1. Elkington SG: Pituitary adenoma: preoperative symptomatology in a series of 260 patients. Br J Ophthalmol 52:322, 1968 2. Nachtigaller H, Hoyt WF: Storungen des Scheindruckes bei bitemporaler Hemianopsie und Verschiebung
der Sehachsen. Klin Monatsbl Augenheilkd 156:821, 1970 3. Chamlin M, Davidoff LM, Feiring EH: Ophthalmologic changes produced by pituitary tumors. Am J Ophthalmol 40:353, 1955 4. Wilson P, Falconer MA: Patterns of visual failure with pituitary tumours: clinical and radiological
correlations. Br J Ophthalmol 52:94, 1968 5. Taylor D: Congenital tumours of the anterior visual system with dysplasia of the
optic discs. Br J Ophthalmol 66:455, 1982 6. Grimson BS, Perry DD: Enlargement of the optic disc in childhood optic nerve tumors. Am J Ophthalmol 97:627, 1993 7. Trobe JD, Acosta PC, Krischer JP: A screening method for chiasmal visual field defects. Arch Ophthalmol 99:264, 1981 8. Frisen L: The earliest visual field defects in midchiasmal compression. Doc Ophthalmol 42:14, 1985 9. Gutman I, Behrens M, Odel J: Bilateral central and centrocaecal scotomata due to mass lesions. Br J Ophthalmol 68:336, 1984 10. Horton JC: Wilbrand's knee of the primate optic chiasm is an artefact of monocular
enucleation. Trans Am Ophthalmol Soc 95:579, 1997 11. Suckling RD: Visual fields in posterior chiasmal angle lesions. Trans Ophthalmol Soc N Z 36:23, 1984 12. Symon L, Jakubowski J: Transcranial management of pituitary tumours with suprasellar extension. J Neurol Neurosurg Psychiatry 42:123, 1979 13. Parravano JG, Toledo A, Kucharczyk W: Dimensions of the optic nerves, chiasm, and tracts: MR quantitative comparison
between patients with optic atrophy and normals. J Comput Assist Tomogr 17:688, 1993 14. Wagner AL, Murtagh FR, Hazlett KS et al: Measurement of the normal optic chiasm on coronal images. AJNR Am J Neuroradiol 18:723, 1997 15. Iwata F, Patronas NJ, Caruso RC et al: Association of visual field, cup-disc ratio, and magnetic resonance imaging
of optic chiasm. Arch Ophthalmol 115:729, 1997 16. Kernohan JW, Sayre GP: Tumors of the pituitary gland and infundibulum. In
Atlas of Tumor Pathology, series 1, fascicle 36. Washington, DC, Armed
Forces Institute of Pathology, 1956 17. Burrow GN, Wortzman G, Rewcastle NB et al: Microadenomas of the pituitary and abnormal sellar tomograms in an unelected
autopsy service. N Engl J Med 304:156, 1981 18. Donovan LE, Corenblum B: The natural history of the pituitary incidentaloma. Arch Intern Med 155:181, 1995 19. Thapar K, Kovacs K, Horvath E: Morphology of the pituitary in health and
disease. In Becker KL, Bilezikian JP, Bremner WJ et al (eds): Principles
and Practice of Endocrinology and Metabolism, p 103. Philadelphia, JB
Lippincott, 1995 20. Wilson CB: A decade of pituitary microsurgery: the Herbert Olivecrona lecture. J Neurosurg 61:814, 1984 21. Ebersold MJ, Quast LM, Laws ER et al: Long-term results in transsphenoidal removal of nonfunctioning pituitary
adenomas. J Neurosurg 64:713, 1986 22. Kearns TP, Rucker CW: Arcuate defects in the visual fields due to chromophobe adenoma of the
pituitary gland. Am J Ophthalmol 45:505, 1958 23. Trobe JD: Chromophobe adenoma presenting with a hemianopic temporal arcuate scotoma. Am J Ophthalmol 77: 388, 1974 24. Ikeda H, Yoshimoto T: Visual disturbances in patients with pituitary adenoma. Acta Neurol Scand 92:157, 1995 25. Katznelson L, Klibanski: Prolactin and its disorders. In Becker KL, Bilezikian
