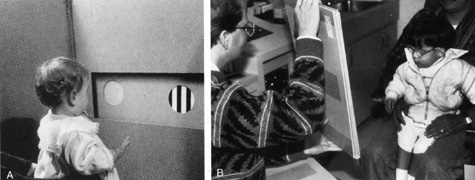
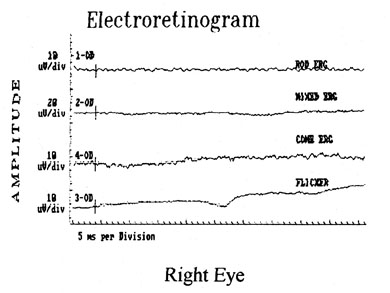
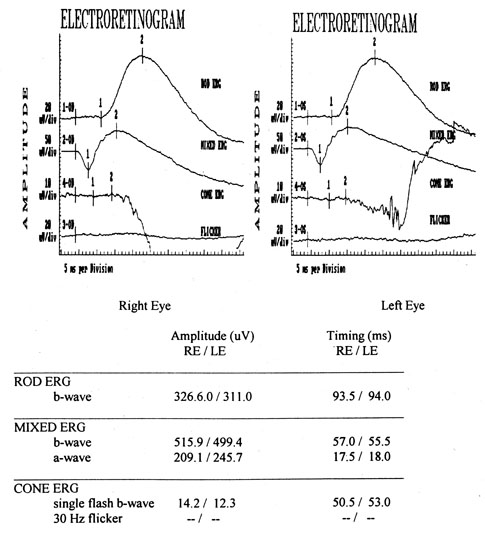
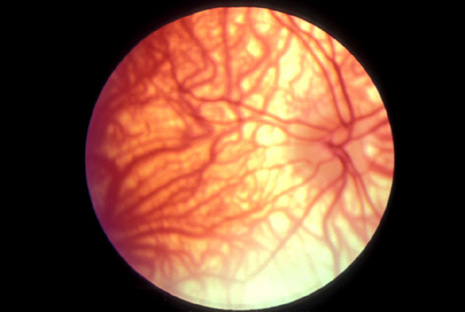
1. Jannerod M: The Neural and Behavioral Organization of Goal-Directed Movements. Oxford: Clarendon Press, 1988 2. Mayer DL, Beiser AS, Warner AF, et al: Monocular acuity norms for the Teller acuity cards between ages one month
and four years. Invest Ophthalmol Vis Sci 36:671, 1995 3. Mash C, Dobson V, Carpenter N: Interobserver agreement for measurement of grating acuity and interocular
acuity differences with the Teller acuity card procedure. Vision Res 35:303, 1995 4. Quinn GE, Berlin JA, James M: The Teller acuity card procedure: three testers in a clinical setting. Ophthalmology 100:488, 1993 5. Kushner BJ, Lucchese NJ, Morton GV: Grating visual acuity with Teller cards compared with Snellen visual acuity
in literate patients. Arch Ophthalmol 113:485, 1995 6. Kushner BJ: Editorial: grating acuity tests should not be used for social service purposes
in preliterate children. Arch Ophthalmol 112:1030, 1994 7. Holmes JM, Archer SM: Vernier acuity cards: a practical method of measuring vernier acuity in
infants. J Pediatr Ophthalmol Strabismus 30:312, 1993 8. Hoyt CS: The clinical usefulness of the visual evoked response. J Pediatr Ophthalmol Strabismus 21:231, 1984 9. Roy MS, Barsoum-Homsy M, Orquin J, Benoit J: Maturation of binocular pattern visual evoked potentials in normal full-term
and preterm infants from 1 to 6 months of age. Pediatr Res 17:140, 1995 10. Mohn G, VanHof-van Duin J: Development of the binocular and monocular visual fields of human infants
during the first year of life. Clin Vision Sci 1:51, 1986 11. American Academy of Pediatrics Policy Statement: Eye examination in infants, children
and young adults. Ophthalmology 110:860, 2003 12. Tychsen L: Development of vision. In Isenberg S (ed): The Eye in Infancy. St. Louis: CV Mosby, 1994:121 13. Thorn F, Gwiazda J, Cruz AAV, et al: The development of eye alignment, convergence, and sensory binocularity
in young infants. Invest Ophthalmol Vis Sci 35:544, 1994 14. Aiello A, Wright KW, Borchert M: Independence of optokinetic nystagmus asymmetry and binocularity in infantile
esotropia. Arch Ophthalmol 112:1580, 1994 15. Fielder AR, Fulton AB, Mayer DL: Visual development of infants with severe ocular disorders. Ophthalmology 98:1306, 1991 16. Bradford GM, Kutschke PJ, Scott WE: Results of amblyopia therapy in eyes with unilateral structural abnormalities. Ophthalmology 99:1616, 1992 17. Willshaw HE: Assessment of nystagmus. Arch Dis Child 69(a):102, 1993 18. Gelbart SS, Hoyt CS: Congenital nystagmus: a clinical perspective in infancy. Graefes Arch Clin Exp Ophthalmol 226:178, 1988 19. Cibis GW, Fitzgerald KM: Electroretinography in congenital idiopathic nystagmus. Pediatr Neurol 9:369, 1993 20. Lambert SR, Taylor D, Kriss A, et al: The infant with nystagmus, normal appearing fundi, but an abnormal ERG. Surv Ophthalmol 34:173, 1989 21. Porto FB, Perrault I, Hicks D, et al: Prenatal human ocular degeneration in Leber's congenital amaurosis (LCA2). J Gene Med 4:390, 2002 22. Heher KL, Traboulsi El, Maumenee IH: The natural history of Leber's congenital amaurosis. Ophthalmology 99:241, 1992 23. Koenekoop RK, Loyer M, Dembinska O, et al: Visual improvement in Leber congenital amaurosis and the CRX genotype. Ophthalmic Genet 1:49, 2002 24. Van Hooser JP, Liang Y, Maeda T, et al: Recovery of visual functions in a mouse model of Leber congenital amaurosis. J Biol Chem 277:19173, 2002 25. Bouzas EA, Caruso RC, Drews-Bankiewica MA, et al: Evoked potential analysis of visual pathways in human albinism. Ophthalmology 101:309, 1994 26. Kushner BJ: Infantile uniocular blindness with bilateral nystagmus: A syndrome. Arch Ophthalmol 113:1298, 1995 27. Tornqvist K, Ericsson A, Kallen B: Optic nerve hypoplasia: risk factors and epidemiology. Acta Ophthalmol Scand 80:300, 2002 28. Brodsky MC, Glasier CM: Optic nerve hypoplasia: clinical significance of associated central nervous
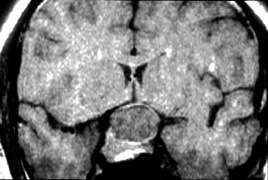
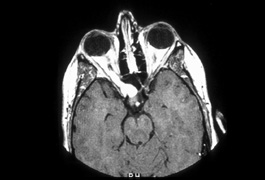
system abnormalities on magnetic resonance imaging. Arch Ophthalmol 111:66, 1993 29. Zeki SM, Hollman AS, Dutton GN: Neuroradiological features of patients with optic nerve hypoplasia. J Pediatr Ophthalmol Strabismus 29:107, 1992 30. Siatkowski RM, Sanchez JC, Andrade R, Alvarez A: The clinical, neuroradiographic, and endocrinologic profile of patients
