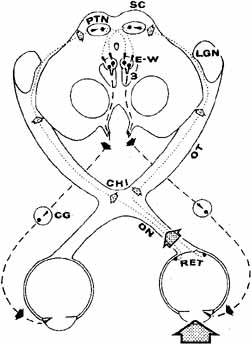
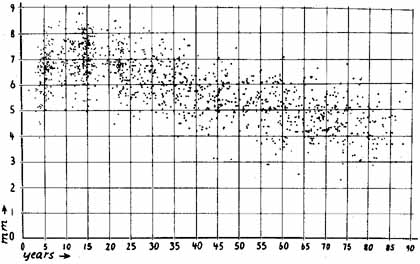
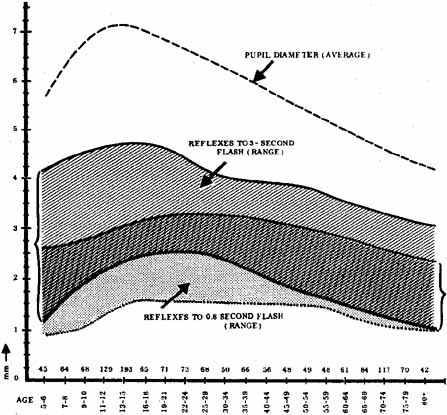
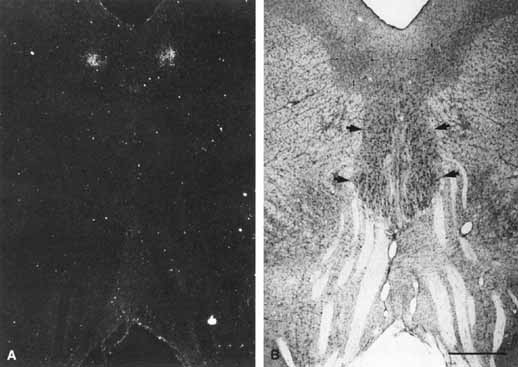
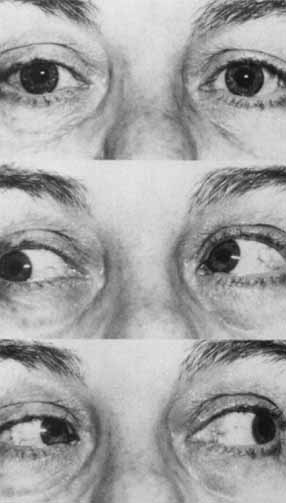
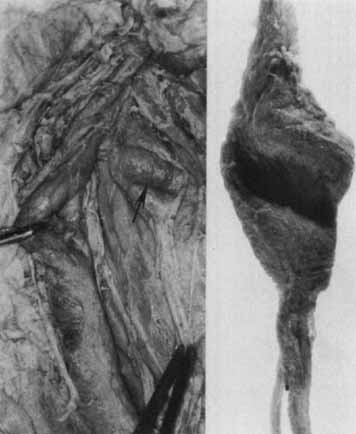
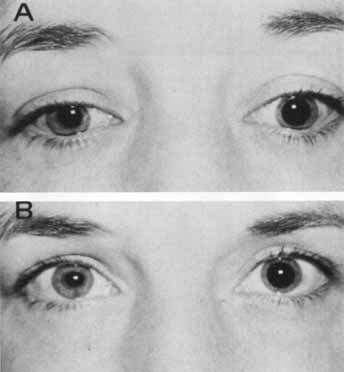
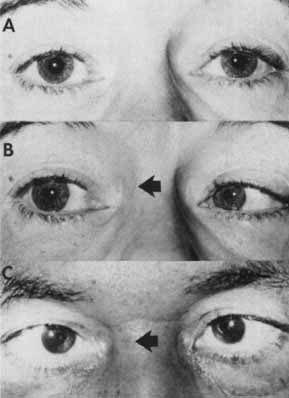
1. Loewenfeld IE: The Pupil: Anatomy, Physiology and Clinical Applications. Boston: Butterworth Heinemann, 1999 2. Wilhelm J, Wilhelm H, Moro S, Barbur JL: Pupil response components: Studies in patients with Parinaud's syndrome. Brain 125(10):2296, 2002 3. Bergamin O, Schoetzau A, Sugimoto K, Zulauf M: The influence of iris color on the pupillary light reflex. Graefes Arch Clin Exp Ophthalmol 236(8):567–570, 1998 4. Thompson HS, Franceschetti AT, Thompson PM: Hippus, semantic and historic considerations of the word. Am J Ophthalmol 71:1116, 1971 5. Loewenfeld IE: Pupillary changes related to age. In Thompson HS (ed): Topics in Neuro-ophthalmology. Baltimore: Williams & Wilkins, 1979:124–150 6. Bourne PR, Smith SA, Smith SE: Dynamics of the light reflex and the influence of age on the human pupil
measured by television pupillometry. J Physiol 293:1P, 1979 7. Campbell FW, Gubisch RW: Optical quality of the human eye. J Physiol 186:558, 1966 8. Bowling DB, Michael CR: Projection patterns of simple physiologically characterized optic tract
fibers in the cat. Nature 286:899, 1980 9. Kupfer C, Chumbley L, Downer J: Quantitative histology of optic nerve, optic tract and lateral geniculate
nucleus of man. J Anat 101:393, 1967 10. Breen L, Burde RM, Loewy AD: Brainstem connections to the Edinger–Westphal nucleus of the cat, a
retrograde tracer study. Br Res 261:303, 1983 11. Smith JD, Masek GA, Ichinose LY, et al: Single neuron activity in the pupillary system. Brain Res 24:219, 1970 12. Carpenter MB, Pierson RJ: Protectal region and the pupillary light reflex. J Comp Neurol 149:271, 1973 13. Benevento LA, Rezak M, Santos-Anderson RM: An autoradiographic study of the projections of the pretectum in the rhesus
monkey (Macaca mulatta): Evidence for sensorimotor links
to the thalamus and oculomotor nuclei. Brain Res 127:197, 1977 14. Burde RM: The visceral nuclei of the oculomotor complex. Trans Am Ophthalmol Soc 81:532, 1983 15. Warwick R: Representation of the extra-ocular muscles in the oculomotor complex. J Comp Neurol 98:449, 1953 16. Jampel RS, Mindel J: The nucleus for accommodation in the midbrain of the macaque. Invest Ophthalmol 6:40, 1967 17. Burde RM, Loewy AD: Central origin of the oculomotor parasympathetic neurons in the monkey. Br Res 198:434, 1980 18. Kourouyan HD, Horton JC: Transneuronal retinal input to the primate Edinger–Westphal nucleus. J Comp Neurol 381:68, 1997 19. Warwick R: The ocular parasympathetic nerve supply and its mesencephalic sources. J Anat 88:71, 1954 20. May PJ, Fratkin JD: Identification of the Edinger–Westphal nucleus in primates by means
of multiple markers: ARVO abstr #1488/B498, May 6, 2002 21. Kerr FWL, Hollowell OW: Location of pupillomotor and accommodation fibers in the oculomotor nerve: Experimental
observations on paralytic mydriasis. J Neurol Neurosurg Psychiatry 27:473, 1964 22. Nadeau SE, Trobe JD: Pupil sparing in oculomotor palsy: A brief review. Ann Neurol 13:143, 1983 23. Ruskell GL: Accommodation and the nerve pathway to the ciliary muscle: A review. Ophthalmic Physiol Opt 10:239, 1990 24. Jaeger RJ, Benevento LA: Innervation of eye internal structures. Invest Ophthalmol Vis Sci 19:575, 1980 25. Parelman JJ, Fay MT, Burde RM: Confirmatory evidence for a direct parasympathetic pathway to internal