JP, Bremner WJ et al (eds): Principles and Practice of Endocrinology
and Metabolism, p 140. Philadelphia, JB Lippincott, 1995 26. Randall RV, Laws ER, Abboud CF et al: Transsphenoidal microsurgical treatment of prolactin-producing pituitary
adenomas: results in 100 patients. Mayo Clin Proc 58: 108, 1983 27. Woodruff WW, Heinz ER, Djang WT et al: Hyperprolactenemia: an unusual manifestation of suprasellar cystic lesions. AJNR Am J Neuroradiol 8:113, 1987 28. Kahn SR, Leblanc R, Sadikot AF et al: Marked hyperprolactinemia caused by carotid aneurysm. Can J Neurol Sci 24:64, 1997 29. Kruse A, Astrup J, Gyldensted C et al: Hyperprolactinemia in patients with pituitary adenomas: the pituitary stalk
compression syndrome. Br J Neurosurg 9:453, 1995 30. Spark RF, Baker R, Bienfang DC, Bergland R: Bromocriptine reduces pituitary tumor size and hypersecretion. JAMA 247: 311, 1982 31. Bonneville JF, Poulignot D, Catin F et al: Computed tomographic demonstration of the effects of bromocriptine on pituitary
microadenoma size. Radiology 143:451, 1982 32. Wollesen F, Anderson T, Karle A: Size reduction of extrasellar pituitary tumors during bromocriptine treatment: quantitation
of effect on different types of tumors. Ann Intern Med 96:281, 1982 33. Moster ML, Savino PJ, Schatz NJ et al: Visual function in prolactinoma patients treated with bromocriptine. Ophthalmology 92:1332, 1985 34. King LW, Molitch ME, Gittinger JW et al: Cavernous sinus syndrome due to prolactinoma: resolution with bromocriptine. Surg Neurol 19:280, 1083 35. Colao A: Prolactinomas resistant to standard dopamine agonists respond to chronic
cabergoline treatment. J Clin Endocrinol Metab 82:876, 1997 36. Grochowski M, Khalfallah Y, Vighetto A et al: Ophthalmic results in patients with macroprolactinomas treated with a new
prolactin inhibitor CV 205–502. Br J Ophthalmol 77:785, 1993 37. Saito K, Kuwayama A, Yamamoto N et al: The transsphenoidal removal of nonfunctioning pituitary adenomas with suprasellar
extensions: the open sella method and intentionally staged
operation. Neurosurgery 36:668, 1995 38. Ciric I, Ragin A, Baumgartner C et al: Complications of transsphenoidal surgery: results of a national survey, review
of the literature, and personal experience. Neurosurgery 40:225, 1997 39. Tsang RW, Brierly JD, Panzarella T et al: Radiation therapy for pituitary adenoma: treatment outcome and prognostic
factors. Int J Radiat Oncol Biol Phys 30:557, 1994 40. Seo Y, Fukuoka S, Takanashi M et al: Gamma knife surgery for Cushing's disease. Surg Neurol 43:170, 1995 41. Raymond J, Hardy J, Czepko R et al: Arterial injuries in transspenoidal surgery for pituitary adenoma: the
role of angiography and endovascular treatment. AJNR Am J Neuroradiol 18:655, 1997 42. Slavin ML, Lam BL, Decker RE et al: Chiasmal compression from fat packing after transsphenoidal resection of
intrasellar tumor in two patients. Am J Ophthalmol 115: 368, 1993 43. Baskin DS: Neurosurgical management of pituitary-hypothalamic neoplasms. In
Becker KL, Bilezikian JP, Bremner WJ et al (eds): Principles and
Practice of Endocrinology and Metabolism, p 238. Philadelphia, JB Lippincott, 1995 44. Valtonen S, Salmi J: Operative management of chromophobe pituitary tumour recurrences. Acta Neurochir (Wien) 62:233, 1982 45. Ebersold MJ, Quast LM, Laws ER et al: Long-term results in transsphenoidal removal of nonfunctioning pituitary