with bilateral optic nerve hypoplasia. Ophthalmology 104:493, 1997 31. Tajima T, Hattorri T, Nakajima T, et al: Sporadic heterozygous frameshift mutation of HESX1 causing pituitary and
optic nerve hypoplasia and a combined pituitary hormone deficiency in
a Japanese patient. J Clin Endocrinol Metab 88:45, 2003 32. Purvin VA. Superior segmental optic nerve hypoplasia. J Neuroophthalmol 22:116, 2002 33. Lempert P. Optic nerve hypoplasia and small eyes in presumed amblyopia. J AAPOS 4:258, 2000 34. Brodsky MC, Hoyt WF, Hoyt CF, et al: Atypical retinochoroidal coloboma in patients with dysplastic optic discs
and transsphenoidal encephalocele. Arch Ophthalmol 113:624, 1995 35. Eustis HS, Sanders MR, Zimmerman T: Morning glory syndrome in children: association with endocrine and central
nervous system anomalies. Arch Ophthalmol 112:204, 1994 36. Good WV, James EJ, DeSa L, et al: Cortical visual impairment in children. Surv Ophthalmol 38:351, 1994 37. Wong VC: Cortical blindness in children: a study of etiology and prognosis. Pediatr Neurol 7:178, 1991 38. Hoyt CS. Visual function in the brain-damaged child. Eye 17:369, 2003 39. Jacobson L, Ek U, Fernell E, et al: Visual impaiment in preterm children with periventricular leukomalacia-visual, cognitive, and
neuropediatric characteristics related to magnetic
resonance imaging. Dev Med Child Neurol 38:724, 1996 40. Brodsky MC: Periventricula leukomalacia: an intracranial cause of pseudoglaucomatous
cupping. Arch Ophthalmol 199:626, 2001 41. Fielder AR, Mayer DL: Delayed visual maturation. Semin Ophthalmol 6:182, 1991 42. Kraemer M, Sjostrom A: Lack of short-latency potentials in the VEP reflects immature extra-geniculate
visual function in delayed visual maturation (DVM). Doc Ophthalmol 97:189, 1998 43. Cocker KD, Moseley MJ, Stirling , et al: Delayed visual maturation: Pupillary responses implicate subcortical and
cortical visual systems. Dev Med Child Neurol 40:160, 1998 44. Harris CM, Kriss A, Shawkat , et al: Delayed visual maturation in infants: a disorder of figure-ground separation? Brain Res Bull 40:365, 1996 44A. Hoyt CS. Delayed visual maturation: the apparently blind infant. J Am Assoc Ped Ophthalmol Strab 8:215, 2004 45. Herzau V, Bleher I, Joos-Kratsch E: Infantile exotropia with homonymous hemianopia: a rare contraindication
for strabismus surgery. Graefes Arch Clin Exp Ophthalmol 226:148, 1988 46. Göte H, Gregersen E, Rindziunski E: Exotropia and panoramic vision compensating for an occult congenital homonymous
hemianopia: a case report. Binocular Vision Eye Muscle Surg Q 8(3):125, 1993 47. Farris BK, Pickard DJ: Bilateral postinfectious optic neuritis and intravenous steroid therapy
in children. Ophthalmology 97:339, 1990 48. Kennedy C, Carroll FD: Optic neuritis in children. Arch Ophthalmol 63:747, 1960 49. Kennedy C, Carter S: Relation of optic neuritis to multiple sclerosis in children. Pediatrics 28:377, 1961 50. Taylor D, Cuendet F: Optic neuritis in childhood. In: Hess RF, Plant GT (eds): Optic Neuritis. Cambridge, UK: Cambridge University Press, 1986:73 51. Brady KM, Brar AS, Lee AG, et al: Optic neuritis in children: clinical features and visual outcome. J AAPOS 3:68, 1999 52. Morales DS, Siatkowski RM, Howard CW, et al: Optic neuritis in children. J Ped Ophthalmol 37:254, 2000 53. Luchinetti CF, Kiers L, et al: Risk factors for developing multiple sclerosis after childhood optic neuritis. Neurology 49:1413, 1997 54. Johns DR, Heher KL, Miller NR, Smith KH: Leber's hereditary optic neuropathy. Arch Ophthalmol 111:495, 1993 55. Dubois LG, Feldon SE: Evidence for a metabolic trigger for Leber's hereditary optic neuropathy: a
case report. J Clin Neuroophthalmol 12:15, 1992 56. Mackey DA, Buttery RG: Leber hereditary optic neuropathy in Australia. Austr NZ J Ophthalmol 20:177, 1992 57. Newman NJ, Lott MT, Wallace DC: The clinical characteristics of pedigrees of Leber's hereditary optic
