a prominent ciliary process of the pars plicata. (× 12.) Fig. 11. Partially enclosed ora bay in a 20-year-old woman. Posteriorly, the ora
bay extends 1.8 mm behind general line of ora serrata, and the retina
shows a large area of typical cystoid degeneration. Anteriorly, the ora
bay is embraced by two long dentate processes that converge toward, but
do not meet, a prominent ciliary process of the pars plicata. (× 12.)
|
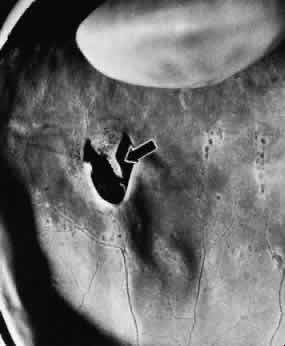
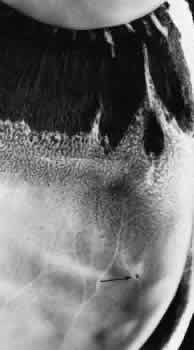 Fig. 12. Enclosed ora bay in a 35-year-old man. Anteriorly, two broad dentate processes
converge and join to enclose a bay (island of pars plana). Posteriorly
there is a focus of retinal thinning (peripheral retinal excavation; arrow). (× 12.) Fig. 12. Enclosed ora bay in a 35-year-old man. Anteriorly, two broad dentate processes
converge and join to enclose a bay (island of pars plana). Posteriorly
there is a focus of retinal thinning (peripheral retinal excavation; arrow). (× 12.)
|
PERIPHERAL RETINAL EXCAVATION Peripheral retinal excavation appears as a rather small oval depression
in the retina. Usually this lesion is aligned meridionally with a meridional
fold or complex and located 1 to 7.2 mm posterior to the ora serrata (Fig. 13; see Figs. 6 and 12). The focal depression may be surrounded by margins that appear to be
elevated; however, microscopic examination reveals that the depression
corresponds to afocal loss of the inner retinal layers and that the surrounding
tissue is normal (Fig. 14). 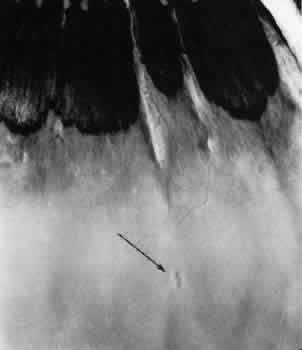 Fig. 13. Meridional complexes with peripheral retinal excavation. Two complexes
can be seen anteriorly; both contain meridional folds (the fold of complex
on the right is discontinuous). Peripheral retinal excavation (arrow) is aligned with the complex on the left (× 12.) Fig. 13. Meridional complexes with peripheral retinal excavation. Two complexes
can be seen anteriorly; both contain meridional folds (the fold of complex
on the right is discontinuous). Peripheral retinal excavation (arrow) is aligned with the complex on the left (× 12.)
|
 Fig. 14. Peripheral retinal excavation. The notable loss of tissue from the retinal
surface extends to the inner nuclear layer. Centrally, the middle
retinal layers show degeneration, but outer layers of retina are intact. (Hematoxylin-eosin; × 250.) Fig. 14. Peripheral retinal excavation. The notable loss of tissue from the retinal
surface extends to the inner nuclear layer. Centrally, the middle
retinal layers show degeneration, but outer layers of retina are intact. (Hematoxylin-eosin; × 250.)