adenomas. J Neurosurg 64:713, 1986 46. Thapar K: Proliferative activity and invasiveness among pituitary adenomas and carcinomas: an
analysis using the MIB-1 antibody. Neurosurgery 38:99, 1996 47. Sammartino A, Bonavolonta G, Pettinato G, Loffredo A: Exophthalmos caused by an invasive pituitary adenoma in a child. Ophthalmologica 179:83, 1979 48. De Divitiis E, De Chiara A, Benvenuti D et al: Adenome invasif hypophysaire chez un enfant. Neurochirurgie 26: 405, 1980 49. Juneau P, Schoene WC, Black P: Malignant tumors in the pituitary gland. Arch Neurol 49:555, 1992 50. Gould TJ, Johnson LN, Colapinto EV et al: Intrasellar vascular malformation mimicking a pituitary macroadenoma. J Neuroophthalmol 16:199, 1996 51. Hollenhorst RW, Younge BR: Ocular manifestations produced by adenomas of
the pituitary gland: analysis of 1,000 cases. In Kohler PO, Ross GT (eds): Diagnosis
and Treatment of Pituitary Tumors, p 53. New York, Elsevier, 1973 52. Merimee TJ, Grant MB: Growth hormone and its disorders. In Becker KL, Bilezikian
JP, Bremner WJ et al (eds): Principles and Practice of Endocrinology
and Metabolism, p 129. Philadelphia, JB Lippincott, 1995 53. Lundin P, Engstom E, Karlsson FA et al: Long-term octrotide therapy in growth hormone secreting pituitary adenomas: evaluation
with serial MR. AJNR Am J Neuroradiol 18:765, 1997 54. Rolih CA, Ober P: Pituitary apoplexy. Endocrinol Metab Clin North Am 22:291, 1993 55. Symon L, Mohanty S: Haemorrhage in pituitary tumours. Acta Neurochir (Wien) 65:41, 1982 56. Fraioli B, Esposito V, Palma L et al: Hemorrhagic pituitary adenomas: clinicopathological features and surgical
treatment. Neurosurgery 27:741, 1990 57. Holness RO, Ogundino FA, Langille RA: Pituitary apoplexy following closed head trauma. J Neurosurg 59:677, 1983 58. Savage EB, Gugino L, Starr PA et al: Pituitary apoplexy following cardiopulmonary bypass: considerations for
a staged cardiac and neurosurgical procedure. Eur J Cardiothorac Surg 8:333, 1994 59. Masago A, Ueda Y, Kanai H et al: Pituitary apoplexy after pituitary function test: a report of two cases
and review of the literature. Surg Neurol 43:158, 1995 60. Reid RL, Quigley ME, Yen SSC: Pituitary apoplexy: a review Arch Neurol 42:712, 1985 61. Poussaint TY, Barnes PD, Anthony DC et al: Hemorrhagic pituitary adenomas in adolescence. AJNR Am J Neuroradiol 17: 1907, 1996 62. McFadzean RM, Doyle D, Rampling R et al: Pituitary apoplexy and its effect on vision. Neurosurgery 29:669, 1991 63. Bonicki W, Kasperlik-Zaluska A, Koszewski W et al: Pituitary apoplexy: endocrine, surgical and oncological emergency. Incidence, clinical
course and treatment with reference to 799 cases of pituitary
adenomas. Acta Neurochir (Wien) 120:118, 1993 64. Brisman MH, Katz G, Post KD: Symptoms of pituitary apoplexy rapidly reversed with bromocriptine: case
report. J Neurosurg 85:1153, 1996 65. Maccagnan P, Macedo CL, Kayath MJ et al: Conservative management of pituitary apoplexy: a prospective study. J Clin Endocrinol Metab 80:2190, 1995 66. Hall WA, Luciano MG, Doppman JL et al: Pituitary magnetic resonance imaging in normal human volunteers: occult
adenomas in the general population. Ann Intern Med 120:817, 1994 67. Wolansky LJ, Rao SB, Schulder M et al: Extrasellar extension of pituitary lesions: comparison of T2 weighted fast
