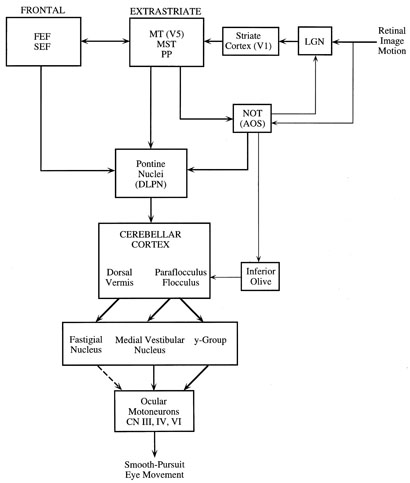
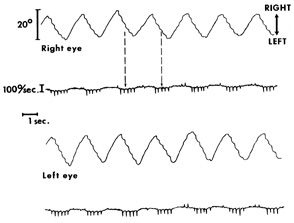
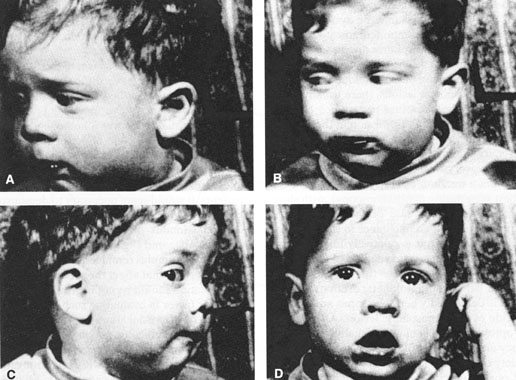
1. Kaminski HJ, Leigh RJ, eds: Neurobiology of Eye Movements—From Molecules to Behavior. New York: New York Academy of Sciences, 2002 2. Leigh R, Zee D: The Neurology of Eye Movements. Third ed. New York: Oxford University Press, 1999 3. Leigh RJ, Kennard C: Using saccades as a research tool in clinical neurosciences. Brain 27:1–18, 2004 4. Demer JL: Pivotal role of orbital connective tissues in binocular alignment and strabismus. Invest Ophthalmol Vis Sci 45:729–738, 2004 5. Büttner-Ennever JA, Horn AK, Graf W, et al:et al: Modern concepts of brainstem anatomy: from extraocular motoneurons to proprioceptive
pathways. Ann NY Acad Sci 956:75–84, 2002 6. Highstein SM, Baker R: Excitatory termination of abducens internuclear neurons on medial rectus
motoneurons: relationship to syndrome of internuclear ophthalmoplegia. J Neurophysiol 41:1647–1661, 1978 7. King W, Lisberger S, Fuchs A: Responses of fibers in medial longitudinal fasciculus (MLF) of
alert monkeys during horizontal and vertical conjugate eye movements
evoked by vestibular or visual stimuli. J Neurophysiol 39:1135–1149, 1976 8. McCrea R, Strassman A, Highstein S: Morphology and physiology of abducens motoneurons and internuclear neurons
intracellularly injected with horseradish peroxidase in alert squirrel
monkeys. J Comp Neurol 243:291–308, 1986 9. Spencer R, Baker R. Histochemical localization of acetycholinesterase in relation to motor
neurons and internuclear neurons of the cat abducens nucleus. J Neurocytol 15:137–154, 1986 10. McCrea R, Strassman A, May E, et al:et al: Anatomical and physiological characteristics of vestibular neurons mediating
the horizontal vestibulo-ocular reflex of the squirrel monkey. J Comp Neurol 264:547–570, 1987 11. Horn AKE, Büttner-Ennever JA, Suzuki Y, et al: Histological identification of premotor neurons for horizontal saccades
in monkey and man by parvalbumin immunostaining. J Comp Neurol 359:350–363, 1997 12. Belknap D, McCrea R: Anatomical connections of the prepositus and abducens nuclei in the squirrel
monkey. J Comp Neurol 268:13–28, 1988 13. Bronstein AM, Morris J, Du B, et al: Abnormalities of horizontal gaze. Clinical, oculographic and magnetic resonance
imaging findings. I Abducens palsy. J Neurol Neurosurg Psychiatry 53:194–199, 1990 14. Hirose G, Furui K, Yoshioka A, et al: Unilateral conjugate gaze palsy due to a lesion of the abducens nucleus. J Clin Neuroophthalmol 13:54–58, 1993 15. Meienberg O, Büttner-Ennever JA, Kraus-Ruppert R: Unilateral paralysis of conjugate gaze due to lesion of the abducens nucleus. Clinico-pathological case report. Neuroophthalmology 2:47–52, 1981 16. Müri RM, Chermann JF, Cohen L, et al: Ocular motor consequences of damage to the abducens nucleus area in humans. J Neuroophthalmol 16:191–195, 1996 17. Gamlin PD, Gnadt JW, Mays LE: Lidocaine-induced unilateral internuclear ophthalmoplegia: effects on convergence
and conjugate eye movements. J Neurophysiol 62:82–95, 1989 18. Zee DS, Hain TC, Carl JR: Abduction nystagmus in internuclear ophthalmoplegia. Ann Neurol 21:383–388, 1987 19. Brandt T, Dieterich M: Vestibular syndromes in the roll plane: topographic diagnosis from brainstem
to cortex. Ann Neurol 36:337–347, 1994 20. Cremer P, Migliaccio A, Halmagyi GM, et al: Vestibulo-ocular reflex pathways in internuclear ophthalmoplegia. Ann Neurol 45:529–533, 1999 21. Ranalli P, Sharpe J: Vertical vestibulo-ocular reflex, smooth pursuit and eye-head tracking
dysfunction in internuclear ophthalmoplegia. Brain 111:1299–1317, 1988 22. Herishanu YO, Sharpe JA. Saccadic intrusions in internuclear ophthalmoplegia. Ann Neurol 14:67–72, 1983 23. Smith JL, David NJ: Internuclear ophthalmoplegia. Two new clinical signs. Neurology 14:307–309, 1964 24. Frohman TC, Frohman EM, O'Suilleabhain P, et al: Accuracy of clinical detection of INO in MS: corroboration with quantitative
infrared oculography. Neurology. 61:848–850, 2003 25. Frohman EM, Frohman TC, O'Suilleabhain P, et al: Quantitative oculographic characterisation of internuclear ophthalmoparesis
in multiple sclerosis: the versional dysconjugacy index Z score. J
Neurol Neurosurg Psychiatry. 73:51–55, 2002 26. Flipse JP, Straathof CS, Van der Steen J, et al: Binocular saccadic acceleration in multiple sclerosis. Neuro-ophthalmology 16:43-46, 1996 27. Ventre J, Vighetto A, Bailly G, et al: Saccade metrics in multiple sclerosis: versional velocity disconjugacy
as the best clue? J Neurol Sci 102:144–149, 1991 28. Frohman EM, Zhang H, Kramer PD, et al: MRI characteristics of the MLF in MS patients with chronic internuclear
ophthalmoparesis. Neurology. 57:762–768, 2001 29. Lutz A. Ueber die Bahnen der Blickwendung und deren Dissoziierung. Klin Monatsble Augenheilkd 70:213–235, 1923 30. Bogousslavsky J, Regli F, Ostinelli B, et al: Paresis of lateral gaze alternating with so-called posterior internuclear
ophthalmoplegia. A partial paramedian pontine reticular formation-abducens
nucleus syndrome. J Neurol 232:38–42, 1985 31. Clendaniel RA, Mays LE: Characteristics of antidromically identified oculomotor internuclear neurons
during vergence and versional eye movements. J Neurophysiol 71:1111–1127, 1994 32. Fisher CM: Some neuro-ophthalmological observations. J Neurol Neurosurg Psychiatry 30:383–392, 1967 33. Pierrot-Deseilligny C, Chain F, Serdaru M, et al: The “one-and-a-half” syndrome. Electro-oculographic analyses
of five cases with deductions about the physiological mechanisms of
lateral gaze. Brain 104:665–699, 1981 34. Wall M, Wray SH: The one-and-a-half syndrome—A unilateral disorder of the pontine