|
Peripheral retinal excavation is present in 10% of patients, is bilateral
in 43%, and therefore is evident in 8% of all eyes (see Table 2). Half of the affected eyes contain two or more areas of focal excavation, and
most of the excavations are located in the superior nasal quadrant. Developmental variations of the peripheral retina are present in about 20% of
eyes. These conditions have certain common features: they are present
at birth, persist throughout life, tend to occur symmetrically
in anatomically corresponding positions in both eyes, and are commonly
associated with an abnormal alignment of a dentate and a ciliary process
in the same meridian.5 Clinically, these developmental abnormalities are readily identified in
the peripheral retina. The ridgelike elevation of a meridional fold is
best seen by scleral depression combined with indirect ophthalmoscopy. A
meridional complex should be suspected (even though the ciliary processes
are not visible) when a large dentate process, a meridional fold, and
peripheral retinal excavation occur in the same meridian. An
enclosed ora bay appears as a depressed, bright red area that simulates
a retinal hole. Although identification of an enclosed ora bay is aided
by a thin epithelial layer extending across the base and by the absence
of any elevation of the surrounding retina, in some instances, distinction
between an enclosed bay and a retinal hole is impossible. Peripheral
retinal excavation presents as a small retinal pit that may
be adjacent to or up to 4 disc diameters posterior to the ora serrata. This
form of focal depression probably has been often mistaken for a
full-thickness retinal break; therefore, knowledge of developmental variations
is essential to the appropriate diagnosis of a peripheral retinal
lesion. In clinical studies, retinal breaks posterior to meridional folds have
been noted in a number of eyes with rhegmatogenous retinal detachment.8 Retinal tears may also develop at or near the posterior margins of enclosed
ora bays.7 Therefore, when retinal detachment is present, the ophthalmologist must
carefully search for meridional folds, meridionalcomplexes, enclosed
ora bays, peripheral retinal excavations, and any associated retinal
breaks. TROPHIC PERIPHERAL RETINAL DEGENERATIONS Various specific peripheral retinal degenerations augment these developmental
variations. Degeneration (i.e., irreversible retrograde change) is
evident in the peripheral retina of every adult. For classification, this
degeneration may be regarded as trophic when the primary process
is a loss of retinal tissue, tractional when the process is related
primarily to tugging or pulling of vitreous or zonule on the retina, and
trophic and tractional when both retinal tissue loss and vitreous-zonule
traction are involved. The principal peripheral retinal degenerations
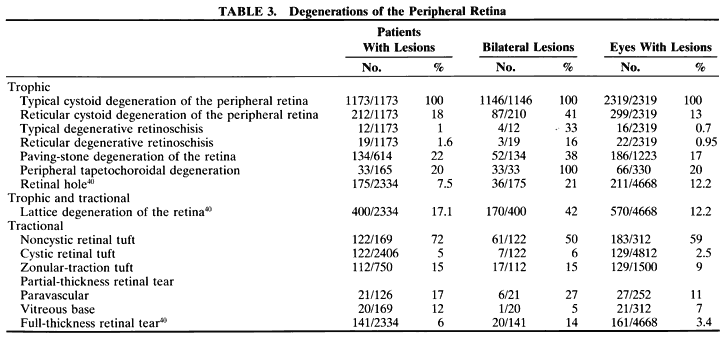
are noted in Table 3.
TYPICAL CYSTOID DEGENERATION The most common form of degeneration of the peripheral retina is typical
cystoid degeneration. Spaces develop in the outer plexiform and inner
nuclear layers and coalesce to form interlacing tunnels; they are separated
by pillars that extend from the inner to the outer retinal layers, giving
the inner surface a uniformly stippled appearance (Fig. 15). The stippled depressions correspond to retinal pillars; the intervening
rounded domes result from the intraretinal cystoid spaces.9 Degeneration begins at the ora serrata, particularly at the base of dentate
processes, and extends posteriorly and circumferentially to form
a band that may encircle the eye and reach from the ora serrata to the
equator (Fig. 16). 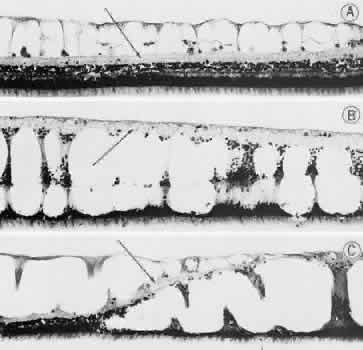 Fig. 15. Typical and retinal cystoid degenerations. A. Degeneration of nerve fiber layer with persistence of delicate vertical
columns of Müller's cells. Superficial capillary plexus courses
through the cystoid cavities, and surviving ganglion cells are subtended