neuropathy with the 11778 mutation. Am J Ophthalmol 111:750, 1991 58. Dutton JJ: Gliomas of the anterior visual pathway. Surv Ophthalmol 38:427, 1994 59. Hoffman HJ, Humphreys RP, Drake JM, et al: Optic pathway/hypothalamic gliomas: a dilemma in management. Pediatr Neurosurg 19(4):186, 1993 60. Gayre GS, Scott IU, Feuer WF, et al: Long-term visual outcome in patients with anterior visual pathway gliomas. J Neuro-ophthalmol 21:1, 2001 61. Liu GT, Lessell S: Spontaneous visual improvement in chiasmal gliomas. Am J Ophthalmol 114:193, 1992 62. Brzowski AE, Bazan C, Mumma JV, et al: Spontaneous regression of optic glioma in a patient with neurofibromatosis. Neurology 42(3 Pt 1):679, 1992 63. Hoyt CS, Billson F, Ouvrier R, et al: Ocular features of Aicardi's syndrome. Arch Ophthalmol 96:291, 1978 64. DelPero RA, Mets MB, Tripathi RC, et al: Anomalies of retinal architecture in Aicardi syndrome. Arch Ophthalmol 104:1659, 1986 65. Rosser TL, Acosta MT, Packer RJ: Aicardi syndrome: spectrum of disease and long-term prognosis in 77 females. Pediatr Neurol 27:343, 2002 66. Cooper JD: Progress towards understanding the neurobiology of Batten disease or neuronal
ceroid lipofuschinosis. Curr Opin Neurol 16:121, 2003 67. Bohra LI, Weizer JS, Lee AG, et al: Vision loss as the presenting sign in juvenile neuronal ceroid lipofuscinosis. J Neurol Ophthalmol 10:111, 2000 68. Hoyt WF, Nachtigaller H: Anomalies of ocular motor nerves: neuroanatomic correlates of paradoxical
innervation in Duane's syndrome and related congenital ocular
motor disorders. Am J Ophthalmol 60:443, 1965 69. Phillips WH, Dirion JK, Graves GO: Congenital bilateral palsy of abducens. Arch Ophthalmol 8:355, 1932 70. Blodi FC: Electromyographic evidence for supranuclear gaze palsies. Trans Ophthalmol See UK 90:451, 1970 71. Demer JL: The orbital pulley system: a revolution in concepts of orbital anatomy. Ann NY Acad Sci 956:17, 2002 72. Demer JL: Ocular kinematics, vergence, and orbital mechanics. Strabismus 11:49, 2003 73. Kono R, Clark RA, Demer JL: Active pulleys: magnetic resonance imaging of rectus muscle paths in tertiary
gazes. Invest Ophthalmol Vis Sci 43:2179, 2002 74. Clark RA, Miller JM, Demer JL: Three-dimensional location of human rectus pulleys by path inflections
in secondary gaze positions. Invest Ophthalmol Vis Sci 41:3787, 2000 75. Demer JL, Oh SY, Poukens V: Evidence for active control of rectus extraocular muscle pulleys. Invest Ophthalmol Vis Sci 41:1280, 2000 76. Clark RA, Miller JM, Demer JL: Locations and stability of rectus muscle pulleys. Muscle paths as a function
of gaze. Invest Ophthalmol Vis Sci 38:227, 1997 77. Oh SY, Clark RA, Velez F, Rosenbaum AL, Demer JL: Incomitant strabismus associated with instability of rectus pulleys. Invest Ophthalmol Vis Sci 43:2169, 2002 78. Huber A: Electrophysiology of the retraction syndromes. Br J Ophthalmol 58:293, 1974 79. Matteucci P: I difetti congeniti di abduzione (“congenital abduction deficiency”) con
particolare riguardo alla patogenesi. Rass Ital Ottal 15:345, 1946 80. Hotchkiss MG, Miller NR, Clark AW, Green WR: Bilateral Duane's retraction syndrome: a clinical-pathologic case
report. Arch Ophthalmol 98:870, 1980 81. Miller NR, Kiel SM, Green WR, Clark AW: Unilateral Duane's retraction syndrome (type 1). 100:1468, 1982 82. Mims JL III: Describing Duane's retraction syndrome. Binoc Vis Strabismus Q 17:86, 2002 82A. Thomas R, Mathai A, Gieser SC, et al: Bilateral synergistic divergence. J Pediatr Ophthalmol Strabismus 30:122, 1993 83. Scott AB, Wong GY: Duane's syndrome: An electromyographic study. Arch Ophthalmol 87:140, 1972 84. Pressman SH, Scott WE: Surgical treatment of Duane's syndrome. Ophthalmology 93:29, 1986 85. Raab EL: Clinical features of Duane's syndrome. J Pediatr Ophthalmol Strabismus 33:64, 1986 86. Hoffman RJ: Monozygotic twins concordant for bilateral Duane's retraction syndrome. Am J Ophthalmol 99:563, 1985 87. Chew CKS, Foster P, Hurst JA, et al: Duane's retraction syndrome associated with chromosome 4g27-31 segment
deletion. Am J Ophthalmol 119:807, 1995 88. Cullen P, Rodgers CS, Callen DF, et al: Association of familial Duane anomaly and urogenital abnormalities with
a bisatellited marker derived from chromosome 22. Am J Med Genet 47:925, 1993 89. Tredici TD, von Noorden GK: Are anisometropia and amblyopia common in Duane's syndrome? J Pediatr Ophthalmol Strabismus 22:23, 1985 90. Osher RH, Schatz NJ, Duane TD: Acquired orbital retraction syndrome. Arch Ophthalmol 98:1798, 1980 91. Miller GR, Glaser JS: The retraction syndrome and trauma. Arch Ophthalmol 76:662, 1966 92. Kivlin JD, Lundergan MK: Acquired retraction syndrome associated with orbital metastasis. J Pediatr Ophthalmol Strabismus 22:109, 1985 93. Rubenstein AE, Lovelace RE, Behrens MM, Weisburg LA: Möbius syndrome in association with peripheral neuropathy and Kallman