tegmentum: a study of 20 cases and review of the literature. Neurology 33:971-980, 1983 35. Sharpe JA, Rosenberg MA, Hoyt WF, et al: Paralytic pontine exotropia. A sign of acute unilateral pontine gaze palsy
and internuclear ophthalmoplegia. Neurology 24:1076-1081, 1974 36. Hanson M, Hamid M, Tomsak R, et al: Selective saccadic palsy caused by pontine lesions: clinical, physiological, and
pathological correlations. Ann Neurol 20:209-217, 1986 37. Henn V, Lang W, Hepp K, et al: Experimental gaze palsies in monkeys and their relation to human pathology. Brain 107:619-636, 1984 38. Johnston JL, Sharpe JA, Ranalli PJ, et al: Oblique misdirection and slowing of vertical saccades after unilateral
lesions of the pontine tegmentum. Neurology 43:2238-2244, 1993 39. Zee DS, Optican LM, Cook JD, et al: Slow saccades in spinocerebellar degeneration. Arch Neurol 33:243-251, 1976 40. Aksay E, Gamkrelidze G, Seung HS, et al: In vivo intracellular recording and perturbation of persistent activity
in a neural integrator. Nat Neurosci 4:184–193, 2001 41. Cannon SC, Robinson DA: Loss of the neural integrator of the oculomotor system from brainstem lesions
in monkey. J Neurophysiol 57:1383–1409, 1987 42. Dieterich M, Brandt T: Wallenberg's syndrome: lateropulsion, cyclorotation, and subjective
visual vertical in thirty-six patients. Ann Neurol 31:399–408, 1992 43. Kirkham TH, Guitton D, Gans M: Task-dependent variations of ocular lateropulsion in Wallenberg's
syndrome. Can J Neurol Sci 8:21–22, 1981 44. Waespe W, Baumgartner R: Enduring dysmetria and impaired gain adaptivity of saccadic eye movements
in Wallenberg's lateral medullary syndrome. Brain 115:1125–1146, 1992 45. Büttner-Ennever JA, Buttner U: A cell group associated with vertical eye movements in the rostral mesencephalic
reticular formation of the monkey. Brain Res 151:31–47, 1978 46. Horn AKE, Büttner-Ennever JA: Premotor neurons for vertical eye-movements in the rostral mesencephalon
of monkey and man: the histological idnetification by parvalbumin immunostaining. J Comp Neurol 392:413–427, 1998 47. Suzuki Y, Büttner-Ennever JA, Straumann D, et al: Deficits in torsional and vertical rapid eye movements and shift of Listing's
plane after uni- and bilateral lesions of the rostral interstitial
nucleus of the medial longitudinal fasciculus. Exp Brain Res 106:215–232, 1995 48. Moschovakis AK, Scudder CA, Highstein SM: Structure of the primate oculomotor burst generator. I. Medium-lead burst
neurons with upward on-directions. J Neurophysiol 65:203–217, 1991 49. Moschovakis AK, Scudder CA, Highstein SM, et al: Structure of the primate oculomotor burst generator. II. Medium-lead burst
neurons with downward on-directions. J Neurophysiol 65:218–229, 1991 50. Kokkoroyannis T, Scudder CA, Balaban CD, et al: Anatomy and physiology of the primate interstitial nucleus of Cajal I. Efferent
projections. J Neurophysiol 75:725–739, 1996 51. Partsalis AM, Highstein SM, Moschovakis AK: Lesions of the posterior commissure disable the vertical neural integrator
of the primate oculomotor system. J Neurophysiol 71:2582–, 19942585 52. Pasik P, Pasik T, Bender MB: The pretectal syndrome in monkeys. I. Disturbances of gaze and body posture. Brain 92:521–534, 1969 53. Fukushima F. The interstitial nucleus of Cajal and its role in the control of movements
of head and eyes. Prog Neurobiol 29:107–192, 1987 54. McCrea R, Strassman A, Highstein S: Anatomical and physiological characteristics of vestibular neurons mediating
the vertical vestibulo-ocular reflexes of the squirrel monkey. J Comp Neurol 264:571–594, 1987 55. Ranalli PJ, Sharpe JA, Fletcher WA: Palsy of upward and downward saccadic, pursuit, and vestibular movements
with a unilateral midbrain lesion: pathophysiologic correlations. Neurology 38:114–122, 1988 56. Bender MB: Brain control of conjugate horizontal and vertical eye movements. A survey
of the structural and functional correlates. Brain 103:23–69, 1980 57. Kompf D, Pasik T, Pasik P, et al: Downward gaze in monkeys. Stimulation and lesion studies. Brain 102:527–558, 1979 58. Bhidayasiri R, Plant GT, Leigh RJ: A hypothetical scheme for the brainstem control of vertical gaze. Neurology 54:1985–1993, 2000 59. Büttner-Ennever JA, Buttner U, Cohen B, et al: Vertical gaze paralysis and the rostral interstitial nucleus of the medial
longitudinal fasciculus. Brain 105:125–149, 1982 60. Crawford JD, Cadera W, Vilis T: Generation of torsional and vertical eye position signals by the interstitial
nucleus of Cajal. Science 252:1551–1553, 1991 61. Westheimer G, Blair SM: The ocular tilt reaction—A brainstem oculomotor routine. Invest Ophthalmol 14:833–839, 1975 62. Lueck CJ, Hamlyn P, Crawford TJ, et al: A case of ocular tilt reaction and torsional nystagmus due to direct stimulation
of the midbrain in man. Brain 114:2069–2079, 1991 63. Evinger LC, Fuchs AF, Baker R: Bilateral lesions of the medial longitudinal fasciculus in monkeys: Effects
on the horizontal and vertical components of voluntary and vestibular
induced eye movements. Exp Brain Res 28:1–20, 1977 64. Keane JR: Sustained upgaze in coma. Ann Neurol 9:409–412, 1981 65. Hoyt CS, Mousel DK: Transient supranuclear disturbances of gaze in healthy neonates. Am J Ophthalmol 89:708–713, 1980 66. Fisher CM: The pathologic and clinical aspects of thalamic hemorrhage. Trans Am Neurol Assoc 84:56–59, 1959 67. Frohman LP, Kupersmith MJ: Reversible vertical ocular deviations associated with raised intracranial
pressure. J Clin Neuroopthalmol 5:158–163, 1985 68. Keane JR, Rawlinson DG, Lu AT: Sustained downgaze deviation. Two cases without structural pretectal lesions. Neurology 26:594–595, 1976 69. Corbett JJ, Schatz NJ, Shults WT, et al: Slowly alternating skew deviation: description of a pretectal syndrome