on the inner aspect of the inner plexiform layer (arrow). Outer retinal layers are well preserved. B. Extensive degeneration of middle retinal layers with broad cellular columns (between
cystoid cavities) composed of Müller's cells
and remnants of outer plexiform layer and inner nuclear layer (vertically
stretched). Outer nuclear layer also shows degeneration. Inner plexiform
layer (arrow) is intact. C. Overlapping reticular (on the left) and typical (on the right) cystoid
degeneration. Note combined degenerative effect on the inner plexiform
layer (arrow) of both types of cystoid degeneration. Superficial small arteriole retards
progression of reticular cystoid degeneration. (Hematoxylin-eosin, × 250.) (Foos RY: Senile retinoschisis: Relationship to cystoid degeneration. Trans
Am Acad Ophthalmol Otolaryngol 1970;74:33.) Fig. 15. Typical and retinal cystoid degenerations. A. Degeneration of nerve fiber layer with persistence of delicate vertical
columns of Müller's cells. Superficial capillary plexus courses
through the cystoid cavities, and surviving ganglion cells are subtended
on the inner aspect of the inner plexiform layer (arrow). Outer retinal layers are well preserved. B. Extensive degeneration of middle retinal layers with broad cellular columns (between
cystoid cavities) composed of Müller's cells
and remnants of outer plexiform layer and inner nuclear layer (vertically
stretched). Outer nuclear layer also shows degeneration. Inner plexiform
layer (arrow) is intact. C. Overlapping reticular (on the left) and typical (on the right) cystoid
degeneration. Note combined degenerative effect on the inner plexiform
layer (arrow) of both types of cystoid degeneration. Superficial small arteriole retards
progression of reticular cystoid degeneration. (Hematoxylin-eosin, × 250.) (Foos RY: Senile retinoschisis: Relationship to cystoid degeneration. Trans
Am Acad Ophthalmol Otolaryngol 1970;74:33.)
|
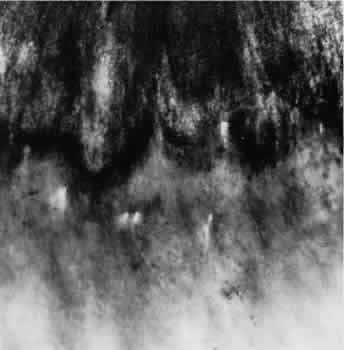
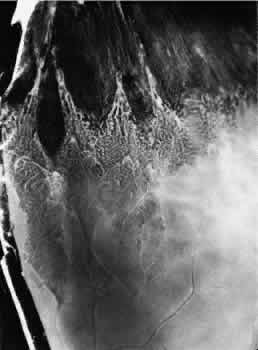 Fig. 16. Typical and reticular cystoid degeneration found immediately behind the
ora serrata and about enclosed ora bay near cut edge of calotte. Posteriorly, note
the conspicuous vascular pattern of degeneration (seen as
gray background), finely stippled surface pattern, and angular free
margins (related to limitation by surface vessels). Fig. 16. Typical and reticular cystoid degeneration found immediately behind the
ora serrata and about enclosed ora bay near cut edge of calotte. Posteriorly, note
the conspicuous vascular pattern of degeneration (seen as
gray background), finely stippled surface pattern, and angular free
margins (related to limitation by surface vessels).
|
This degenerative process may be noted in infants at 1 year of age; it
is always present in both eyes of patients over 8 years of age, usually
increases in area with advancing age, and is most extensive in the superior
and temporal quadrants (see Table 3).10,11 RETICULAR CYSTOID DEGENERATION Reticular cystoid degeneration of the peripheral retina is almost invariably
located posterior to and continuous with typical cystoid degeneration. It
is characterized by a prominent linear or reticular pattern
that corresponds to the retinal vessels and by a finely stippled internal
surface. Areas of involvement are single or multiple, form the shape
of an irregular angle, and are often demarcated posteriorly by retinal
blood vessels (see Fig. 16).12 Spaces develop in the nerve fiber layer and are divided by delicate retinal
pillars (see Fig. 15). Reticular cystoid degeneration is present in 18% of adult patients, is
bilateral in 41% of affected patients, and is thus evident in 13% of all
adult eyes (see Table 3). The process is most prevalent in the inferior temporal quadrant.13 Clinically reticular cystoid degeneration is detected on contact lens biomicroscopy
by noting the location, shape, prominent reticular pattern, and
finely stippled appearance of the affected area. DEGENERATIVE RETINOSCHISIS Degenerative retinoschisis, a more extensive trophic process, presents
as a round or ovoid area of retinal splitting with a smooth fusiform elevation