syndrome. Arch Neurol 32:480, 1975 94. Wishnick MM, Nelson LB, Huppert L, Reich EW: Möbius syndrome and limb abnormalities with dominant inheritance. Ophthalmic Pediatr Genet 2:77, 1983 95. Miller MT, Stromland K: The Mobius sequence: a relook. J AAPOS 3:199, 1999 96. Croemberger MF, de Castro Moreira JB, Brunoni D, et al: Ocular and clinical manifestations of Mobius syndrome. J Pediatr Ophthalmol Strabismus 38:156, 2001 97. Johansson M, Wentz E, Fernell E, et al: Autistic spectrum disorders in Mobius sequence: a comprehensive study of 25 individuals. Dev Med Child Neurol 43:338, 2001 98. Elsahy N: Möbius syndrome associated with mother taking thalidomide during gestation. Plast Reconstr Surg 51:93, 1973 99. Pedraza S, Gemez J, Rovira A, et al: MRI findings in Mobius syndrome: correlation with clinical features. Neurology 55:1058, 2000 100. Konkol RJ, D'Cruz O, Splaingard M: Central respiratory dysfunction in Möbius syndrome. J Child Neurol 6:278, 1991 101. Gilmore RL, Falace P, Kanga J, et al: Sleep-disordered breathing in Möbius syndrome. J Child Neurol 6:73, 1991 102. Szajnberg NM: Möbius syndrome: alternatives in affective communication. Dev Med Child Neurol 36:459, 1994 103. Terzis JK, Noah Em: Dynamic restoration in Mobius and Mobius-like patients. Plast Reconstr Surg 111:40, 2003 104. Terzis JK, Noah EM: Mobius and Mobius-like patients: etiology, diagnosis, and treatment options. Clin Plast Surg 29:497, 2002 105. Zuker RM, Goldberg CS, Lanktelow RT: Facial animation in children with Mobius syndrome after segmental bracilis
muscle transplant. Plast Reconstr Surg 106:1, 2000 106. Goldberg C, DeLorie R, Zuker RM, et al: The effects of gracilis muscle transplantaiton on speech in children with
Moebisu syndrome. J Craniofac Surg 14:687, 2003 107. Ziffer AJ, Rosenbaum AL, Demer JL, et al: Congenital double elevator palsy: vertical saccadic velocity utilizing
the scleral search coil technique. J Pediatr Ophthalmol Strabismus 29:142, 1992 108. Jampel RS, Fells P: Monocular elevation paresis caused by a central nervous system lesion. Arch Ophthalmol 80:45, 1968 109. Ford CS, Schwartze GM, Weaver RG, Troost BT: Monocular elevation paresis caused by an ipsilateral lesion. Neurology 34:1264, 1984 110. Munoz M, Page LK: Acquired double elevator palsy in a child with a pineocytoma. Am J Ophthalmol 118:810, 1994 111. von Noorden GK: Burian and von Noorden's Binocular Vision and Ocular Motility: Theory
and Management of Strabismus, 6th ed. St. Louis: CV Mosby, 2001 112. Jacobs L, Heffner RR, Newman RP: Selective paralysis of downward gaze caused by bilateral lesions of the
mesencephalic periaqueductal gray matter. Neurology 35:516, 1985 113. Scassellati-Sforzolini G: Una sindroma motto rare: diffeto congenito monolaterale della elevazione
con retrazione del globe. Riv Otoneurooftalmol 33:431, 1958 114. Khodadoust AA, von Noorden GK: Bilateral vertical retraction syndrome: a family study. Arch Ophthalmol 78:606, 1967 115. Pruksacholawit K, Ishikawa S: Atypical retraction syndrome: a case study. J Pediatr Ophthalmol 13:215, 1976 116. Wartenberg R: Inverted Marcus Gunn phenomenon. Arch Neurol Psychiatry 60:584, 1948 117. Carmicheal EA, Critchley M: The relations between eye movements and other cranial muscles. Br J Ophthalmol 9:49, 1925 118. Sano K: Trigemino-oculomotor synkineses. Neurologia 1:29, 1959 119. Wartenberg R: Associated movements in the oculomotor and facial muscles. Arch Neurol Psychiatry 55:439, 1946 120. Beyer-Machule CK, Johnson CC, Pratt SG, Smith BR: The Marcus-Gunn phenomenon. Orbit 4:15, 1985 121. Doucet TW, Crawford JS: The quantification, natural course, and surgical results in 57 eyes with
Marcus Gunn (jaw-winking) syndrome. Am J Ophthalmol 92:702, 1981 122. Mrabet A, Oueslati S, Gazzah H, et al: Clinical and electrophysiological study of 2 familial cases of Marcus Gunn
phenomenon [transl. French]. Rev Neurol 147:215, 1991 123. Dryden RM, Fleming JC, Quickert MH: Levator transposition and frontalis sling procedure in severe unilateral