in three patients. Ann Neurol 10:540–546, 1981 70. Daroff RB, Hoyt WF: Supranuclear disorders of ocular control systems in man: clinical, anatomical
and physiological correlations—1969. In: Bach-y-Rita P, Collins CC, Hyde JE, eds. The Control of Eye Movements. New York: Academic Press, 1971:175–235 71. Büttner-Ennever JA, Acheson JF, Büttner U, et al: Ptosis and supranuclear downgaze palsy. Neurology 39:385–389, 1989 72. Sand JJ, Biller J, Corbett JJ, et al: Partial dorsal mesencephalic hemorrhages: report of three cases. Neurology 36:529–533, 1986 73. Keane JR: Bilateral ocular motor signs after tentorial herniation in 25 patients. Arch Neurol 43:806–807, 1986 74. Seaber JH, Nashold BS: Comparison of ocular motor effects of unilateral stereotactic midbrain
lesions in man. Neuroophthalmology 1:95–99, 1980 75. Rambold H, Kompf D, Helmchen C: Convergence retraction nystagmus: a disorder of vergence? Ann Neurol 50:677–681, 2001 76. Ochs AL, Stark L, Hoyt WF, et al: Opposed adducting saccades in convergence-retraction nystagmus. A patient
with sylvian aqueduct syndrome. Brain 102:479–508, 1979 77. Zee DS, FitzGibbon EJ, Optican LM: Saccade-vergence interactions in humans. J Neurophysiol 68:1624–1641, 1992 78. Brenner RP, Carlow TJ: PLEDs and nystagmus retractorius. Ann Neurol 5:403, 1979 79. Keane JR: Ocular skew deviation. Arch Neurol 32:185–190, 1975 80. Moster ML, Schatz NJ, Savino PJ, et al: Alternating skew on lateral gaze (bilateral abducting hypertropia). Ann Neurol 23:190–192, 1988 81. Zee DS: Considerations on the mechanisms of alternating skew deviation in patients
with cerebellar lesions. J Vestibular Res 6:1–7, 1996 82. Brandt T, Dieterich M: Pathological eye-head coordination in roll: tonic ocular tilt reaction
in mesencephalic and medullary lesions. Brain 110:649–666, 1987 83. Halmagyi GM, Brandt T, Dieterich M, et al: Tonic contraversive ocular tilt reaction due to unilateral meso-diencephalic
lesion. Neurology 40:1503–1509, 1990 84. Rabinovitch HE, Sharpe JA, Sylvester TO: The ocular tilt reaction. A paroxysmal dyskinesia associated with elliptical
nystagmus. Arch Ophthalmol 95:1395–1398, 1977 85. Hedges TR, Hoyt WF: Ocular tilt reaction due to an upper brainstem lesion: paroxysmal skew
deviation, torsion, and oscillation of the eyes with head tilt. Ann Neurol 11:537–540, 1982 86. Lewis JM, Kline LB: Periodic alternating nystagmus associated with periodic alternating skew
deviation. J Clin Neuroopthalmol 13:115–117, 1983 87. Gamlin PDR, Mays LE: Dynamic properties of medial rectus motoneurons during vergence eye movements. J Neurophysiol 67:64–74, 1992 88. Mays LE, Gamlin PDR: Neuronal circuitry controlling the near response. Curr Opin Neurobiol 5:763–768, 1995 89. Mays LE: Neural control of vergence eye movements: convergence and divergence neurons
in the midbrain. J Neurophysiol 51:1091–1108, 1984 90. Zhang Y, Mays LE, Gamlin PDR: Characteristics of near response cells projecting to the oculomotor nucleus. J Neurophysiol 67:944–960, 1992 91. Judge SJ, Cumming BG: Neurons in the monkey midbrain with activity related to vergence eye movements
and accommodation. J Neurophysiol 55:915–930, 1986 92. Mays LE, Porter JD, Gamlin PDR, et al: Neural control of vergence eye movements: neurons encoding vergence velocity. J Neurophysiol 56:1007–1021, 1986 93. Gamlin PDR, Clarke RJ: Single-unit activity in the primate nucleus reticularis tegmenti pontis
related to vergence and accommodation. J Neurophysiol 73:2115–2119, 1995 94. Kitthaweesin K, Riley DE, Leigh RJ: Vergence disorders in progressive supranuclear palsy. Ann NY Acad Sci 956:504–507, 2002 95. Rambold H, Neumann G, Helmchen C: Vergence deficits in pontine lesions. Neurology 62:1850–1853, 2004 96. Keane JR: Neuro-ophthalmological signs and symptoms of hysteria. Neurology 32, 1982 97. Guiloff RJ, Whiteley A, Kelly RE: Organic convergence spasm. Acta Neurol Scand 61:252–259, 1980 98. Krohel GB, Kristan RW, Simon JW, et al: Divergence paralysis. Am J Ophthalmol 94:506–510, 1982 99. Roper-Hall G, Burde RM: Diagnosis and management of divergence paralysis. Am Orthop J 37:113, 1987 100. Langer T, Fuchs A, Scudder C, et al: Afferents to the flocculus of the cerebellum in the rhesus macaque as revealed
by retrograde transport of horseradish peroxidase. J Comp Neurol 235:1–25, 1985 101. Langer T, Fuchs A, Chubb M, et al: Floccular efferents in the rhesus macaque as revealed by autoradiography
and horseradish peroxidase. J Comp Neurol 235:26–37, 1985 102. Belton T, McCrea RA: Role of the cerebellar flocculus region in the coordination of eye and
head movements during gaze pursuit. J Neurophysiol 84:1614–1626, 2000 103. Waespe W, Henn V: Visual-vestibular interaction in the flocculus of the alert monkey. II. Purkinje
cell activity. Exp Brain Res 43:349–360, 1981 104. Raymond JL, Lisberger SG, Mauk MD: The cerebellum: a neuronal learning machine? Science 272:1126–1131, 1996 105. Walberg F, Dietrichs E: The interconnection between the vestibular nuclei and the nodulus: a study
of reciprocity. Brain Res 449:47–53, 1988 106. Cohen H, Cohen B, Raphan T, et al: Habituation and adaptation of the vestibulo-ocular reflex: a model of differential
control by the vestibulo-cerebellum. Exp Brain Res 110:110–120, 1993 107. Yamada J, Noda H: Afferent and efferent connections of the oculomotor cerebellar vermis in
the macaque monkey. Journal of Comparative Neurology 265:224–241, 1987 108. Ikeda Y, Noda H, Sugita S: Olivocerebellar and cerebelloolivary connections of the oculomotor region