of the inner layer (Fig. 17). The schisis is surrounded on all sides by typical cystoid degeneration; the
retinal pillars of the cystoid degeneration as well as the broken
pillars near the margin of the schists are prominent. Vessels are
located in the inner retinal layer, the intraretinal cavity is optically
empty, and the outer retinal layer is moderately irregular in contour.13,14 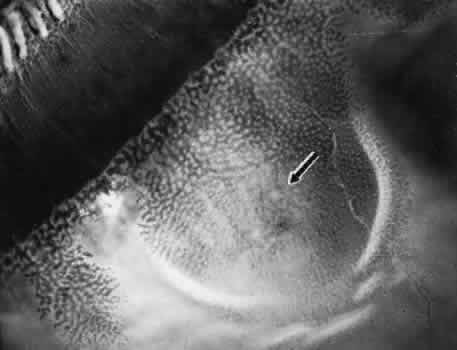 Fig. 17. Typical degenerative retinoschisis.Note extensive region of typical cystoid
degeneration with a rounded and elevated posterior margin. In the
center (arrow), radial columns are randomly disrupted, causing a disturbance in coarse
surface pattern. (× 18.) Fig. 17. Typical degenerative retinoschisis.Note extensive region of typical cystoid
degeneration with a rounded and elevated posterior margin. In the
center (arrow), radial columns are randomly disrupted, causing a disturbance in coarse
surface pattern. (× 18.)
|
In one type of degenerative retinoschisis, the thin inner wall is composed
of the internal limiting membrane, the nerve fiber layer, and retinal
vessels (Fig. 18). The irregular outer wall contains portions of the inner nuclear, outer
plexiform, outer nuclear, external limiting, and rod and cone layers. At
the margin of the cavity, the retinoschisis blends with typical
cystoid degeneration and may be relatively flat. Lesions with this appearance
have been termed typical degenerative retinoschisis.  Fig. 18. Typical degenerative retinoschisis. There is extensive tissue loss in middle
layers of the retina (typical cystoid degeneration); on the left, degeneration
has also destroyed radial supporting columns. (Hematoxylin-eosin; × 250.) Fig. 18. Typical degenerative retinoschisis. There is extensive tissue loss in middle
layers of the retina (typical cystoid degeneration); on the left, degeneration
has also destroyed radial supporting columns. (Hematoxylin-eosin; × 250.)
|
Typical degenerative retinoschisis is present in 1% of adult patients and
is bilateral in 33% of these patients; therefore, it is evident in 0.7% of
adult eyes (see Table 3), with a predilection for location in the inferior temporal quadrant. A
narrow band of typical cystoid degeneration is always present between
the ora serrata and the anterior border of the schisis cavity; the involved
area may extend to or somewhat posterior to the equator. On clinical examination, typical degenerative retinoschisis appears as
round or ovoid areas of retinal splitting with fusiform elevation of the
inner layer (Fig. 19). The stippled pattern of surrounding typical cystoid degeneration extends
on the inner layer for a variable distance; centrally the inner layer, which
contains the blood vessels, is thin and smooth. On contact
lens ophthalmoscopy, the inner layer is finely textured, some of the
retinal vessels are attenuated, and there is a variable number of tiny, glistening, white
dot opacities on the vitreous side. The outer layer, found
external to the optically empty cavity, is best seen with indirect
ophthalmoscopy when it becomes white on scleral depression. It is
somewhat uneven, giving an appearance of finely hammered or beaten metal. Typical
degenerative retinoschisis does not extend posteriorly to
threaten the macula, and it is not often associated with breaks in either
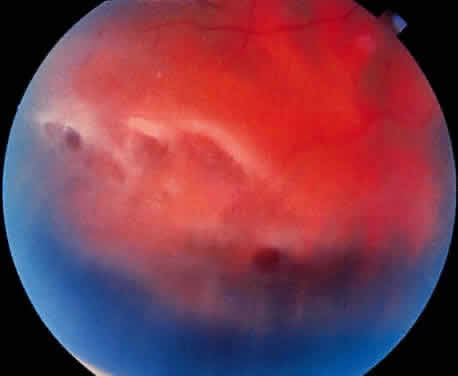
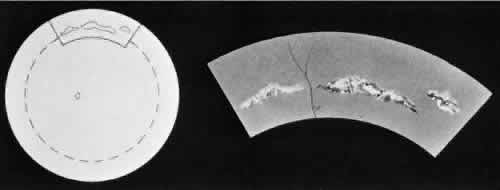
retinal layer; it rarely requires treatment.  Fig. 19. Clinical appearance of typical degenerative retinoschisis: diagram of involved
area and ocular fundus photographs showing optic disc, macula, and
posterior portion of schisis. Within the schisis and adjacent to
the margin is coarse stippling related to broken retinal pillars. Fig. 19. Clinical appearance of typical degenerative retinoschisis: diagram of involved
area and ocular fundus photographs showing optic disc, macula, and
posterior portion of schisis. Within the schisis and adjacent to
the margin is coarse stippling related to broken retinal pillars.