eye structures. Trans Am Ophthalmol Soc 84:371, 1985 26. Burde RM: Direct parasympathetic pathways to the eye: Revisited. Brain Res 463:158, 1988 27. Ponsford JR, Bannister R, Paul EA: Methacholine pupillary responses in third nerve palsy and Adie's syndrome. Brain 105:583, 1982 28. Slamovits TL, Miller RN, Burde RM: Intracranial oculomotor nerve paresis with anisocoria and pupillary parasympathetic
hypersensitivity. Am J Ophthalmol 104:401, 1987 29. Jacobson DM: Pupillary responses to dilute pilocarpine in preganglionic 3rd nerve disorders. Neurology 40:804, 1990 30. Franklin AI, Cruz-Flores S, Chung S, et al: Cholinergic supersensitivity associated with inferior division oculomotor
nerve palsies. Abstract presented at the 23rd Annual North American Neuro-Ophthalmology
Society Meeting, Keystone, CO, February 1997 31. Jampel RS: Representation of the near-response on the cerebral cortex of the
Macaque. Am J Ophthalmol 48:573, 1959 32. Mays LE: Neurophysiological correlates of vergence eye movements. In: Schor CM, Ciuffreda KJ (eds): Vergence Eye Movements: Basic and Clinical Aspects. Woburn, MA: Butterworth Publishers, 1983:649–670 33. Kestenbaum A: Clinical Methods of Neuro-Ophthalmologic Examination. New York: Grune & Stratton, 1961:344–357 34. Levatin P: Pupillary escape in disease of the retina or optic nerve. Arch Ophthalmol 62:768, 1959 35. Kawasaki A, Moore P, Kardon RH: Variability of the relative afferent pupillary defect. Am J Ophthalmol 120:622, 1995 36. Enyedi LB, Dev S, Cox TA: A comparison of the Marcus Gunn and alternating light tests for afferent
pupillary defects. Ophthalmology 105(5):871–873, 1998 37. Finberg E, Thompson HS: Quantitation of the afferent pupillary defect. In Smith JL (ed): Neuro-Ophthalmology Focus 1980. New York: Masson, 1979:25–29 38. McCormick A, Bhola R, Brown L, et al: Quantifying relative afferent pupillary defects using Sbisa bar. Br J Ophthalmol 86(9):985, 2002 39. Browning DJ, Tiedeman JS: The test light affects quantitation of the afferent pupillary defect. Ophthalmology 94:53, 1987 40. Thompson HS, Jiang MQ: Letter to the editor. Ophthalmology 94:1360, 1987 41. Thompson JS, Corbett JJ, Cox TA: How to measure the relative afferent pupillary defect. Surv Ophthalmol 26:39, 1981 42. Wilhelm H, Wilhelm B: Clinical applications of pupillography. J Neuro-Ophthalmol 23(1):42, 2003 43. Bergamin O, Kardon RH: Latency of the pupil light reflex: Sample rate, stimulus intensity, and
variation in normal subjects. Invest Ophthalmol Vis Sci 44:1546, 2003 44. Alford MA, Nerad JA, Carter KD: Predictive value of the initial quantified relative afferent pupillary
defect in 19 consecutive patients with traumatic optic neuropathy. Ophthal Plast Reconstr Surg 17(5):323–327, 2001 45. Newman SA, Miller NR: Optic tract syndrome: Neuro-ophthalmologic considerations. Arch Ophthalmol 101:1241, 1983 46. Ellis CJK: Afferent pupillary defect in pineal region tumour. J Neurol Neurosurg Psychiatry 47:739, 1984 47. Johnson RE, Bell RA: Relative afferent pupillary defect in a lesion of the pretectal afferent
pupillary pathway. Can J Ophthalmol 22:282, 1987 48. Jiang MQ, Thompson HS: Pupillary defects in retinitis pigmentosa. Am J Ophthalmol 99:607, 1985 49. Thompson HS, Montague P, Cox TA, et al: The relationship between visual acuity, pupillary defect and visual loss. Am J Ophthalmol 93:681, 1982 50. Johnson LN, Hill RA, Bartholomew MJ: Correlation of afferent pupillary defect with visual field loss on automated
perimetry. Ophthalmology 95:1649, 1988 51. Thompson HS, Watsky RC, Weinstein JM: Pupillary dysfunction and macular disease. Trans Am Ophthalmol Soc 78:311, 1980 52. Folk JC, Thompson HS, Han DP, et al: Visual function abnormalities and central serous retinopathy. Arch Ophthalmol 102:1299, 1984 53. Han DP, Thompson HS, Folk JC: Differentiation between recently resolved optic neuritis and central serous