of the fastigial nucleus in the macaque monkey. J Comp Neurol 284:463–488, 1989 109. Thier P, Dicke PW, Haas R, et al: The role of the oculomotor vermis in the control of saccadic eye movements. Ann NY Acad Sci 978:50–62, 2002 110. Fuchs AF, Robinson FR, Straube A: Role of the caudal fastigial nucleus in saccade generation: I. Neuronal
discharge patterns. J Neurophysiol 70:1723–1740, 1993 111. Buttner U, Glasauer S, Glonti L, et al: Multimodal signal integration in vestibular neurons of the primate fastigial
nucleus. Ann NY Acad Sci 1004:241–251, 2003 112. Zee D, Yamazaki A, Butler P, et al: Effects of ablation of flocculus and paraflocculus on eye movements in
primate. J Neurophysiol 46:878–899, 1981 113. Rambold H, Churchland A, Selig Y, et al: Partial ablations of the flocculus and ventral paraflocculus in monkeys
cause linked deficits in smooth pursuit eye movements and adaptive modification
of the VOR. J Neurophysiol 87:912–924, 2002 114. Lisberger S, Miles F, Zee D: Signals used to compute errors in monkey vestibuloocular reflex: possible
role of flocculus. J Neurophysiol 52:1140–1153, 1984 115. Waespe W, Cohen B, Raphan T: Dynamic modification of the vestibulo-ocular reflex by the nodulus and
uvula. Science 228:199–202, 1985 116. Hain T, Zee D, Maria B: Tilt suppression of vestibulo-ocular reflex in patients with cerebellar
lesions. Acta Otolaryngol (Stockh) 105:13–20, 1988 117. Furman JM, Wall C III, Pang D: Vestibular function in periodic alternating nystagmus. Brain 113:1425–1439, 1990 118. Takagi M, Zee DS, Tamargo R: Effects of lesions of the oculomotor vermis on eye movements in primate: saccades. J Neurophysiol 80:1911–1930, 1998 119. Robinson FR, Straube A, Fuchs AF. Role of the caudal fastigial nucleus in saccade generation. II. Effects
of muscimol inactivation. J Neurophysiol 70:1741–1758, 1993 120. Swartz BE, Bespavola IN, Burmeister M, et al: Pathogenesis of clinical signs in recessive cerebellar ataxia with saccadic
intrusions and sensorimotor neuropathy (SCASI). Ann Neurol 54:824–828, 2003 121. Pierrot-Deseilligny C, Amarenco P, Roullet E, et al: Vermal infarct with pursuit eye movement disorders. J Neurol Neurosurg Psychiatry 53:519–521, 1990 122. Buttner U, Straube A, Spuler A: Saccadic dysmetria and “intact” smooth pursuit eye movements
after bilateral deep cerebellar nuclei lesions. J Neurol Neurosurg Psychiatry 57:832–834, 1994 123. Duncan GW, Parker SW, Fisher CM. Acute cerebellar infarction in the PICA territory. Arch Neurol 32:364–368, 1975 124. Grad A, Baloh RW: Vertigo of vascular origin. Clinical and electronystagmographic features
in 84 cases. Arch Neurol 46:281–284, 1989 125. Benjamin EE, Zimmerman CF, Troost BT: Lateropulsion and upbeat nystagmus are manifestations of central vestibular
dysfunction. Arch Neurol 43:962–964, 1986 126. Ranalli P, Sharpe J: Contrapulsion of saccades and ipsilateral ataxia: a unilateral disorder
of the rostral cerebellum. Ann Neurol 20:311–316, 1986 127. Uno A, Mukuno K, Sekiya H, et al: Lateropulsion in Wallenberg's syndrome and contrapulsion in the proximal
type of the superior cerebellar artery syndrome. Neuroophthalmology 9:75–80, 1998 128. Pierrot-Deseilligny C, Muri RM, Ploner CJ, et al: Cortical control of ocular saccades in humans: a model for motricity. Prog Brain Res 142:3–17, 2003 129. Pierrot-Deseilligny C, Ploner CJ, Muri RM, et al: Effects of cortical lesions on saccadic: eye movements in humans. Ann N Y Acad Sci. 956:216–229, 2002 130. Andersen R: Visual and eye movement functions of the posterior parietal cortex. Ann Rev Neurosci 12:377–403, 1989 131. Zeki S: A Vision of the Brain. London: Blackwell Scientific, 1993 132. Barash S, Bracewell RM, Fogassi L, et al: Saccade-related activity in the lateral intraparietal area. I. Temporal
proper comparison with area 7a. J Neurophysiol , ties 66:1095–1108, 1991 133. Dieterich M, Bense S, Lutz S, et al: Dominance for vestibular cortical function in the non-dominant hemisphere Cereb Cortex 13:1007, 2003 134. Ocraven KM, Rosen BR, Kwong KK, et al: Voluntary attention modulates fMRI activity in human MT-MST. Neuron 18:591–598, 1997 135. Dursteler M, Wurtz R: Pursuit and optokinetic deficits following chemical lesions of cortical
areas MT and MST. J Neurophysiol 60:940–965, 1988 136. Newsome W, Wurtz R, Komatsu H: Relation of cortical areas MT and MST to pursuit eye movements. II. Differentiation
of retinal from extraretinal inputs. J Neurophysiol 60:604–620, 1988 137. Goldberg ME, Bisley J, Powell KD, et al: The role of the lateral intraparietal area of the monkey in the generation
of saccades and visuospatial attention. Ann NY Acad Sci 956:205–215, 2002 138. Lynch J, Graybiel A, Lobeck L: The differential projection of two cytoarchitectonic subregions of the
inferior parietal lobule of macaque upon the deep layers of the superior
colliculus. J Comp Neurol 235:241–254, 1985 139. Lynch JC: Saccade initiation and latency deficits after combined lesions of the frontal
and posterior eye fields in monkeys. J Neurophysiol 68:1913–1916, 1992 140. Müri RM, Iba-Zizen MT, Derosier C, et al: Location of the human posterior eye field with functional magnetic resonance
imaging. J Neurol Neurosurg Psychiatry 60:445–448, 1996 141. Pierrot-Deseilligny C, Rivaud S, Gaymard B, et al: Cortical control of reflexive visually-guided saccades. Brain 114:1473-1485, 1991 142. Duhamel J, Goldberg ME, FitzGibbon EJ, et al: Saccadic dysmetria in a patient with a right frontoparietal lesion: the
importance of corollary discharge for accurate spatial-behavior. Brain 115:1387-1402, 1992 143. Heide W, Blankenburg M, Zimmermann E, et al: Cortical control of double-step saccades: Implications for spatial orientation. Ann Neurology 38:739-748, 1995 144. Huerta M, Krubitzer L, Kaas J. Frontal eye field as defined by intracortical microstimulation in squirrel