|
Retinoschisis associated with a bullous architecture and prominent reticular
cystoid degeneration has been termed reticular degenerative retinoschisis. Reticular
degenerative retinoschisis can be distinguished from
typical degenerative retinoschisis by the large extent of retinal
involvement, a round or ovoid configuration with bullous elevation of
the extremely thin inner layer, and an irregular, pitted outer layer
(Figs. 20 and 21). Typical cystoid degeneration is always present anterior to the schisis; reticular
cystoid degeneration is usually prominent at some site in
the involved eye. Blood vessels coursing through the inner layer give
it an arborizing reticular pattern on contact lens biomicroscopy. The
intraretinal cavity is optically empty; the outer wall is irregularly
excavated to produce a pocked or honeycomb appearance. Round or ovoid
holes are often present in the outer retinal layer; they are single
or multiple, frequently large, and usually associated with a rolled posterior
edge.13 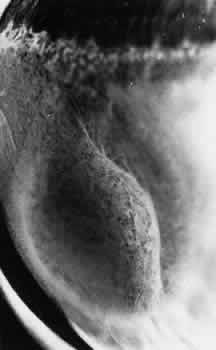 Fig. 20. Reticular degenerative retinoschisis. Note reticulated, highly elevated, inner
wall with a conspicuous delicate vascular pattern. Radial columns
of the retina are completely disrupted within the region of bullous
elevation, and the retinoschisis extends posterior to the equator. (× 18.) Fig. 20. Reticular degenerative retinoschisis. Note reticulated, highly elevated, inner
wall with a conspicuous delicate vascular pattern. Radial columns
of the retina are completely disrupted within the region of bullous
elevation, and the retinoschisis extends posterior to the equator. (× 18.)
|
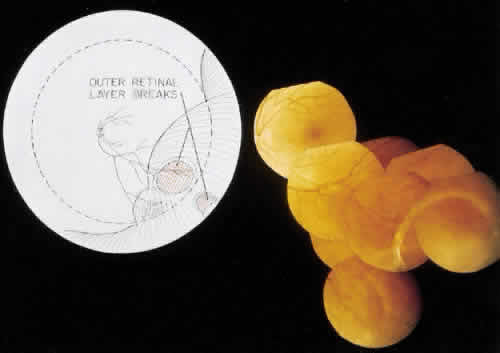 Fig. 21. Clinical appearance of reticulardegenerative retinoschisis: diagram of
involved area and photographs of ocular fundus showing optic disc, macula, and
posterior portion of the schisis. These illustrate outer layer
retinal breaks, adjacent retinal pigment epithelium abnormality, and
a lo-calized nonrhegmatogenous retinal detach-ment. Fig. 21. Clinical appearance of reticulardegenerative retinoschisis: diagram of
involved area and photographs of ocular fundus showing optic disc, macula, and
posterior portion of the schisis. These illustrate outer layer
retinal breaks, adjacent retinal pigment epithelium abnormality, and
a lo-calized nonrhegmatogenous retinal detach-ment.