retinopathy. Arch Ophthalmol 103:394, 1985 54. Servais EG, Thompson HS, Hayreh SS: Relative afferent pupillary defect in central retinal vein occlusion. Ophthalmology 93:301, 1986 55. Portnoy JZ, Thompson HS, Lennarson L, Corbett JJ: Pupillary defects in amblyopia. Am J Ophthalmol 96:609, 1983 56. Greenwald MJ, Folk ER: Afferent pupillary defects in amblyopia. J Pediatr Ophthalmol Strabismus 20:63, 1983 57. Kawasaki A, Moore P, Kardon RH: Long-term fluctuation of relative afferent pupillary defect in subjects
with normal visual function. Am J Ophthalmol 22:875, 1996 58. Girkin CA, Perry JD, Miller NR: A relative afferent pupillary defect without any visual sensory defect. Arch Ophthalmol 116(11):1544, 1998 59. Lam BL, Thompson HS: Relative afferent pupillary defect induced by patching. Am J Ophthalmol 107:305, 1989 60. DuBois LG, Sadun AA: Occlusion-induced contralateral afferent pupillary defect. Am J Ophthalmol 107:306, 1989 61. Lam BL, Thompson HS: A unilateral cataract produces a relative afferent pupillary defect in
the contralateral eye. Ophthalmology 97:334, 1990 62. Lam BL, Thompson HS, Corbett JJ: The prevalence of simple anisocoria. Am J Ophthalmol 104:69, 1987 63. Loewenfeld IE: “Simple central” anisocoria: A common condition seldom recognized. Trans Am Acad Ophthalmol/Otolaryngol 83:832, 1977 64. Roarty JD, Keltner JL: Normal pupil size and anisocoria in newborn infants. Arch Ophthalmol 108:94, 1996 65. Lam BL, Thompson HS, Walls RC: Effect of light on the prevalence of simple anisocoria. Ophthalmology 103:790, 1996 66. Thompson BM, Corbett JJ, Kline LB, et al: Pseudo-Horner's syndrome. Arch Neurol 39:108, 1982 67. Safran AB, Roth A: Using a slit lamp for the neuroophthalmologic evaluation of old photographs. Am J Ophthalmol 95:558, 1983 68. Caccamise WC, Townes PL: Congenital mydriasis. Am J Ophthalmol 81:515, 1976 69. Seybold ME, Yoss RE, Hollenhorst RW, et al: Pupillary abnormalities associated with tumors of the pineal region. Neurology 21:232, 1971 70. Cahill M, Eustace P, de Jesus V: Pupillary autonomic denervation with increasing duration of diabetes mellitus. Br J Ophthalmol 85(10):1225, 2002 71. Patel JI, Jenkins L, Benjamin L, Webber S: Dilated pupils and loss of accommodation following diode panretinal photocoagulation
with sub-tenons local aneasthetic in four cases. Eye 16(5):628, 2002 72. Said G, Goulon-Goeau C, Slama G, Tchobroutsky G: Severe early-onset polyneuropathy in insulin-dependent diabetes
mellitus: A clinical and pathological study. N Engl J Med 326(19):1257, 1992 73. Loewenfeld IE: The Argyll Robertson pupil, 1869–1969: A critical survey of the literature. Surv Ophthalmol 14:199, 1969 74. Carrasco JR, Bilyk JR: Primary aberrant oculomotor nerve regeneration from a posterior communicating
artery aneurysm. Arch Ophthalmol 120:663, 2002 75. Olsen T, Jakobsen J: Abnormal pupillary function in third nerve regeneration (the pseudo
Argyll Robertson pupil): A case report. Acta Ophthalmol 62:163, 1984 76. Kardon RH, Corbett JJ, Thompson HS: Segmental denervation and re-innervation of the iris sphincter as
shown by infrared videographic transillumination. Ophthalmology 105:313, 1998 77. Cox TA: Czarnecki's sign as the initial finding in acquired oculomotor synkinesis. Am J Ophthalmol 102:543, 1986 78. Ohno S, Mukumo K: Studies on synkinetic pupillary phenomena resulting from aberrant regeneration
of the third nerve. Jpn J Clin Ophthalmol 27:229, 1973 79. Spiegel P, Kardon RH: Features of eye, eyelid and pupil movement critical to the recognition
of aberrant regeneration of the oculomotor nerve revealed by infrared
and color videography. Abstract presented at the 23rd Annual North American Neuro-Ophthalmology