monkeys, owl monkeys, and macaque monkeys. II. Cortical connections. J Comp Neurol 265:332-361, 1987 145. Bruce CJ, Goldberg ME. Primate frontal eye fields. I. Single neurons discharging before saccades. J Neurophysiol 53:603-635, 1985 146. Dias EC, Kiesau M, Segraves MA: Acute activation and inactivation of macaque frontal eye field with GABA-related
drugs. J Neurophysiol 74:2744–2748, 1995 147. Rosano C, Krisky CM, Welling JS, et al: Pursuit and saccadic eye movement subregions in human frontal eye field: a
high-resolution fMRI investigation. Cereb Cortex 12:107–115, 2002 148. Rivaud S, Müri RM, Gaymard B, et al: Eye movement disorders after frontal eye field lesions in humans. Exp Brain Res 102:110–120, 1994 149. Pierrot-Deseilligny C, Israël I, Berthoz A, et al: Role of the different frontal lobe areas in the control of the horizontal
component of memory-guided saccades in man. Exp Brain Res 95:166–171, 1993 150. Schlag J, Schlag-Rey M: Evidence for a supplementary eye field. J Neurophysiol 57:179–200, 1987 151. Huerta M, Kaas J: Supplementary eye fields as defined by intracortical microstimulation: connections
in macaques. J Comp Neurol 293:299–330, 1990 152. Husain M, Parton A, Hodgson TL, et al: Self-control during response conflict by human supplementary eye field. Nat Neurosci 6:117-118, 2003 153. Gaymard B, Rivaud S, Pierrot-Deseilligny C: Role of the left and right supplementary motor areas in memory-guided saccade
sequences. Ann Neurol 34:404–406, 1993 154. Procyk E, Tanaka YL, Joseph JP: Anterior cingulate activity during routine and non-routine sequential behaviors
in macaques. Nat Neurosci 3:502–508, 2000 155. Tanji J: Sequential organization of multiple movements: Involvement of cortical
motor areas. Annu Rev Neurosci 24:631–651 , 2001 156. Goldmanrakic PS: Cellular basis of working memory. Neuron 14:477–485, 1995 157. Sawaguchi T, Iba M: Prefrontal cortical representation of visuospatial working memory in monkeys
examined by local inactivation with muscimol. J Neurophysiol 86:2041–2053, 2001 158. Sawaguchi T: The effects of dopamine and its antagonists on directional delay-period
activity of prefrontal neurons in monkeys during an oculomotor delayed-response
task. Neurosci Res 41:115–128, 2001 159. Iwatsubo T, Kuzuhara S, Kanemitsu A, et al: Corticofugal projections to the motor nuclei of the brainstem and spinal
cord in humans. Neurology 40:309–312, 1990 160. Stanton GB, Goldberg ME, Bruce CJ: Frontal eye field efferents in the macaque monkey. I. Subcortical pathways
and topography of striatal and thalamic terminal fields. J Comp Neurol 271:473–492, 1988 161. Stanton GB, Goldberg ME, Bruce CJ: Frontal eye field efferents in the macaque monkey. II. Topography of terminal
fields in midbrain and pons. J Comp Neurol 271:493–506, 1988 162. Pierrot-Deseilligny C, Rivaud S, Penet C, et al: Latencies of visually guided saccades in unilateral hemispheric cerebral
lesions. Ann Neurol 21:138–148, 1987 163. Gonzalo-Ruiz A, Leichnetz G, Smith D: Origin of cerebellar projections to the region of the oculomotor complex, medial
pontine reticular formation, and superior colliculus in new
world monkeys: a retrograde horseradish peroxidase study. J Comp Neurol 268:508–526, 1988 164. Leichnetz G: The prefrontal cortico-oculomotor trajectories in the monkey. J Neurol Sci 49:387–396, 1981 165. Shook B, Schlag-Rey M, Schlag J: Direct projection from the supplementary eye field to the nucleus raphe
interpositus. Exp Brain Res 73:215–218, 1988 166. Selemon L, Goldman-Rakic P: Common cortical and subcortical targets of the dorsolateral prefrontal
and posterior parietal cortices in the rhesus monkey: evidence for a distributed
neural network subserving spatially guided behavior. J Neurosci 8:4049–4068, 1988 167. Hikosaka O, Takikawa Y, Kawagoe R: Role of the basal ganglia in the control of purposive saccadic eye movements. Physiol Rev 80:953–978, 2000 168. Lasker AG, Zee DS, Hain TC, et al: Saccades in Huntington's disease: initiation defects and distractability. Neurology 37:364–370, 1987 169. Robinson DA. Eye movements evoked by collicular stimulation in the alert monkey. Vision Res 12:1795–1808, 1972 170. Everling S, Paré M, Dorris MC, et al: Comparison of the discharge charactersitics of brainstem omipause neurons
and superior colliculus fixation neurons in monkey: implications for
control of fixation and saccade behavior. J Neurophysiol 79:511–528, 1998 171. Bergeron A, Matsuo S, Guitton D. Superior colliculus encodes distance to target, not saccade amplitude, in
multi-step gaze shifts. Nat Neurosci 6:404–413, 2003 172. Waitzman DM, Silakov VL, DePalma-Bowles S, et al: Effects of reversible inactivation of the primate mesencephalic reticular
formation. I. Hypermetric goal-directed saccades. J Neurophysiol 83:2260–2284, 2000 173. Waitzman DM, Silakov VL, DePalma-Bowles S, et al: Effects of reversible inactivation of the primate mesencephalic reticular
formation. II. Hypometric vertical saccades. J Neurophysiol 83:2285–2299, 2000 174. Hanes DP, Wurtz RH: Interaction of the frontal eye field and superior colliculus for saccade
generation. J Neurophysiol 85:804–815, 2001 175. Pierrot-Deseilligny C, Rosa A, Masmoudi K, et al: Saccade deficits after a unilateral lesion affecting the superior colliculus. J Neurol Neurosurg Psychiatry 54:1106–1109, 1991 176. Schiller PH, Sandell JH, Maunsell JHR: The effect of frontal eye field and superior colliculus lesions on saccadic