ptosis and the paradoxically innervated levator. Arch Ophthalmol 100:462, 1982 124. Dillman DB, Anderson RL: Levator myectomy in synkinetic ptosis. Arch Ophthalmol 102:422, 1984 125. Lubkin V: The inverse Marcus-Gunn phenomenon: An electromyographic contribution. Arch Neurol 35:249, 1978 126. McLead AR, Glaser JS: Deglutition-trochlear synkinesis. Arch Ophthalmol 92:171, 1974 127. Pang MP, Zweifach PH, Goodwin J: Inherited levator-medial rectus synkinesis. Arch Ophthalmol 104:1489, 1986 128. Hwang J. A case of Marcus Gunn jaw winking and speudo inferior oblique overaction. Am J Ophthalmol 131:148, 2001 129. Brown HW: True and simulated superior oblique tendon sheath syndromes. Doe Ophthalmol 34:123, 1973 130. Katz NNK, Whitmore PV, Beauchamp GR: Brown's syndrome in twins. J Pediatr Ophthalmol Strabismus 18:32, 1981 131. Catford GV, Dean-Hart JC: Superior oblique tendon sheath syndrome: an electromyographic study. Br J Ophthalmol 55:155, 1971 132. Sevel D: Brown's syndrome: a possible etiology explained embryologically. J Pediatr Ophthalmol Strabismus 18:26, 1981 133. Beck M, Hickling P: Treatment of bilateral superior oblique tendon sheath syndrome complicating
rheumatoid arthritis. Br J Ophthalmol 64:358, 1980 134. Wang FM, Wertenbaker C, Behrens MM, Jacobs JC: Acquired Brown's syndrome in children with rheumatoid arthritis. Ophthalmology 91:23, 1984 135. Moore AT, Morin JD: Bilateral acquired inflammatory Brown's syndrome. J Pediatr Ophthalmol Strabismus 22:26, 1985 136. Clarke WN, Noel LP, Agapitos PJ: Traumatic Brown's syndrome. Am Orthoptic J 37:100, 1987 137. Blanchard CL, Young LA: Acquired inflammatory superior oblique tendon sheath (Brown's) syndrome. Arch Otolaryngol 110:120, 1984 138. Wesley RE, Pollard ZF, McCord CD: Superior oblique paresis after blepharoplasty. Plast Reconstr Surg 66:283, 1980 139. Hermann JS: Acquired Brown's syndrome of inflammatory origin. Arch Ophthalmol 96:1228, 1978 140. Booth-Mason S, Kyle GM, Rosser M, Bradbury P: Acquired Brown's syndrome: an unusual cause. Br J Ophthalmol 69:791, 1985 141. Hertle RW, Katowitz JA, Young TL, et al: Congenital unilateral fibrosis, blepharoptosis, and enophthalmos syndrome. Ophthalmology 99:347, 1992 142. Engle EC: The molecular basis of the congenital fibrosis syndromes. Strabismus 10:125, 2002 143. Wang SM, Zwaan J, Mullaney PB, et al: Congenital fibrosis of the extraocular muscles Type 2, an inherited exotropic
strabismus fixus, maps to distal 11q13. Am J Hum Genet 63:517, 1998 144. Engle EC, McIntosh N, Yamada K, et al: CFEOM1, the classic familial form of congenitla fibrosis of the extraocular
muscles, is genetically heterogeneous but does not result from mutations
in ARIX. BMC Genetics 3:3, 2002 145. Pieh C, Goebel HH, Engle EC, et al: Congenital fibrosis syndrome associated with central nervous system abnormalities. Graefe Arch Clin Exp Ophthalmol 241:546, 2003 146. Good WV, Hou C, Carden SM: Transient idiopathic nystagmus in infants. Dev Ned Child Neurol 45:304, 2003 147. Dell'Osso LF, van der Steen J, Steinman RM, et al: Foveation dynamics in congenital nystagmus: I. Fixation. Doe Ophthahmol 79:1, 1992 148. Dell'Osso LF, van der Steen J, Steinman RM, et al: Foveation dynamics in congenital nystagmus: II. Smooth pursuit. Doe Ophthalmol 79:25, 1992 149. Dell'Osso LF, Weissman BM, Leigh RJ, et al: Hereditary congenital nystagmus and gaze-holding failure: the role of the
neural integrator. Neurology 43:1741, 1993 150. Hertle RW, Chan CC, Galita DA, et al: Neuroanatomy of the extraocular muscle tendon enthesis in macaque, normal
human, and patients with congenital nystagmus. J AAPOS 6:319, 2002 151. Abel LA, Williams IM, Levi L: Intermittent oscillopsia in a case of congenital nystagmus: dependence
upon waveform. Invest Ophthalmol Vis Sci 32:3104, 1991 152. Gresty MA, Bronstein AM, Page NG, et al: Congenital-type nystagmus emerging in later life. Neurology 41:653, 1991 153. Hertle RS, FitzGibbon EJ, Avallone JM, et al: Onset of oscillopsia after visual maturation in patients with congenital
nystagmus. Ophthalmology 108:2301, 2001 154. Safran AB, Gambazzi Y: Congenital nystagmus: rebound phenomenon following removal of contact lenses. Br J Ophthalmol 76:497, 1992 155. Zubcov AA, Stark N, Weber A, et al: Improvement of visual acuity after surgery for nystagmus. Ophthalmology 100:1488, 1993 156. Helveston EM, Ellis FD, Plager DA: Large recession of the horizontal recti for treatment of nystagmus. Ophthalmology 98:1302, 1991 157. Dell'Osso LF: Development of new treatments for congenital nystagmus. Ann NY Acad Sci 956:361, 2002 158. Hertle RW, Dell'Osso LF, FitzGibbon EJ, et al: Horizontal rectus tenotomy in patients with congenital nystagmus: results