Society Meeting, Keystone, CO, 1997 80. Martinelli P: Holmes-Adie syndrome. Lancet 356:1760, 2000 81. Thompson HS: Adie's syndrome: Some new observations. Trans Am Ophthalmol Soc 75:587, 1977 82. Thompson HS, Bell RA, Bougon P: The natural history of Adie's syndrome. In: Thompson HS, Daroff RB, Frisen L, et al (eds): Topics in Neuro-Ophthalmology. Baltimore: Williams & Wilkins, 1979:96–99 83. Pilley SFJ, Thompson HS: Cholinergic supersensitivity in Adie's syndrome: Pilocarpine vs. mecholyl. Am J Ophthalmol 80:955, 1975 84. Ramsay DA: Dilute solutions of phenylephrine and pilocarpine in the diagnosis of disordered
autonomic innervation of the iris. J Neurol Sci 73:125, 1986 85. Leavitt JA, Wyman LL, Hodge DO, Brubaker RF: Pupillary response to four concentrations of pilocarpine in normal subjects: Application
to testing for Adie tonic pupil. Am J Ophthalmol 135(3):333, 2002 86. Babikian PV, Thompson HS: Arecoline miosis. Am J Ophthalmol 98:514, 1984 87. Loewenfeld IE, Thompson HS: The tonic pupil: A reevaluation. Am J Ophthalmol 63:46, 1967 88. Harriman DGF, Garland H: The pathology of Adie's syndrome. Brain 91:401, 1968 89. Selhorst JB, Madge G, Ghatak M: The neuropathology of the Holmes-Adie syndrome. Ann Neurol 16:138, 1984 90. Firth AY: Adie syndrome: Evidence for refractive error and accommodative asymmetry
as the cause of amblyopia. Am J Ophthalmol 128(1):118, 1999 91. Anzai T, Uematsu D, Takahashi K, Katayama T: Guillian-Barre syndrome with bilateral tonic pupil. Intern Med 33(4):248, 1994 92. Bowie EM, Givre SJ: Tonic pupil and sarcodosis. Am J Ophthalmol 135(3):417, 2003 93. Currie J, Lessell S: Tonic pupil with giant cell arteritis. Br J Ophthalmol 68:135, 1984 94. Bennett JL, Pelak VA, Mourelatos Z, et al: Acute sensorimotor polyneuropathy with tonic pupil and an abduction deficit: An
unusual presentation of polyarteritis nodosa. Surv Ophthalmol 43(4):341, 1999 95. Jacome DE: Status migrainosus and Adie's syndrome. Headache 42(8):793, 2002 96. Goldstein SM, Liu GT, Edmond JC, et al: Orbital neural-glial hamartoma associated with a congenital tonic
pupil. JAAPOS 6(1):54, 2002 97. Lambert SR, Yang LL, Stone C: Tonic pupil associated with congenital neuroblastoma, Hirschsprung disease, and
central hypoventilation syndrome. Am J Ophthalmol 130(2):238, 2000 98. Lucy DD, Van Allen MW, Thompson HS: Holmes-Adie syndrome with segmental hypohidrosis. Neurology 17:763, 1967 99. Spector RH, Bachman DL: Bilateral Adie's tonic pupil with anhidrosis and hyperthermia. Arch Neurol 41:342, 1984 100. Shin RK, Galetta SL, Ting TY, et al: Ross syndrome plus: Beyond Horner, Holmes-Adie, and Harlequin. Neurol 55:1861, 2000 101. Perretti A, Nolano M, De Joanna G, et al: Is Ross syndrome a dysautonomic disorder only? An electrophysiologic
and histologic study. Clin Neurophysiol 114(1):7, 2003 102. Kimber J, Mitchell D, Mathias CJ: Chronic cough in the Holmes-Adie syndrome: Association in five cases
with autonomic dysfunction. J Neurol Neurosurg Psychiatry 65:583, 1998 103. Esterly NB, Cantolino SJ, Alter BP, et al: Pupillotonia, hyporeflexia, and segmental hypohidrosis: Autonomic dysfunction
in a child. J Pediatr 73:852, 1968 104. Goldberg MF, Payne JW, Brunt PW: Ophthalmologic studies of familial dysautonomia: The Riley-Day syndrome. Arch Ophthalmol 80:732, 1968 105. Thompson HS: Pupillary light near dissociation: A new classification. Surv Ophthalmol 19:290, 1975 106. Maitland GC, Scherokman BJ, Schiffman J, et al: Paraneoplastic tonic pupils. J Clin Neuro-Ophthalmol 5:99, 1985 107. Van Lieshout JJ, Wieling W, Van Montfrans GA, et al: Acute dysautonomia associated with Hodgkin's disease. J Neurol Neurosurg Psychiatry 49:830, 1986 108. Bell TAG: Adie's tonic pupil in a patient with carcinomatous neuromyopathy. Arch Ophthalmol 104:331, 1986 109. Bruno MK, Winterkorn JM, Edgar MA, et al: Unilateral Adie pupil as sole ophthalmic sign of anti-Hu paraneoplastic