spin echo MRI with T1 weighted sequences. Int J Neuroradiol 2:147, 1996 68. Scotti G, Yu CY, Dillon WP et al: MR imaging of cavernous sinus involvement by pituitary adenomas. AJNR Am J Neuroradiol 9:657, 1988 69. Destrieux C, Kakou MK, Velut S et al: Microanatomy of the hypophyseal fossa boundaries. Neurosurgery 88:743, 1998 70. Teng MMH, Huang CI, Chang T: The pituitary mass after transsphenoidal hypophysectomy. AJNR Am J Neuroradiol 9:23, 1988 71. Hsu DW, Efird JT, Hedley-White ET: Progesterone and estrogen receptors in meningiomas: prognostic considerations. J Neurosurg 86:113, 1997 72. Rucker CW, Kearns TP: Mistaken diagnosis in some cases of meningioma. Am J Ophthalmol 51:15, 1961 73. Slavin ML: Acute, severe, symmetric visual loss with cecocentral scotomas due to olfactory
groove meningioma. J Clin Neuroophthalmol 6:224, 1986 74. Gregorius FK, Hepler RS, Stern WE: Loss and recovery of vision with suprasellar meningiomas. J Neurosurg 42:69, 1975 75. Ehlers N, Malmros R: The suprasellar meningioma: a review of the literature and presentation
of a series of 31 cases. Acta Ophthalmol Suppl 121:1, 1973 76. Yeakley JW, Kulkarni MV, McArdle CB et al: High-resolution MR imaging of juxtasellar meningiomas with CT and angiographic
correlation. AJNR Am J Neuroradiol 9:279, 1988 77. Chan RC, Thompson GB: Morbidity, mortality and quality of life following surgery for intracranial
meningiomas: a retrospective study in 257 cases. J Neurosurg 60:52, 1984 78. Mirimanoff RO, Dosoretz DE, Linggood RM et al: Analysis of recurrence and progression following neurosurgical resection. J Neurosurg 62:18, 1985 79. Rosenberg LF, Miller NR: Visual results after microsurgical removal of meningiomas involving the
anterior visual system. Arch Ophthalmol 102:1019, 1984 80. Petty AM, Kun LE, Meyer GA: Radiation therapy for incompletely resected meningiomas. J Neurosurg 62:502, 1985 81. Carella RJ, Ransohoff J, Newall J: Role of radiation therapy in the management of meningioma. Neurosurgery 10:332, 1982 82. Maire JP, Caudry M, Guerin J et al: Fractionated radiation therapy in the treatment of intracranial meningiomas: local
control, functional efficacy, and tolerance in 91 patients. Int J Radiat Oncol Biol Phys 33:315, 1995 83. Haie-Meder C, Brunel P, Cioloca C et al: Role of radiotherapy in the treatment of meningioma. Bull Cancer Radiother 82:35, 1995 84. Black PM: Hormones, radiosurgery and virtual reality: new aspects of meningioma management. Can J Neurol Sci 24:302, 1997 85. Kupersmith MJ, Warren FA, Newall J et al: Irradiation of meningiomas of the intracranial anterior visual pathway. Ann Neurol 21:131, 1987 86. Kennerdell JS, Maroon JC, Malton M et al: The management of optic nerve sheath meningiomas. Am J Ophthalmol 106:450, 1988 87. Grunberg SM, Weiss MH, Spitz IM et al: Treatment of unresectable meningiomas with the antiprogesterone agent mifepristone. J Neurosurg 74:861, 1991 88. Kyritsis AP: Chemotherapy for meningiomas. J Neurooncol 29: 269, 1996 89. Olivero WC, Lister R, Elwood PW: The natural history and growth rate of asymptomatic meningiomas: a review
of 60 patients. J Neurosurg 83:222, 1995 90. Burger PC, Scheithauer BW, Vogel FS: Surgical Pathology of the Nervous