in 10 adults. Ophthalmology 110:2097, 2003 159. Gresty MA, Metcalfe T, Timms C, et al: Neurology of latent nystagmus. Brain 115(Pt 5):1303, 1992 160. Kommerell G, Zee DS: Latent nystagmus: release and suppression at will. Invest Ophthalmol Vis Sci 34:1785, 1993 161. Liu C, Gresty M, Lee J: Management of symptomatic latent nystagmus. Eye 7(Pt 4):550, 1993 162. Abadi RV, Pascal E: Periodic alternating nystagmus in humans with albinism. Invest Ophthalmol Vis Sci 35:4080, 1994 163. Shalloh-Hoffman J, Faldon M, Tusa RJ: The incidence and waveform characteristics of periodic alternating nystagmus
in congenital nystagmus. Invest Ophthalmol Vis Sci 40:2546, 1999 164. Gradstein L, Reinecke RD, Wizov SS, et al: Congenital periodic alternating nystagmus. Diagnosis and management. Ophthalmology 104:918, 1997 165. Smith JL, Flynn JT, Spiro HJ: Monocular vertical oscillations of amblyopia: the Bielschowsky phenomenon. J Clin Neuroophthalmol 2:85, 1982 166. Pritchard C, Flynn JT, Smith JL: Wave form characteristics of vertical oscillations in long-standing vision
loss. J Pediatr Ophthalmol Strabismus 25:233, 1988 167. Prasad P, Nair S: Congenital ocular motor apraxia: sporadic and familial: support for natural
resolution. J Neuroophthalmol 14:102, 1994 168. Jan JE, Kearney S, Groenveld M, et al: Speech, cognition, and imaging studies in congenital ocular motor apraxia. Dev Med Child Neurol 40:95, 1998 169. Sargent MA, Poskitt KJ, Jan JE: Congenital ocular motor apraxia: imaging findings. Am J Neuroradiol 18:1915, 1997 170. Anteby I, Lee B, Noetzel M, et al: Variants of congenital ocular motor apraxia: associations with hydrocephalus, pontocerebellar
tumor, and a deficit of vertical saccades. J AAPOS 1:201, 1997 171. Punal JE, Rodriguez E, Pintos E, et al: Congenital ocular motor apraxia associated with myopathy, external hydrocephalus
and NADH dehydrogenase deficiency. Brain Dev 20:175, 1998 172. Churchyard A, Stell R, Mastaglia FL: Ataxia telangiectasia presenting as an extrapyramidal movement disorder
and ocular motor apraxia without overt telangiectasia. Clin Exp Neurol 28:90, 1991 173. Steinlin M, Thun-Hohenstein L, Boltshauser E: Congenital oculomotor apraxia. Presentation developmental problems: differential
diagnosis [transl. German]. Klin Monatsbl Augenheilkd 200:623, 1992 174. Young TL, Weis JR, Summer G, Esbert JE: The association of strabismus, amblyopia, and refractive errors in spasmus
nutans. Ophthalmology 104:112, 1997 175. Gottlob IF, Zubcov AA, Wizov SS, et al: Head nodding is compensatory in spasmus nutans. Ophthalmology 99: 1024, 1992 176. Gottlob I, Wizov SS, Reinecke RD: Spasmus nutans. Invest Ophthalmol Vis Sci 36:2768, 1995 177. Wizov SS, Reinecke RD, Bocarnea M, et al: A comparative demographic and socioeconomic study of spasums nutans and
intantile nystagmus. Am J Ophthalmol 133:256, 2002 178. Gottlob I, Wizov SS, Reinecke RD: Quantitative eye and head movement recordings of retinal disease mimicking
spasmus nutans. Am J Ophthalmol 119:374, 1995 179. Lambert SR, Newman NJ: Retinal disease masquerading as spasmus nutans. Neurology 43:1607, 1993 180. Shawkat FS, Harris CM, Wilson J, et al: Eye movements in children with opsoclonus-polymyoclonus. Neuropediatrics 24:218, 1993 181. Wang PY: Acute cerebellitis with ocular flutter and truncal ataxia: a case report. Chin Med J (Engl) 50(2):169, 1992 182. De Graaf JH, Tamminga RY, Kamps WA: Paraneoplastic manifestations in children. Eur J Pediatr 153:784, 1994 183. Koh PS, Raffensperger JR, Berry S, et al: Long-term outcome in children with opsoclonus-myoclonus and ataxia and
coincident neuroblastoma. J Pediatr 125(5 Pt 1):712, 1994 184. Pranzatelli MR, Tate ED, Wheeler A, et al: Screening for autoantibodies in children with opsoclonus-myoclonus-ataxia. Pediatr Neurol 27:384, 2002 185. Fisher PG, Wechsler DS, Singer HS: Anti-Hu antibody in a neuroblastoma-associated paraneoplastic syndrome. Pediatr Neurol 10:309, 1994 186. Wilfong AA, Parke JT, McCrary JA III: Opsoclonusmyoclonus with Beckwith-Wiedemann syndrome and hepatoblastoma. Pediatr Neurol 8:77, 1992 187. Pranzatelli MR, Kao PC, Tate ED, et al: Antibodies to ACTH in opsoclonusmyoclonus [review]. Neuropediatrics 24:131, 1993 188. Mitchell WG, Davalos-Gonzalez Y, Brumm VL, et al: Opsoclonus-ataxia caused by childhood neuroblastoma: developmental and