syndrome. J Neuro-Ophthalmol 20(4):248, 2000 110. Keanen JR: Tonic pupils with acute ophthalmoplegic polyneuritis. Ann Neurol 2:393, 1977 111. Igarashi Y, Takeda M, Maekawa H, et al: Fisher's syndrome without total ophthalmoplegia. Ophthalmologica 205(3):163, 1992 112. Bachmeyer C, Zuber M, Dupont S, et al: Adie syndrome as the initial sign of primary Sjogren syndrome. Am J Ophthalmol 123(5):691, 1997 113. Sundaram MBM: Pupillary abnormalities in congenital neurosyphilis. Can J Neurol Sci 12:134, 1985 114. Fletcher WA, Sharpe JA: Tonic pupils in neurosyphilis. Neurology 36:188, 1986 115. Wirtschafter JD, Horman WK: Low concentration eserine therapy for the tonic pupil (Adie) syndrome. Ophthalmology 87:1037, 1980 116. Thompson HS: Discussion of Wirtschafter JD, Herman WK: Low concentration eserine therapy
for the tonic pupil. Ophthalmology 87:1043, 1980 117. Chou SY, Digre KB: Neuro-ophthalmic complications of raised intracranial pressure, hydrocephalus, and
shunt malfunction. Neurosurg Clin N Am 10(4):587, 1999 118. Kitthaweesin K: Upgaze paralysis as the initial manifestation of HIV-infected patient: A
case report. J Med Assoc Thai 85(6):728, 2002 119. Deramo VA, Jayamanne GR, Auerbach DB, Danes-Meyer H: Acute bilateral ophthalmoplegia in a young woman. Surv Ophthalmol 44(6):513, 2000 120. Salisachs P, Lapresle J: Argyll-Robertson pupils in the neural type of Charcot-Marie-Tooth
disease. Eur Neurol 16(1–6):172, 1977 121. Said G, Goulon-Goeau C, Slama G, Tchbioutsky G: Severe early-onset polyneuropathy in insulin dependent diabetes
mellitus: A clinical and pathological study. N Engl J Med 326(19):1257, 1992 122. Kirkham TH, Kline LB: Monocular elevator paresis, Argyll–Robertson pupils and sarcoidosis. Can J Ophthalmol 11(4):330, 1976 123. Van der Wiel H: Johann Friedrich Horner (1831–1886). J Neurol 249:636, 2002 124. Lapore FE: Enophthalmos and Horner's syndrome. Arch Neurol 40:460, 1983 125. Van der Wiel HL, Van Gijn J: Letter to the editor: No enophthalmos in Horner's syndrome. J Neurol Neurosurg Psychiatry 50:498, 1987 126. Smith SA, Smith SE: Bilateral Horner's syndrome: Detection and occurrence. J Neurol Neurosurg Psychiatry 66:48, 1999 127. Makley MA, Abbott K: Neurogenic heterochromia: A report of an interesting case. Am J Ophthalmol 59:927, 1965 128. Diesenhouse MC, Palay DA, Newman NJ, et al: Acquired heterochromia with Horner's syndrome in two adults. Ophthalmology 99:1815, 1992 129. Thompson HS, Mensher JM: Adrenergic mydriasis of Horner's syndrome: Hydroxyamphetamine test
for diagnosis of postganglionic defects. Am J Ophthalmol 72:472, 1971 130. Kardon RH, Dennison CE, Brown CK, et al: Critical evaluation of the cocaine test in the diagnosis of Horner's
syndrome. Arch Ophthalmol 108:384, 1990 131. Wilhelm H, Ochsner H, Kopycziok E, et al: Horner's syndrome: A retrospective analysis of 90 cases and recommendation
for clinical handling. J Ophthalmol 1:76, 1992 132. Jacobson DM, Berg R, Grinstead GF, Kruse JR: Duration of positive urine for cocaine metabolite after ophthalmic administration: Implications
for testing patients with suspected Horner syndrome
using ophthalmic cocaine. Am J Ophthalmol 13(6):742, 2001 133. Cremer SA, Thompson HS, Digre KB, Kardon RH: Hydroxyamphetamine mydriasis in normal subjects. Am J Ophthalmol 110(1):66, 1990 134. Cremer SA, Thompson HS, Digre KB, et al: Hydroxyamphetamine mydriasis in Horner's syndrome. Am J Ophthalmol 110(1):71, 1990 135. Grimson BS, Thompson HS: Drug testing in Horner's syndrome. In Glaser JS, Smith JL (eds): Neuro-Ophthalmology Symposium,vol 8. St. Louis: CV Mosby, 1975:265–270 136. Morrison DA, Biddy K, Woodruff G: The “harlequin” sign and congenital Horner's syndrome. J Neurol Neurosurg Psychiatry 62(6):626, 1997 137. Weinstein JM, Zweifel TJ, Thompson HS: Congenital Horner's syndrome. Arch Ophthalmol 98:1074, 1980 138. Donahue SP, Lavin PJ, Digre K: False-negative hydroxyamphetamine (Paredrine) test in