latencies in the rhesus monkey. J Neurophysiol 57:1033–, 19871049 177. Bhidayasiri R, Riley DE, Somers JT, et al: Pathophysiology of slow vertical saccades in progressive supranuclear palsy. Neurology 57:2070–2077, 2001 178. Schiller P, Sandell J, Maunsell J: The effect of frontal eye field and superior colliculus lesions on saccadic
latencies in the rhesus monkey. J Neurophysiol 57:1033–1049, 1987 179. Deng S-Y, Goldberg ME, Segraves MA, et al: The effect of unilateral ablation of the frontal eye fields on saccadic
performance in the monkey. In: Keller EL, Zee DS, eds. Adaptive Processes in Visual and Oculomotor Systems. Oxford:Pergamon, 1986:201–208 180. Schiller PH, True SD, Conway JL: Deficits in eye movements following frontal eye-field and superior colliculus
ablations. J Neurophysiol 44:1175–1189, 1980 181. Lynch JC, McLaren J: Deficits of visual attention and saccadic eye movements after lesions of
parietoocipital cortex in monkeys. J Neurophysiol 61:74–90, 1989 182. Tusa RJ, Zee DS, Herdman SJ: Effect of unilateral cerebral cortical lesions on ocular motor behavior
in monkeys: saccades and quick phases. J Neurophysiol 56:1590–1625, 1986 183. Pierrot-Deseilligny C, Gautier JC, Loron P: Acquired ocular motor apraxia due to bilateral fronto-parietal infarcts. Ann Neurol 23:199-202, 1988 184. Maunsell J, Van Essen D: Functional properties of neurons in middle temporal visual area of the
macaque monkey. I. Selectivity for stimulus direction, speed, and orientation. J Neurophysiol 49:1127-1147, 1983 185. Gottlieb J, MacAvoy M, Bruce C: Unit activity related to smooth pursuit eye movements in rhesus monkey
frontal eye fields. Soc Neurosci Abstr 15:1203, 1989 186. Glickstein M, Cohen J, Dixon B, et al: Corticopontine visual projections in macaque monkeys. J Comp Neurol 190:209-229, 1980 187. Suzuki D, Noda H, Kase M: Visual and pursuit eye movement-related activity in posterior vermis of
monkey cerebellum. J Neurophysiol 46:1120-1139, 1981 188. Newsome W, Pare E: A Selective impairment of motion perception following lesions of the middle
temporal visual area (MT). J Neurosci 8:2201-2211, 1988 189. Morrow MJ, Sharpe JA: Retinoptic and directional deficits of smooth pursuit initiation after
posterior cerebral hemispheric lesions. Neurology 43:595–603, 1993 190. Thurston SE, Leigh RJ, Crawford TJ, et al: Two distinct deficits of visual tracking caused by unilateral lesions of
cerebral cortex in humans. Ann Neurol 23:266–273, 1988 191. Barton JJ, Sharpe JA, Raymond JE: Directional defects in pursuit and motion perception in humans with unilateral
cerebral lesions. Brain 119:1535–1550, 1996 192. Barton JJ, Simpson T, Kiriakopoulos E, et al: Functional MRI of lateral occipitotemporal cortex during pursuit and motion
perception. Ann Neurol 40:387–398, 1996 193. May JG, Keller EL, Suzuki DA: Smooth-pursuit eye movement deficits with chemical lesions in the dorsolateral
pontine nucleus of the monkey. J Neurophysiol 59:952–977, 1988 194. Thier P, Bachor A, Faiss J, et al: Selective impairment of smooth-pursuit eye movements due to an ischemic
lesion of the basal pons. Ann Neurol 29:443–448, 1991 195. Keller EL, Heinen SJ: Generation of smooth-pursuit eye movements: neuronal mechanisms and pathways. Neurosci Res 11:79–107, 1991 196. Johnston JL, Sharpe JA, Morrow MJ: Paresis of contralateral smooth pursuit and normal vestibular smooth eye
movements after unilateral brainstem lesions. Ann Neurol 31:495–, 1992502 197. Büttner U, Straube A: The effect of cerebellar midline lesions on eye movements. Neuroophthalmology 15:75–82, 1995 198. Kaufman SR, Abel LA: The effects of distraction on smooth pursuit in normal subjects. Acta Otolaryngol (Stockh) 102:57–64, 1986 199. Leigh RJ, Tusa RJ: Disturbance of smooth pursuit caused by infarction of occipitoparietal
cortex. Ann Neurol 17:185–187, 1985 200. Levin S, Luebke A, Zee DS, et al: Smooth pursuit eye movements in schizophrenics. Quantitative measurements
with the search-coil technique. J Psychiatr Res 22:195–206, 1988 201. Huebner WP, Leigh RJ, Seidman SH, et al: An investigation of horizontal combined eye-head tracking in patients with
abnormal vestibular and smooth pursuit eye movements. J Neurol Sci 116:152–164, 1993 202. Andersen RA, Bracewell RM, Barash S, et al: Eye position effects on visual, memory, and saccade-related activity in
areas LIP and 7a of macaque. J Neurosci 10:1176–1196, 1990 203. De Renzi E, Colombo A, Faglioni P, et al: Conjugate gaze paresis in stroke patients with unilateral damage. An unexpected
instance of hemispheric asymmetry. Arch Neurol 39:482–486, 1982 204. Kömpf D, Gmeiner H-J: Gaze palsy and visual hemineglect in acute hemisphere lesions. Neuroophthalmology 9:49–53, 1989 205. Sharpe JA, Bondar RL, Fletcher WA: Contralateral gaze deviation after frontal lobe haemorrhage. J Neurol Neurosurg Psychiatry 48:86–88, 1985 206. Steiner I, Melamed E: Conjugate eye deviation after acute hemispheric stroke: delayed recovery
after previous contralateral frontal lobe damage. Ann Neurol 16:509–511, 1984 207. Sullivan HC, Kaminski HJ, Maas EF, et al: Lateral deviation of the eyes on forced lid closure in patients with cerebral
lesions. Arch Neurol 48:310–311, 1991 208. Sharpe JA, Lo AW, Rabinovitch HE: Control of the saccadic and smooth pursuit systems after cerebral hemidecortication. Brain 102:387–403, 1979 209. Troost BT, Daroff RB, Weber RB, et al: Hemispheric control of eye movements. II. Quantitative analysis of smooth