|
Microscopic sections demonstrate the extremely attenuated, blood vessel-containing
inner layer composed of the internal limiting membrane and
remnants of the nerve fiber layer (Fig. 22). The honeycomb appearance of the outer layer corresponds to irregular
excavations. In some areas, the outer layer is made up of outer plexiform, outer
nuclear, external limiting, and rod and cone layers; in other
areas it is reduced to only the external limiting and the rod and
cone layers; round or ovoid holes may be present (Fig. 23). 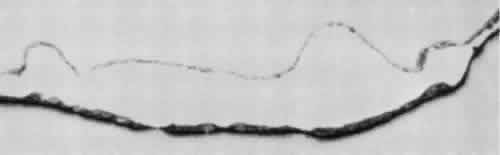 Fig. 22. Reticular degenerative retinoschisis. Note complete loss of radial supporting
columns of retina and marked elevation of delicate inner wall, which
contains fine blood vessels. Outer wall shows periodic exaggerated
thinning. (Hematoxylin-eosin; × 60.) Fig. 22. Reticular degenerative retinoschisis. Note complete loss of radial supporting
columns of retina and marked elevation of delicate inner wall, which
contains fine blood vessels. Outer wall shows periodic exaggerated
thinning. (Hematoxylin-eosin; × 60.)
|
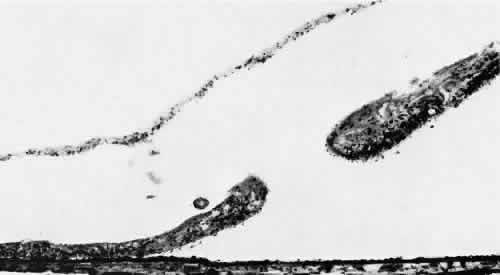 Fig. 23. Reticular degenerative retinoschisis with hole in outer wall and localized
retinal detachment. Margins of hole are rolled and covered by a garland
of degenerating photoreceptor outer segments. (Hematoxylin-eosin; × 250.) Fig. 23. Reticular degenerative retinoschisis with hole in outer wall and localized
retinal detachment. Margins of hole are rolled and covered by a garland
of degenerating photoreceptor outer segments. (Hematoxylin-eosin; × 250.)
|
Reticular degenerative retinoschisis is evident in 1.6% of adult patients, is
bilateral in only 16% of these, and thus is noted in 0.95% of adult
eyes (see Table 3). The lesion is found most commonly in the inferior temporal quadrant. A
band of typical cystoid degeneration always separates the schisis from
the ora serrata; the schists usually reaches the equator and often
extends appreciably into the posterior retina. On contact lens biomicroscopy, many retinal blood vessels present irregular
contours, telangiectases, occluded segments, and microaneurysms. Between
these vessels, the inner wall has a finely textured appearance
and variable white, glistening particles on the vitreous side. The outer
retinal wall is best seen when scleral depression produces a “white
with pressure” phenomenon and reveals the honeycomb appearance. The
retinal pigment epithelium often has a granular, salt-and-pepper
appearance, and outer-layer retinal breaks are common. These breaks
are particularly likely near the anterior and posterior margins of
the schisis. Treatment for reticular degenerative retinoschisis is rarely necessary.15 Treatment is indicated when the schisis cavity is symptomatic or progressive
and associated with a rhegmatogenous component from both inner
and outer wall retinal holes.15 Surgical management has included: external drainage ofsubretinal fluid