acute Horner's syndrome. Am J Ophthalmol 122:900, 1996 139. Moster ML, Galiani D, Garfinkle W: False negative hydroxyamphetamine test in Horner syndrome caused by acute
internal carotid artery dissection. J Neuro-Ophthalmol 23(1):22, 2003 140. Wilhelm H, Schaffer E: Pholedrine for determining the site of Horner's syndrome. Klin Monatsbl Augenheilk 204:169, 1994 141. Bates AT, Chamberlain SY, Champion M, et al: Pholedrine: A substitute for hydroxyamphetamine as a diagnostic eyedrop
test in Horner's syndrome. J Neurol Neurosurg Psychiatry 58:215, 1995 142. Smith SA, Smith SE: Reduced pupillary light reflexes in diabetic autonomic neuropathy. Diabetica 24(5):330, 1983 143. Stone WM, de Toledo J, Romanul FCA: Horner's syndrome due to hypothalamic infarction: Clinical radiologic
and pathologic correlation. Arch Neurol 43:199, 1986 144. Schiffter R: Letter to the editor: Telodiencephalic ischemic syndrome. Arch Neurol 44:1218, 1987 145. Schiffter R, Reinhart K: The telodiencephalic ischemic syndrome. J Neurol 222:265, 1980 146. Kori SH, Foley KM, Posner JB: Brachial plexus lesions in patients with cancer: 100 cases. Neurology 31:45, 1981 147. Yang PT, Seeger JF, Carmody RF, et al: Horner's syndrome secondary to traumatic pseudoaneurysms. Am J Neuroradiol 7:913, 1986 148. Teich SA, Halprin SL, Tay S: Horner's syndrome secondary to Swan-Ganz catheterization. Am J Med 78:168, 1985 149. O'Doherty DS, Green JB: Diagnostic value of Horner's syndrome in thrombosis of the carotid
artery. Neurology 111:842, 1958 150. Mokri B, Sundt TMJr , Houser OW: Spontaneous internal carotid dissection, hemicrania, and Horner's
syndrome. Arch Neurol 36:877, 1979 151. Kline LB, Vitek JJ, Raymon BC: Painful Horner's syndrome due to spontaneous carotid artery dissection. Ophthalmology 94:226, 1987 152. Francis KR, Williams DP, Troost BT: Facial numbness and dyesthesia: New features of carotid artery dissection. Arch Neurol 44:345, 1987 153. Bougousslavsky J, Deepland PA, Regli F: Spontaneous carotid dissection with acute stroke. Arch Neurol 44:137, 1987 154. Biousse V, Touboul P-J, D'Anglejan-Chatillon J, et al: Ophthalmoplegic manifestations of internal carotid artery dissection. Am J Ophthalmol 126(4):565, 1998 155. Goldberg HI, Grossman RI, Gomori JM, et al: Cervical internal carotid artery dissecting hemorrhage: Diagnosis using
MR. Radiology 158:157, 1986 156. Assaf M, Sweeney PJ, Kosmorsky G, et al: Horner's syndrome secondary to angiogram negative, subadventitial
carotid artery dissection. Can J Neurol Sci 20:62, 1993 157. Reader ALIII , Massey EW: Fibromuscular dysplasia of the carotid artery: A cause of congenital Horner's
syndrome? Ann Ophthalmol 12:326, 1980 158. Jeffrey AR, Ellis FJ, Repka MX, Buncic JR: Pediatric Horner syndrome. J AAPOS 2(3):129, 1998 159. Ryan FH, Kline LB, Gomez C: Congenital Horner's syndrome resulting from agenesis of the internal
carotid artery. Ophthalmol 107(1):185, 2000 160. Muira J, Doita M, Miyata K, et al: Horner's syndrome caused by a thoracic dumbbell-shaped schwannoma: Sympathetic
chain reconstruction after a one-stage removal
of the tumor. Spine 28(2):E33, 2003 161. Gilmer-Hill HS, Kline DG: Neurogenic tumors of the cervical vagus nerve: Report of four cases and