System and its Coverings, 3rd ed, p 536. New York, Churchill Livingstone, 1991 91. McLone DG, Raimondi AJ, Naidich TP: Craniopharyngiomas. Childs Brain 9:188, 1982 92. Savino PJ, Paris M, Schatz NJ et al: Optic tract syndrome: a review of 21 patients. Arch Ophthalmol 96:656, 1978 93. Eldevik OP, Blaivas M, Gabrielson TO et al: Craniopharyngioma: radiologic and histologic findings and recurrence. AJNR Am J Neuroradiol 17:1427, 1996 94. Brodsky MC, Hoyt WF, Barnwell SL et al: Intrachiasmatic craniopharyngioma: a rare cause of chiasmal thickening. Case
report. J Neurosurg 68:300, 1988 95. Cushing HW: Papers Relating to the Pituitary Body, Hypothalamus, and Parasympathetic
Nervous System. Springfield, IL, Charles C Thomas, 1932 96. Katz E: Late results of radical excision of craniopharyngioma in children. Neurosurgery 42:86, 1975 97. Carmel PW, Antunes JL, Chang CH: Craniopharyngiomas in children. Neurosurgery 11:382, 1982 98. Fischer EG, Welsh K, Belli JA et al: Treatment of craniopharyngioma in children 1972-1981. J Neurosurg 62:496, 1985 99. Laws ER: Transsphenoidal microsurgery in the management of craniopharyngioma. J Neurosurg 52:661, 1980 100. Halstead AE: The operative treatment of tumors of the hypophysis. Surg Gynecol Obstet 10:494, 1910 101. Alvord EC Jr, Lofton S: Gliomas of the optic nerve or chiasm: outcome by patient's age, tumor
site, and treatment. J Neurosurg 68:85, 1988 102. Hoyt WF, Baghdassarian SA: Optic glioma of childhood: natural history and rationale for conservative
management. Br J Ophthalmol 53:793, 1969 103. Blatt J, Jaffe R, Deutsch M et al: Neurofibromatosis and childhood tumors. Cancer 57:1225, 1986 104. Lewis RA, Gerson LP, Axelson KA et al: Von Recklinghausen neurofibromatosis. II. Incidence of optic gliomata. Ophthalmology 91:929, 1984 105. Bognanno JR, Edwards MK, Lee TA et al: Cranial MR imaging in neurofibromatosis. AJNR Am J Neuroradiol 9:461, 1988 106. Miller NR, Iliff WJ, Green WR: Evaluation and management of gliomas of the anterior visual pathways. Brain 97:743, 1974 107. McCullough DC, Epstein F: Optic pathway tumors: a review with proposals for clinical staging. Cancer 56:1789, 1985 108. Layden WE, Edwards WC: Ocular manifestations of the dien-cephalic syndrome. Am J Ophthalmol 73:78, 1972 109. Arnoldi KA, Tychsen L: Prevalence of intracranial lesions in children originally diagnosed with
disconjugate nystagmus (spasmus nutans). J Pediatr Ophthalmol Stabismus 32:296, 1995 110. DeSousa AC, Kalsbeck JE, Mealey J, Fitzgerald J: Diencephalic syndrome and its relation to optico-chiasmatic glioma: review
of twelve cases. Neurosurgery 4:207, 1979 111. Poussaint TY, Barnes PD, Nichols K et al: Diencephalic syndrome: clinical features and imaging findings. AJNR Am J Neuroradiol 18:1499, 1997 112. Perlongo G, Carollo C, Salviati L et al: Diencephalic syndrome and disseminated juvenile pilocytic astrocytomas
of the hypothalamic-optic chiasm region. Cancer 80:142, 1997 113. Rieth KG, Comite F, Dwyer AJ et al: CT of cerebral abnormali-ties in precocious puberty. AJR Am J Roentgenol 148:1231, 1987 114. Bronen RA, Fulbright RK, Reynders CS et al: Magnetic resonance imaging of central precocious puberty: the importance
of hypothalamic abnormalities. Int J Neuroradiol 1:145, 1995 115. Habiby R, Silverman B, Listernick R et al: Precocious puberty in children with neurofibromatosis type 1. J Pediatr 126:364, 1995 116. Lavery MA, O'Neill JF, Chu FC et al: Acquired nystagmus in early childhood: a presenting sign of intracranial