with simultaneous intraocular gas injection,16 pars plana vitrectomy with either limited inner wall retinectomy and internal
drainage through preexistent outer wall holes17 or with perfluorocarbon liquid assisted anterior displacement of subretinal
fluid.18 No treatment is indicated for nonprogressive reticular degenerative retinoschisis
that does not extend toward the macula. Photographic documentation
of the posterior borders of the retinoschisis cavity may be useful
in clinical monitoring for progression. PAVING-STONE DEGENERATION Paving-stone degeneration of the retina is characterized by one or more
discrete, rounded foci of depigmentation and retinal thinning located
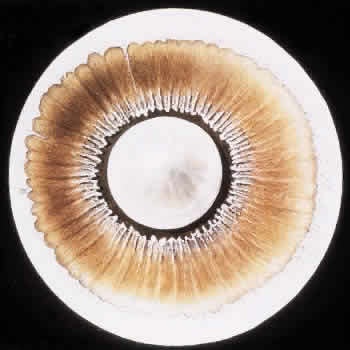
between the ora serrata and the equator (Fig. 24). The lesions appear yellow-white, frequently reveal prominent underlying
choroidal vessels, and often have a pigmented margin
(Fig. 25). The basic lesion is rounded and 0.1 to 1.5 mm in diameter. Clusters
of these rounded foci may merge to form larger lesions with convexly scalloped
margins and incomplete pigmented septa;19 these features aid in distinguishing paving-stone degeneration from postinflammatory
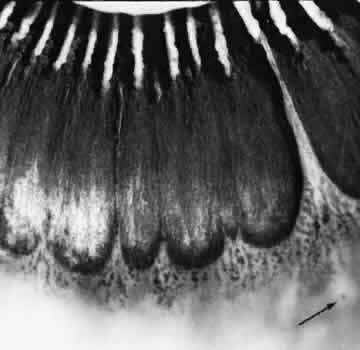
chorioretinal lesions. 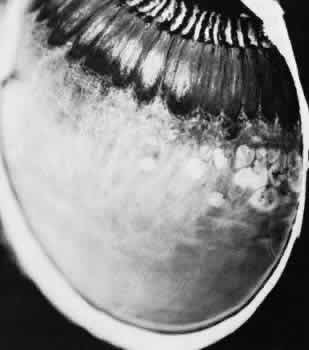 Fig. 24. Paving-stone degeneration. Note multiple round foci of depigmentation in
temporal sector. Larger lesions show scalloped margins and linear pigmentation
resulting from confluence of several smaller lesions. (× 65.) Fig. 24. Paving-stone degeneration. Note multiple round foci of depigmentation in
temporal sector. Larger lesions show scalloped margins and linear pigmentation
resulting from confluence of several smaller lesions. (× 65.)
|
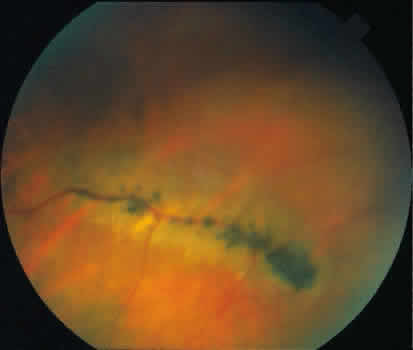
 Fig. 25. Multiple discrete yellow-white lesions in the peripheral fundus in paving-stone
degeneration. Fig. 25. Multiple discrete yellow-white lesions in the peripheral fundus in paving-stone
degeneration.
|
Paving-stone degeneration is associated with sharply circumscribed retinal
thinning due to loss of the rods and cones and external limiting membrane, absence
of the pigment epithelium, adherence of the retina to
Bruch's membrane, and alteration of the choriocapillaris (Fig. 26). The hyperpigmented cuffs and septa consist of proliferated pigment epithelial
cells.  Fig. 26. Paving-stone degeneration. Within the lesion, there is selective loss of
outer retinal layers,including photoreceptor cells, outer plexiform
layer, and to a lesser extent, inner nuclear layer. The choroidand Bruch's
membrane are intact. Retinal pigment epithelium cannot be discerned. (Hematoxylin-eosin; × 250.) Fig. 26. Paving-stone degeneration. Within the lesion, there is selective loss of
outer retinal layers,including photoreceptor cells, outer plexiform
layer, and to a lesser extent, inner nuclear layer. The choroidand Bruch's
membrane are intact. Retinal pigment epithelium cannot be discerned. (Hematoxylin-eosin; × 250.)