review of the literature. Neurosurgery 46(6):1498, 2000 162. Moukheiber AK, Nicollas R, Roman S, et al: Primary pediatric neuroblastic tumors of the neck. J Pediatr Otorhinolaryngol 60(2):155, 2001 163. George ND, Gonzalez G, Hoyt CS: Does Horner's syndrome in infancy require investigation. Br J Ophthalmol 82(1):51, 1998 164. Birchler MT, Landau K, Went PT, Stoeckli SJ: Paraganglioma of the cervical sympathetic trunk. Ann Otol Rhinol Laryngol 111(12 Pt 1):1087, 2002 165. Moyer JS, Bradford CR: Sympathetic paraganglioma as an unusual cause of Horner's syndrome. Head Neck 23(4):338, 2001 166. Leone A, Cerase A, Tarquini E, Mule A: Chordoma of the low cervical spine presenting with Horner's syndrome. Eur Radiol Suppl 4:S43, 2002 167. Bell RL, Atweh N, Ivy ME, Possenti PA-C: Traumatic and iatrogenic Horner syndrome: Case reports and review of literature. J Trauma 51(2):400, 2001 168. Ringer AJ, Fessler RD, Qureshi AI, et al: Horner's syndrome after carotid artery stenting: Case report. Surg Neurol 54(6):439, 2000 169. Taskapan H, Oymak O, Dogukan A, Utas C: Horner's syndrome secondary to internal jugular catheterization. Clin Nephrol 56(1):78, 2001 170. Perry C, James D, Wixon C, et al: Horner's syndrome after carotid endarterectomy: A case report. Vasc Surg 35(4):325, 2001 171. Kerrison JB, Biousse V, Newman NJ: Isolated Horner's syndrome and syringomyelia. J Neurol Neurosurg Psychiatry 69(1):131, 2000 172. Parkinson D: Bernard Mitchell, Horner's syndrome and others? Surg Neurol 11:221, 1979 173. Abad JM, Alvarez F, Munoz J, et al: Un sindrome neurologico no reconocido: Paralisis del sexto par y sindrome
de Bernard Horner's debido a lesiones traumaticas intracavernosas. Rev Clin Esp 165:135, 1982 174. Gutman I, Levartovski S, Goldhammer Y, et al: Sixth nerve palsy and unilateral Horner's syndrome. Ophthalmology 93:913, 1986 175. Havelius ULF, Hindfelt B: Minor vessels leaving the extracranial internal carotid artery: Possible
clinical implications. Neuro-Ophthalmology 5:51, 1985 176. Grimson BS, Thompson HS: Raeder's syndrome: A clinical review. Surg Ophthalmol 24:199, 1980 177. Grimson BS, Thompson HS: Drug testing in Horner's syndrome. In: Glaser JS, Smith JL (eds): Neuro-Ophthalmology Symposium, vol 8. St Louis: CV Mosby, 1975:265–270 178. Giles CL, Henderson JW: Horner's syndrome: An analysis of 216 cases. Am J Ophthalmol 46:289, 1958 179. Grimson BS, Thompson HS: Horner's syndrome: Overall view of 120 cases. In: Thompson HS, Daroff RB, Frisen L, et al (eds): Topics in Neuro-Ophthalmology. Baltimore: Williams & Wilkins, 1979:151–156 180. Keane JR: Oculosympathetic paresis: Analysis of 100 hospitalized patients. Arch Neurol 36:13, 1979 181. Maloney WF, Younge BR, Moyer NJ: Evaluation of the causes and accuracy of pharmacologic localization in
Horner's syndrome. Am J Ophthalmol 90:394, 1980 182. Musarella MA, Chan HS, deBoer G, et al: Ocular involvement—Neuroblastoma: Prognostic implications. Ophthalmology 91:936, 1984 183. Sauer T, Levinsohn M: Horner's syndrome in childhood. Neurology 26:216, 1976 184. Woodruff G, Buncic JR, Morin JD: Horner's syndrome. J Pediatr Ophthalmol Strabismus 25:40, 1988 185. Digre KB, Smoker WRK, Johnston P, et al: Selective MR imaging approach for evaluation of patients with Horner's
syndrome. Am J Neuroradiol 13:223, 1992 186. Lee AG, Hayman LA, Tang RA, et al: An imaging guide for Horner's syndrome. Int J Neuroradiol 2:196, 1996 187. Rayatt S, Khanna A: Unilateral mydriasis during blepharoplasty (Letter). Br J Plastic Surg 54(7):648, 2001 188. Berger I, Steinberger A, Schlesinger Y, et al: Neonatal mydriasis: Intravenous lidocaine adverse reaction. J Child Neurol 17(5):400, 2002 189. Bond DW, Vyas H, Venning HE: Mydriasis due to self-administered inhaled ipratopium bromide. Eur J Pediatr 161(3):178, 2002 190. Havelius U, Asman P: Accidental mydriasis from exposure to Angel's trumpet (Datura
suaveolens). Acta Ophthalmol Scand 80(3):332, 2002 191. Atabek ME, Aydin K, Erkul I: Different clinical features of amitraz poisoning in children. Hum Exper Toxicol 21:13, 2002 192. Burns JD, Muller LT, Jenkins PF, Gunderson CA: Unilateral mydriasis associated with exposure to flea spray. Arch Ophthalmol 120(5):665, 2002 193. Ulukaya S, Demirag K, Moral AR: Acute amitraz intoxication in human. Intensive Care Med 27:930, 2001 194. Yilmaz HL, Yildizdas DR: Amitraz poisoning, an emerging problem: Epidemiology, clinical features, management, and