pursuit in a hemispherectomy patient. Arch Neurol 27:449–452, 1972 210. Sharpe JA, Morrow MJ: Cerebral hemispheric smooth pursuit disorders. Neuro-ophthalmology 11:87–98, 1991 211. Cogan DG, Loeb DR: Optokinetic response and intracranial lesions. Arch Neurol Psychiatry 61:183–187, 1949 212. Ebner R, Lopez L, Ochoa S, et al: Vertical ocular motor apraxia. Neurology 40:712–, 1990713 213. Husain M, Stein J: Rezso Balint and his most celebrated case. Arch Neurol 45:89–93, 1988 214. Holmes G: Spasm of fixation. Trans Ophthalmol Soc UK 50:253–262, 1930 215. Johnston JL, Sharpe JA, Morrow MJ: Spasm of fixation: a quantitative study. J Neurol Sci 107:166-171, 1992 216. Fielder AR, Gresty MA, Dodd KL, et al: Congenital ocular motor apraxia. Trans Ophthalmol Soc UK 105:589–598, 1986 217. Harris CM, Shawkat F, Russell-Eggitt I, et al: Intermittent horizontal saccade failure (‘ocular motor apraxia’) in
children. Br J Ophthalmol 80:151–158, 1996 218. Zee DS, Yee RD, Singer HS: Congenital ocular motor apraxia. Brain 100:581–599, 1977 219. Cogan DG, Chu FC, Reingold DR, et al: A long-term follow-up of congenital ocular motor apraxia. Neuroophthalmology 1:145–147, 1980 220. Hsu HN, Yang ML, Lai HC: Familial congenital ocular motor apraxia. Chang Gung Med J 25:411–414, 2002 221. Phillips PH, Brodsky MC, Henry PM: Congenital ocular motor apraxia with autosomal dominant inheritance. Am J Ophthalmol 129:820–822, 2000 222. Shawkat FS, Kingsley D, Kendall B, et al: Neuroradiological and eye movement correlates in children with intermittent
saccade failure: “Ocular motor apraxia.“rdquo; Neuropediatrics 26:298–305, 1995 223. Rascol O, Sabatini U, Simonetta-Moreau M, et al: Square wave jerks in Parkinsonian syndromes. J Neurol Neurosurg Psychiatry 54:599–602, 1991 224. O'Sullivan JD, Maruff P, Tyler P, et al: Unilateral pallidotomy for Parkinson's disease disrupts ocular fixation. J Clin Neurosci 10:181–185, 2003 225. White OB, Saint-Cyr JA, Tomlinson RD, et al: Ocular motor deficits in Parkinson's disease. II. Control of saccadic
and smooth pursuit systems. Brain 106:571–587, 1983 226. Chamberlain W: Restriction of upward gaze with advancing age. Am J Ophthalmol 71:341–346, 1971 227. Demer JL: Pivotal role of orbital connective tissues in binocular alignment and strabismus: the
Friedenwald lecture. Invest Ophthalmol Vis Sci 45 :729–738, 2004 228. Rottach KG, Riley DE, DiScenna AO, et al: Dynamic properties of horizontal and vertical eye movements in parkinsonian
syndromes. Ann Neurol 39:368–377, 1996 229. Repka MX, Claro MC, Loupe DN, et al: Ocular motility in Parkinson's disease. Pediatr Ophthalmol Strabismus 33:144–147, 1996 230. Crawford T, Goodrich S, Henderson L, et al: Predictive responses in Parkinson's disease: manual key presses and
saccadic eye movements to regular stimulus events. J Neurol Neurosurg Psychiatry 52:1033–1042, 1989 231. Lueck CJ, Crawford TJ, Henderson L, et al: Saccadic eye movements in Parkinson's disease. II. Remembered saccades—towards
a unified hypothesis? Q J Exp Psychol 45A:211–233, 1992 232. Nakamura T, Bronstein AM, Lueck CJ, et al: Vestibular, cervical and visual remembered saccades in Parkinson's
disease. Brain 117:1423–1432, 1994 233. Vermersch AI, Rivaud S, Vidailhet M, et al: Sequences of memory-guided saccades in Parkinson's disease. Ann Neurol 35:487–490, 1994 234. Kimmig H, Haussmann K, Mergner T, et al: What is pathological with gaze shift fragmentation in Parkinson's
disease? J Neurol 249:683–692, 2002 235. Vidailhet M, Rivaud S, Gouider-Khouja N, et al: Eye movements in parkinsonian syndromes. Ann Neurol 35:420–426, 1994 236. MacAskill MR, Anderson TJ, Jones RD: Saccadic adaptation in neurological disorders. Prog Brain Res 140:417–431, 2002 237. Bronstein AM, Kennard C: Predictive ocular motor control in Parkinson's disease. Brain 108:925–940, 1985 238. Rascol O, Clanet M, Montastruc JL, et al: Abnormal ocular movements in Parkinson's disease. Evidence for involvement
of dopaminergic systems. Brain 112:1193–1214, 1989 239. Waterston JA, Barnes GR, Grealy MA, et al: Abnormalities of smooth eye and head movement control in Parkinson's
disease. Ann Neurology 39:749–760, 1996 240. White OW, Saint-Cyr JA, Sharpe JA: Ocular motor deficits in Parkinson's disease. I. The horizontal vestibulo-ocular
reflex and its regulation. Brain 106:555–570, 1983 241. Reichert WH, Doolittle J, McDowell FH: Vestibular dysfunction in Parkinson's disease. Neurology 32:1133–1138, 1982 242. Gibson JM, Pimlott R, Kennard C: Ocular motor and manual tracking in Parkinson's disease and the effect
of treatment. J Neurol Neurosurg Psychiatry 50:853–860, 1987 243. Blekher T, Siemers E, Abel LA, et al: Eye movements in Parkinson's disease: before and after pallidotomy. Invest Ophthalmol Vis Sci 41:2177–2183, 2000 244. Rivaud-Pechoux S, Vermersch AI, Gaymard B, et al: Improvement of memory guided saccades in parkinsonian patients by high
frequency subthalamic nucleus stimulation. J Neurol Neurosurg Psychiatry. 68:381–384, 2000 245. Averbuch-Heller L, Stahl JS, Hlavin ML, et al: Square wave jerks induced by pallidotomy in parkinsonian patients. Neurology 52:185–188, 1999 246. Hotson JR, Langston EB, Langston JW. Saccade responses to dopamine in human MTPT-induced parkinsonism. Ann Neurol 20:456–463, 1986 247. Schultz W, Romo R, Scarnati E, et al: Saccadic reaction times, eye-arm coordination and spontaneous eye movements
in normal and MTPT-treated monkeys. Exp Brain Res 78:253–267, 1989 248. Brooks BA, Fuchs AF, Finochio D: Saccadic eye movement deficits in the MTPT monkey model of Parkinson's
disease. Brain Res 383:402–407, 1986 249. Leigh RJ, Foley JM, Remler BF, et al: Oculogyric crisis: a syndrome of thought disorder and ocular deviation. Ann Neurol 22:13–17, 1987 250. Liu GT, Carrazana EJ, Macklis JD, et al: Delayed oculogyric crises associated with striatocapsular infarction. J Clin Neuro-ophthalmol 11:198–201, 1991 251. Frankel M, Cummings JL: Neuro-ophthalmic abnormalities in Tourette's syndrome. Functional