neurolgoic sequelae. Pediatrics 109:86, 2002 189. Gambini IG, Conte M, Bernini G, et al: Neuroblastic tumors associated with opsoclonus-myoclonus syndrome: histological, immunohistochemicl and molecular features of 15 Italian cases. Virchows Arch 44w:555, 2003 190. Yee RD, Spiegel PH, Yamada T, et al: Voluntary saccadic oscillations, resembling ocular flutter and opsoclonus. J Neuroophthalmol 14:95, 1994 191. Shaywitz SE, Shaywitz BA: The science of reading and dyslexia. J AAPOS 7:158, 2003 192. Irlen H: Improving reading problems due to symptoms of scotopic sensitivity syndrome
using Irlen lenses and overlays. Education 109:413, 1989 193. Menacker SJ, Breton ME, Breton ML, et al: Do tinted lenses improve the reading performance of dyslexic children? Arch Ophthalmol 111:213, 1993 194. Metsahonkala L, Sillanpaa M: Migraine in children: an evaluation of the IHS criteria. Cephalalgia 14:285, 1994 195. Katerji MA, Painter MJ: Infantile migraine presenting as colic. J Child Neurol 9:336, 1994 196. Norton G: Letter to the Editor: migraine and somnambulism. Austr NZ J Med 23:715, 1993 197. Shevell MI, Silver K, Watters GV, et al: Transient oculosympathetic paresis (group II Raeder paratrigeminal
neuralgia) of childhood: migraine variant. Pediatr Neurol 9:289, 1993 198. Ottman R, Lipton RB: Comorbidity of migraine and epilepsy. Neurology 44:2105, 1994 199. Lessell S: Pediatric pseudotumor cerebri (idiopathic intracranial hypertension) [review]. Surv Ophthalmol 37:155, 1992 200. Phillips PH, Repka MX, Lambert SR: Pseudotumor cerebri in children. J AAPOS 2:33, 1998 201. Kesler A, Fattal-Valevski A: Idiopathic intracranial hypertension in the pediatric population. J Child Neurol 17:745, 2002 202. Balcer LJ, Liu GT, Forman S, Pun K, et al: Idiopathic intracranial hypertension: relation of age and obesity in children. Neurology 52:870, 1999 203. Cinciripini GS, Donahue S, Borchert MS: Idiopathic intracranial hypertension in prepubertal pediatric patients: characteristics, treatment, and outcome. Am J Ophthalmol 127:178, 1999 204. Scott IU, Siatkowski RM, Eneyni M, et al: Idiopathic intracranial hypertension in children and adolescents. Am J Ophthalmol 124:253, 1997 205. Quinn AG, Singer SB, Buncic JR: Pediatric tetracycline-induced pseudotumor cerebri. J AAPOS 3:53, 1999 206. Mochizuki K, Takahashi T, Kano M, et al: Pseudotumor cerebri induced by minocycline therapy for acne vulgaris. Jpn H Ophthalmol 46:668, 2002 207. Rogers AH, Rogers GL, Bremer DL, et al: Pseudotumor cerebri in children receiving recombinant human growth hormone. Ophthalmology 106:1186, 1999 208. Weig SG. Asymptomatic idiopathic intracranial hypertension in young children. J Child Neurol 17:239, 2002 209. Jeng MR, Rieman M, Bhakta M, et al: Pseucotumor cerebri in two adolesecnts with acquired aplastic anemia. J Pediatr Hematol Oncol 24:765, 2002 210. Nazir SA, Siatkowski RM: Pseudotumor cerebri in idiopathic aplastic anemia. J AAPOS 7:71, 2003 211. Kodsi SR, Younge BR: Acquired oculomotor, trochlear, and abducens cranial nerve palsies in pediatric
patients. Am J Ophthalmol 114:568, 1992 212. Norman AA, Farris BK, Siatkowski RM: Neuroma as a chase of oculomotor palsy in infancy and early childhood. J AAPOS 5:9, 2001 213. Hamed LM: Associated neurologic and ophthalmologic findings in congenital oculomotor
nerve palsy. Ophthalmology 98:708, 1991 214. Ing EB, Sullivan TJ, Clarke MP, et al: Oculomotor nerve palsies in children. J Pediatr Ophthalmol Strabismus 29:331, 1992 215. Wolin MJ, Saunders RA: Aneurysmal oculomotor nerve palsy in an 11-year-old boy. J Clin Neuroophthalmol 12:178, 1992 216. Branley MG, Wright KW, Borchert MS: Third nerve palsy due to cerebral artery aneurysm in a child. Austr NZ J Ophthalmol 20:137, 1992 217. Nazir SA, Murphy SA, Siatkowski RM: Recurrent para-infectious third nerve palsy with cisternal enhancement
on MRI. J Neurol Ophthalmol 24:96, 2004 218. Hamed LM, Fang EN, Fanous MM, et al: The prevalence of neurologic dysfunction in children with strabismus who
have superior oblique overaction. Ophthalmology 100:1483, 1993 219. Named LM, Maria BL, Quisling RG, et al: Alternating skew on lateral gaze. Ophthalmology 100:281, 1993 220. Davitt BD, Fenton GA, Cruz OA: Childhood myasthenia. J Ped Ophthalmol Strab 37:5, 2000 221. Mullaney P, Vajsar J, Smith R, Buncic JR: The natural history and ophthalmic involvement in childhood myasthenia