preventive strategies. Arch Dis Child 88(2):130, 2003 195. Velhurst L, Waggie Z, Hatherill M, et al: Presentation and outcome of severe anticholinesterase insecticide poisoning. Arch Dis Child 86(5):352, 2002 196. Lepore FE: Letter to the editor: More on cycloplegia from transdermal scopolamine. N Engl J Med 307:284, 1982 197. Verdier D, Kennerdell J: Letter to the editor: Fixed dilated pupil resulting from transdermal scopolamine. Am J Ophthalmol 93:803, 1982 198. Johnson SF, Moore RJ: Transderm pupil and confusion in a 10 year old. Ann Neurol 13:583, 1983 199. Thompson HS, Newsome DA, Loewenfeld IE: The fixed dilated pupil: Sudden iridoplegia or mydriatic drops? A
simple diagnostic test. Arch Ophthalmol 86:21, 1971 200. American Hospital Formulary Source (AHFS), American Society of
Health Systems Pharmacies. Bethesda, MD: AHFS, 2003:2682–2694 201. Bartlett J, James SD: Clinical Ocular Pharmacology, 4th ed. Boston: Butterworth Heinemann, 2001:135–148, 149–166 202. Monteiro ML, Ulrich RF, Imes RK, et al: Iron mydriasis. Am J Ophthalmol 97:794, 1984 203. Lam S, Beck RW, Hall D, et al: Atonic pupil after cataract surgery. Ophthalmology 96:589, 1989 204. Minasian M, Ayliffe W: Fixed dilated pupil following deep lamellar keratoplasty (Urrets–Zavalia
syndrome). Br J Ophthalmol 86(1):115, 2002 205. Geerling G, Neppert B, Wirbelauer C, Laqua H: Relative mydriasis after photorefractive keratotomy. J Refract Surg 16(1):69, 2000 206. Hallett M, Cogan DG: Episodic unilateral mydriasis in otherwise normal patients. Arch Ophthalmol 84:130, 1970 207. Thompson HS, Zackon DH, Czarnecki JSC: Tadpole-shaped pupils caused by segmental spasm of the iris dilator
muscle. Am J Ophthalmol 96:467, 1983 208. Edelson RN, Levy DE: Transient benign unilateral pupillary dilation in young adults. Arch Neurol 31:12, 1974 209. Woods D, O'Connor PS, Fleming R: Episodic unilateral mydriasis and migraine. Am J Ophthalmol 98:229, 1984 210. Purvin VA: Adie's tonic pupil secondary to migraine. J Neuro-Ophthalmol 15:43, 1995 211. Miller NR: Intermittent pupillary dilation in a young woman: Comments by Keltner JL, Gittinger JW Jr , Burde RM. Surv Ophthalmol 31:65, 1986 212. Jacobson DM: Benign episodic unilateral mydriasis-clinical characteristics. Ophthalmology 102:1623, 1995 213. Kline LB, McCluer SM, Bonikowski FP: Oculosympathetic spasm with cervical spinal cord injury. Arch Neurol 41:61, 1984 214. Pant SS, Benton JW, Dodge PR: Unilateral pupillary dilatation during and immediately following seizures. Neurology 16:337, 1966 215. Gadoth N, Margalith D, Bechar M: Unilateral pupillary dilatation during focal seizures. J Neurol 225:227, 1981 216. Zee DS, Griffin J, Price DL: Unilateral pupillary dilatation during adversive seizures. Arch Neurol 30:403, 1974 217. Chia JK, Clark JB, Ryan GA, et al: Botulism in an adult associated with food-borne intestinal infection
with clostridium botulinum. N Engl J Med 315:239, 1986 218. Tyler HR: Botulism. Arch Neurol 9:652, 1963 219. Cherington M: Botulism: Ten-year experience. Arch Neurol 30:432, 1974 220. Dagi LR, Chrousos GA, Cogan DC: Spasm of the near reflex associated with organic disease. Am J Ophthalmol 103:582, 1987 221. Tijssen CC, Goor C, Van Woerkom TCAM: Spasm of the near reflex: Functional or organic disorder? Neuro-Ophthalmology 3:59, 1983 222. Safran AB, Roth A, Gauthier G: Le syndrome des spasmes de convergence “plus.” Klin Monatsbl Augenheilkd 108:471, 1982 223. Maddalena MA: Transient myopia associated with acute glaucoma and retinal edema. Arch Ophthalmol 80:186, 1968 224. Dangel ME, Weber PA, Leier CB: Transient myopia following isosorbide dinitrate. Ann Ophthalmol 15:1156, 1983 |