tumor. Ophthalmology 91:425, 1984 117. Glaser JS, Hoyt WF, Corbett J: Visual morbidity with chiasmal glioma: long-term studies of visual fields
in untreated and irradiated cases. Arch Ophthalmol 85:3, 1971 118. Fletcher WA, Imes RK, Hoyt WF: Chiasmal gliomas: appearance and long-term changes demonstrated by computerized
tomography. J Neurosurg 65:154, 1986 119. Lourie GL, Osborne DR, Kirks DR: Involvement of posterior visual pathways by optic nerve gliomas. Pediatr Radiol 16:271, 1986 120. Patronas NJ, Dwyer AJ, Papathanasiou M et al: Contributions of magnetic resonance imaging in the evaluation of optic
gliomas. Surg Neurol 28:367, 1987 121. Menor F, Marti-Bonmati L: CT detection of basal ganglion lesions in neurofibromatosis type 1: correlation
with MRI. Neuro-radiology 34:305, 1992 122. Albright AL, Sclabassi RJ: Use of the Cavitron ultrasonic aspirator (CUSA) and visual evoked potentials
for chiasmal gliomas of children. J Neurosurg 63:138, 1985 123. Coppeto JR, Monteiro ML, Uphoff DF: Exophytic suprasellar gliomas: a rare cause of chiasmatic compression. Case
report. Arch Ophthalmol 105:28, 1987 124. Massry GG, Morgan CF, Chung SM: Evidence of optic pathway gliomas after previously negative neuroimaging. Ophthalmology 104:930, 1997 125. Parazzini C, Triulzl F, Bianchini E et al: Spontaneous involution of optic pathway lesions in neurofibromatosis type 1: serial
contrast MR evaluation. AJNR Am J Neuroradiol 16:1711, 1995 126. Leisti EL, Pyhtinen J, Poyhonen M: Spontaneous decrease of a pilocytic astrocytoma in neurofibromatosis type 1. AJNR Am J Neuroradiol 17:1691, 1996 127. Imes RK, Hoyt WF: Childhood chiasmal gliomas: update on the fate of patients in the 1969 San
Francisco study. Br J Ophthalmol 70:179, 1986 128. Pierce SM, Barnes PD, Loeffler JS et al: Definitive radiation therapy in the management of symptomatic patients
with optic glioma: survival and long-term effects. Cancer 65: 45, 1990 129. Kovalic JJ, Grigsby PW, Shepard MJ et al: Radiation therapy for gliomas of the optic nerve and chiasm. Int J Radiat Oncol Biol Phys 18:927, 1990 130. Tao ML, Barnes PD, Billet AL et al: Childhood optic chiasm gliomas: radiographic response following radiotherapy
and long-term clinical outcome. Int J Radiat Oncol Biol Phys 39:579, 1997 131. Erkal HS, Serin M, Cakmak A: Management of optic pathway and chiasmatic-hypothalmic gliomas in children
with radiation therapy. Radiother Oncol 45:11, 1997 132. Danoff BF, Cowchock FS, Marquette C et al: Assessment of the long-term effects of primary radiation therapy for brain
tumors in children. Cancer 49:1580, 1982 133. Packer RJ, Savino PJ, Bilaniuk LT et al: Chiasmatic gliomas of childhood: a reappraisal of the natural history and
effectiveness of cranial irradiation. Childs Brain 10:393, 1983 134. Davis PC, Hoffman JC, Pearl GS et al: CT evaluation of effects of cranial radiation therapy in children. AJNR Am J Neuroradiol 7:639, 1986 135. Beyer RA, Paden P, Sobel DF et al: Moyamoya pattern of vascular occlusion after radiotherapy for gliomas of
the optic chiasm. Neurology 36:1173, 1986 136. Packer RJ, Ater J, Allen J et al: Carboplatin and vincristine chemotherapy for children with newly diagnosed