|
Paving-stone degeneration is present in 22% of adult patients, is bilateral
in 38% of these, and thus is evident in 17% of adult eyes (see Table 3). Prevalence increases markedly with advancing age. In distribution, there
is a preference for the inferior temporal and inferior nasal quadrants, with
more than half of the lesions located in the inferior retina
between the 5- and 7-o'clock positions. Lesions are most numerous immediately posterior to the ora serrata; they
rarely extend posterior to the equator. Paving-stone degeneration does not predispose to retinal breaks or to retinal
detachment and thus does not warrant any form of treatment. However, if
a retinal detachment from some other cause involves an area of
paving-stone degeneration, the retina may be torn at the site of the
chorioretinal adherence. The resultant retinal breaks are often small, irregular
in shape, inconspicuous, and best detected with contact lens
biomicroscopy. PERIPHERAL TAPETOCHOROIDAL DEGENERATION With advancing age, the peripheral fundus usually develops a granular appearance, due
to irregularity of the retinal pigment epithelium. Peripheral
tapetochoroidal degeneration, a pronounced degree of this alteration, presents
as a diffusely depigmented circumferential band that extends
from the ora serrata to the equator. The anterior border is irregular
because of splotchy pigmentation that remains within the area of
the vitreous base, but the posterior border is usually smooth and well
defined.11 Peripheral tapetochoroidal degeneration is associated with degeneration
and loss of pigment granules in the retinal pigment epithelium, some
loss of photoreceptor cells, diffuse thickening of Bruch's membrane, and
a diminution of capillaries in the choriocapillaris. Although minor degrees of degeneration of the retinal pigment epithelium
are common in older patients, extensive alterations that meet the criteria
of peripheral tapetochoroidal degeneration are present in 20% of
patients over 40 years of age, are bilateral in all cases, and thus
are present in 20% of eyes in this group (see Table 3). This age-related process does not predispose to retinal tear formation
and requires no treatment. RETINAL HOLE Most retinal holes (76%) are secondary to lattice degeneration.20 Primary retinal holes, unrelated to lattice degeneration or other identifiable
disorders, are rounded, full-thickness retinal breaks without
a flap or a free operculum
(Fig. 27). Usually located anteriorly within the area of the vitreous base, the
holes have smooth margins; the adjacent retina usually appears normal; proliferative
reactions are absent; and vitreous attachments are not
unusual.11 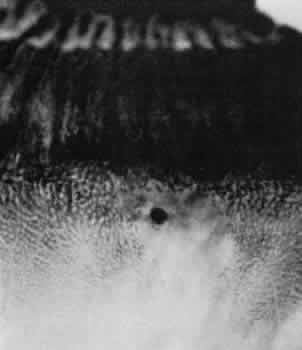 Fig. 27. Retinal hole with round, full-thickness break immediately behind the ora
serrata in an area of relatively normal retina. Typical cystoid degeneration
extensively involves retina on both sides of retinal hole. (× 16.) Fig. 27. Retinal hole with round, full-thickness break immediately behind the ora
serrata in an area of relatively normal retina. Typical cystoid degeneration
extensively involves retina on both sides of retinal hole. (× 16.)
|
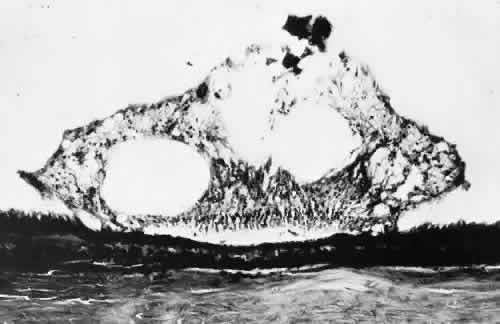
Microsections of retinal holes confirm the complete retinal discontinuity
and the smooth, rounded margins (Fig. 28). There is minimal reactive gliosis; no significant alteration is found
in the adjacent vitreous body or in the pigment epithelium. 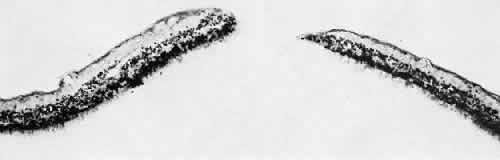 Fig. 28. Full-thickness retinal break in otherwise normal-appearing peripheral retina. Outer
retinal layers are intact. (Hematoxylin-eosin; × 250.) Fig. 28. Full-thickness retinal break in otherwise normal-appearing peripheral retina. Outer
retinal layers are intact. (Hematoxylin-eosin; × 250.)
|
Primary retinal holes are present in 7.5% of adults, they are bilateral
in 21% of patients, and thus are evident in 12% of adult eyes (see Table 3). Virtually all retinal holes occur within the vitreous base and give
no evidence of quadrant predilection. Retinal holes are detected clinically by indirect ophthalmoscopy and by
contact lens biomicroscopy. Scleral depression is helpful in differentiating
a retinal hole from a round retinal hemorrhage: the retinal hole
develops a changing reddish color as it moves over the crest of the
scleral depression, and the round retinal hemorrhage maintains a stable
red color as it moves over the crest of the scleral depression. Asymptomatic
round retinal holes with or without free opercula do not generally
require treatment. A rhegmatogenous retinal detachment rarely originates
from an untreated retinal hole.21 However, retinal holes located within a rhegmatogenous retinal detachment
should be treated during the surgical repair as these can be a source
of persistent subretinal fluid postoperatively. |