and anatomical implications. Neurology 34:359–361, 1984 252. FitzGerald PM, Jankovic J, Glaze DG, et al: Extrapyramidal involvement in Rett's syndrome. Neurology 40:293–295, 1990 253. FitzGerald PM, Jankovic J: Tardive oculogyric crisis. Neurology 39:1434–1437, 1989 254. Kim JS, Kim HK, Im JH, et al: Oculogryic crisis and abnormal magnetic resonance imaging signals in bilateral
lentiform nuclei. Mov Disord 11:756–758, 1996 255. Nath U, Ben-Shlomo Y, Thomson R, et al: Clinical features and natural history of progressive supranuclear palsy. A
clinical cohort study. Neurology 60:910–916, 2003 256. Davis PH, Bergeron C, McLachlan DR: Atypical presentation of progressive supranuclear palsy. Ann Neurol 17:337–343, 1985 257. Quinn N: The “round the houses” sign in progressive supranuclear palsy. Ann Neurol 40:951 , 2003 258. Das V, Leigh R: Visual-vestibular interaction in progressive supranuclear palsy. Vision Res 40:2077–2081, 2000 259. Seemungal B, Faldon M, Revesz T, et al: Influence of target size on vertical gaze palsy in a pathologically proven
case of progressive supranuclear palsy. Mov Disord 18:818–822, 2003 260. Garbutt S, Han Y, Kumar A, et al: Vertical optokinetic nystagmus and saccades in normal human subjects. Invest Ophthalmol Vis Sci 44:3833–3841, 2003 261. Mastaglia FL, Grainger KMR: Internuclear ophthalmoplegia in progressive supranuclear palsy. J Neurol Sci 25:303–308, 1975 262. Kuniyoshi S, Riley DE, Zee DS, et al: Distinguishing progressive supranuclear palsy from other forms of Parkinson's
disease: evaluation of new signs. Ann NY Acad Sci 956:484–, 2002486 263. Pierrot-Deseilligny C, Rivaud S, Fournier E, et al: Lateral visually-guided saccades in progressive supranuclear palsy. Brain 112:471–487, 1989 264. Goffinet AM, DeVolder AG, Gillian C, et al: Positron tomography demonstrates frontal lobe hypometabolism in progressive
supranuclear palsy. Ann Neurol 25:131–139, 1989 265. Litvan I, Dickson D, Büttner-Ennever J, et al: Research goals in progressive supranuclear palsy. Movement Disorders. Mov Disord 15:446–458, 2000 266. Moses HI, Zee DS: Multi-infarct PSP. Neurology 37:1819, 1987 267. Averbuch-Heller L, Paulson G, Daroff R, et al: Whipple's disease mimicking progressive supranuclear palsy: the diagnostic
value of eye movement recording. J Neurol Neurosurg Psychiatry 66:532–535, 1999 268. Knox DL, Green WR, Troncosa JC, et al: Cerebral ocular Whipple's disease: a 62 year-old odyssey from death
to diagnosis. Neurology 45:617–625, 1995 269. Schwartz MA, Selhorst JB, Ochs AL, et al: Oculomasticatory myorhythmia: a unique movement disorder occurring in Whipple's
disease. Ann Neurol 20:677–683, 1986 270. Lowsky R, Archer GL, Fyles G, et al: Diagnosis of Whipple's disease by molecular analysis of peripheral
blood. N Engl J Med 331:1343–1346, 1994 271. Fleming JL, Wiesner RH, Shorter RG: Whipple's disease: clinical, biochemical, and histopathological features
and assessment of treatment in 29 patients. Mayo Clin Proc 63:539–551, 1988 272. Riley DE, Fogt N, Leigh RJ. The syndrome of ‘pure akinesia’ and its relationship to progressive
supranuclear palsy. Neurology 44:1025–1029, 1994 273. Vidailhet M, Rivaud-Pechoux S: Eye movement disorders in corticobasal degeneration. Adv Neurol 82:161–167, 2000 274. Riley DE, Lang AE, Lewis A, et al: Cortical-basal ganglionic degeneration. Neurology 40:1203–1212, 1990 275. Gibb WRG, Luthert PJ, Marsden CD: Corticobasal degeneration. Brain 112:1171–, 19891192 276. Bertholon P, Bronstein AM, Davies RA, et al: Positional down beating nystagmus in 50 patients: cerebellar disorders
and possible anterior semicircular canalithiasis. J Neurol Neurosurg Psychiatry 72:366–372 , 2002 277. Brett F, Henson C, Staunton H: Familial diffuse Lewy body disease, eye movement abnormalities, and distribution
of pathology. Arch Neurol 59:464–467, 2003 278. de Bruin VM, Lees AJ, Daniel SE: Diffuse Lewy body disease presenting with supranuclear gaze palsy, parkinsonism, and
dementia: a case report. Mov Disord 7:355–358, 1992 279. Grant MP, Cohen M, Petersen RB, et al: Abnormal eye movements in Creutzfeldt-Jakob disease. Ann Neurol 34:192–197, 1993 280. Helmchen C, Büttner U: Centripetal nystagmus in a case of Creutzfeldt-Jakob disease. Neuroophthalmology 15:187–192, 1995 281. Collewijn H, Went LN, Tamminga EP, et al: Oculomotor defects in patients with Huntington's disease and their
offspring. J Neurol Sci 86:307–320, 1988 282. Lasker AG, Zee DS: Ocular motor abnormalities in Huntington's disease. Vision Res 37:3639–3645, 1997 283. Leigh RJ, Newman SA, Folstein SE, et al: Abnormal ocular motor control in Huntington's disease. Neurology 33:1268–1275, 1983 284. Zangemeister WH, Mueller-Jensen A: The coordination of gaze movements in Huntington's disease. Neuroophthalmology 5:193–206, 1985 285. Rubin AJ, King WM, Reinbold KA, et al: Quantitative longitudinal assessment of saccades in Huntington's disease. J Clin Neuroopthalmol 13:59–66, 1993 286. Kirkwood SC, Siemers E, Bond C, et al: Confirmation of subtle motor changes among presymptomatic carriers of the
Huntington disease gene. Arch Neurol 57:1040–1044, 2000 287. Rothlind JC, Brandt J, Zee D, et al: Verbal memory and oculomotor control are unimpaired in asymptomatic adults
with the genetic marker for Huntington's disease. Arch Neurol 50:799–802, 1993 288. Reveley MA, Dursun SM, Andrews H: Improvement of abnormal saccadic eye movements in Huntington's disease
by sulpiride: A case study. J Psychopharmacol 8:262–265, 1994 289. Troost BT, Troost EG: Functional paralysis of horizontal gaze. Neurology 29:82, 1979 |