gravis at the Hospital for Sick Children. Ophthalmology 107:504, 2000 222. Kim J, Hwang J, Hwang Y, et al: Childhood ocular myasthenia gravis. Ophthalmology 110:1458, 2003 223. Skelly CL, Jackson CC, Wu Y, et al: Thorascopic thymectomy in children with myasthenia gravis. Am Surg 69:1087, 2003 224. Seaber JH: The effect of supranuclear midbrain lesions on ocular motility. In Ravault AP, Lenk M (eds): Transactions of the Fifth International Orthoptic Congress. Lyon, France: LIPS, 1984:105 225. Lepore FE: Disorders of ocular motility following head trauma. Arch Neurol 52:924, 1995 226. Pratt-Johnson JA, Tillson G: Acquired central disruption of fusional amplitude. Ophthalmology 86:2140, 1979 227. Stanworth A, Mein J: Loss of fusion after removal of cerebellar tumor. Br J Orthopt 28:97, 1971 228. Pratt-Johnson JA, Tillson G: The loss of fusion in adults with intractable diplopia (central fusion
disruption). Aust NZ J Ophthalmol 16:81, 1988 229. Lee MC: Acquired central fusional disruption with spontaneous recovery. Strabismus 6:175, 1998 230. Pratt-Johnson JA: Fusion ability lost and regained in visual adults. Graefes Arch Clin Exp Ophthalmol 226:111, 1988 231. MacLellan AV: A case of recovery from traumatic loss of fusion. Br J Orthopt 31:102, 1974 232. Kushner BJ: Unexpected cyclotropia simulating disruption of fusion. Arch Ophthalmol 110:1415, 1992 233. Miller MM, Guyton DL: Loss of fusion and the development of A or V patterns. J Pediatr Ophthalmol Strabismus 31:220, 1994 234. Morris RJ, Scott WE, Dickey CF: Fusion after surgical alignment of long-standing strabismus in adults. Ophthalmology 100:135, 1993 235. Kushner BJ, Morton GV: Postoperative binocularity in adults with long-standing strabismus. Ophthalmology 99:316, 1992 236. Daw NW: Mechanisms of plasticity in the visual cortex. Invest Ophthalmol Vis Sci 34:4168, 1994 237. Boyd TAS, Budd GE: The monofixation syndrome: heterophoria and asthenopia. Am J Orthopt 26:43, 1976 238. Parks MM: Monofixation syndrome. In Tasman W, Jaeger EA (eds): Duane's Clinical Ophthalmology. Philadelphia: JB Lippincott, 1990:1 239. Burgess D, Roper-Hall G, Burde RM: Binocular diplopia associated with subretinal neovascular membranes. Arch Ophthalmol 98:311, 1980 240. Bixenman WW, Joffe L: Binocular diplopia associated with retinal wrinkling. J Pediatr Ophthalmol Strabismus 21:215, 1984 241. Goldman HD, Nelson LB: Acute acquired comitant esotropia. Ann Ophthalmol 17:777, 1985 242. Burian HM, Miller JE: Comitant convergent strabismus with acute onset. Am J Ophthalmol 45:55, 1958 243. Biexnman WW, Laguna JF: Acquired esotropia as initial manifestation of Arnold-Chiari malformation. J Pediatr Ophthalmol Strabismus 24:83, 1987 244. Lewis AR, Kline LR, Sharp JA: Acquired esotropia due to Arnold Chiari I malformation. J Neuro-ophthalmol 16:49, 1996 245. Biousse V, Newman NJ, Petermann Sh, et al: Isolated comitant esotropia and Chiaria I malformation. Am J Ophthalmol 130:216, 2000 246. Deefort-Dhellemmes S, Denion E, Bouvet-Drumare I, et al: Resoluation of acute acquired comitant esotropia after suboccipital decompression
for Chiara I malformation. Am J Ophthalmol 133:723, 2002 247. Pokharel D, Siatkowski RM: Progressive cerebellar tonsillar herniation with recurrent divergence insufficiency
esotropia. J AAPOS 8:286, 2004 248. Stark N, Walther C: Cyclische esotropiebei einem erwaschsenen. Klin Monatsbl Augenheilkd 184:553, 1984 249. Troost BT, Abel L, Noreika J, Genovese FM: Acquired cyclic esotropia in an adult. Am J Ophthalmol 91:8, 1981 250. Friendly DS, Manson RA, Albert DG: Cyclic strabismus: a case study. Doe Ophthalmol 34:189, 1973 251. Roper-Hall MJ, Yapp JMS: Alternate day squint. In The First International Congress of Orthoptics. St. Louis: CV Mosby, 1968:262 252. Windsor CE, Berg EF: Circadian heterotropia. Am J Ophthalmol 67:656, 1969 253. Hugonnier R, Maynard P: Les desequilibres oculomoteur observes en cas de myopie forte. Ann Ocul 202:713, 1969 254. Krzizok TH, Kaulmann H, Traupe H: New approach in strabismus surgery in high myopia. Br J Ophthalmol 81:617, 1997 255. Lieppman ME: Accommodative and convergence insufficiency after decompression sickness. Arch Ophthalmol 99:453, 1981 256. von Noorden GK, Jenkins RH, Rosenbaum AL: Horizontal transposition of the vertical rectus muscles for treatment of
ocular torticollis. J Pediatr Ophthalmol 30:8, 1993 257. Rubin SE, Wagner RS: Ocular torticollis. Surv Ophthalmol 30:366, 1986 258. Catalano RA, Simon JW, Krohel GB, et al: Functional visual loss in children. Ophthalmology 93:385, 1986 259. Jacobson AM, de Groot M: Psychology of visual loss. In Principles and Practice of Ophthalmology. Philadelphia: WB Saunders |