progressive low-grade gliomas. J Neurosurg 86:747, 1997 137. Chamberlain MC: Recurrent chiasmatic-hypothalamic glioma treated with oral etoposide. Arch Neurol 52:509, 1995 138. Safneck JR, Napier LB, Halliday WC: Malignant astrocytoma of the optic nerve in a child. Can J Neurol Sci 19:498, 1992 139. Aroichane M, Miller NR, Eggenberger ER: Glioblastoma multiforme masquerading as pseudotumor cerebri: case report. J Clin Neuroophthalmol 13:105, 1993 140. Hoyt WF, Meshel LG, Lessell S et al: Malignant optic glioma of adulthood. Brain 96:121, 1973 141. Taphoorn MJB, de Vries-Knoppert WAEJ, Ponssen H et al: Malignant optic glioma in adults. J Neurosurg 70:277, 1989 142. Millar WS, Tartaglino LM, Sergott RC et al: MR of malignant optic glioma of adulthood. AJNR Am J Neuroradiol 16:1673, 1995 143. Albers GW, Hoyt WF, Forno LS et al: Treatment response in malignant optic glioma of adulthood. Neurology 38: 1071, 1988 144. Matsutani M, Sano K, Takakura K et al: Primary intracranial germ cell tumors: a clinical analysis of 153 histologically
verified cases. J Neurosurg 86:446, 1997 145. Camins MB, Mount LA: Primary suprasellar atypical teratoma. Brain 97:447, 1974 146. Wilson JT, Wald SL, Aitken PA et al: Primary diffuse chiasmatic germinomas: differentiation from optic chiasm
gliomas. Pediatr Neurosurg 23:1, 1995 147. Lumsden CE: The neuropathology of multiple sclerosis. In Vinken PJ, Bruyn
GW (eds): Handbook of Neurology, Vol 9, p 217. Amsterdam, North Holland, 1970 148. Traquair HM: Acute retrobulbar neuritis affecting the optic chiasm and tract. Br J Ophthalmol 9:433, 1925 149. Spector RH, Glaser JS, Schatz NJ: Demyelinative chiasmal lesions. Arch Neurol 37:757, 1980 150. Lindenberg R, Walsh FB, Sacks JG: Neuropathology of Vision: An atlas, p 250. Philadelphia, Lea & Febiger, 1973 151. Lefkowitz D, Angelo JN: Neuromyelitis optica with unusual vascular changes. Arch Neurol 41:1103, 1984 152. Waespe W, Haenny P: Bitemporal hemianopia due to chiasmal optic neuritis. Neuroophthalmology 7:69, 1987 153. Purvin V, Herr GJ, De Myer W: Chiasmal neuritis as a complication of Epstein-Barr virus infection. Arch Neurol 45:458, 1988 154. Reynolds WD, Smith JL, McCrary JA: Chiasmal optic neuritis. J Clin Neuroophthalmol 2:93, 1982 155. Cant JS, Harrison MT: Chiasmatic arachnoiditis with growth failure. Am J Ophthalmol 65:432, 1968 156. Oliver M, Beller AJ, Behar A: Chiasmal arachnoiditis as a manifestation of generalized arachnoiditis
in systemic vascular disease: clinico-pathological report of 2 cases. Br J Ophthalmol 52:227, 1968 157. Iraci G, Giordano R, Gerosa M et al: Cystic suprasellar and retrosellar arachnoiditis. Ann Ophthalmol 8:1175, 1979 158. Takahashi T, Isayama Y: Chiasmal meningitis. Neuroophthalmology 1:19, 1980 159. Scott IU, Silva-Lepe A, Siatkowski RM: Chiasmal optic neuritis in Lyme disease. Am J Ophthalmol 123:136, 1997 160. Decker RE, Mardayat M, Marc J et al: Neurosarcoidosis with computerized tomographic visualization and transsphenoidal
excision of a supra- and intrasellar granuloma. J Neurosurg 50: 814, 1979 161. Tang RA, Grotta JC, Lee KF et al: Chiasmal syndrome in sarcoidosis. Arch Ophthalmol 101:1069, 1983 162. Agbogu BN, Stern BJ, Sewell C et al: Therapeutic considerations in patients with refractory neurosarcoidosis. Arch Neurol 52: 875, 1995 163. Stelzer KJ, Thomas CR, Berger MS et al: Radiation therapy for sarcoid of the thalamus/posterior third ventricle: case
report. Neurosurgery 36:1188, 1995 164. Navarro IM, Peralta VR, Leon JM et al: Tuberculous optochiasmatic arachnoiditis. Neurosurgery 9:654, 1981 165. Kuroda Y, Miyahara M, Sakemi T et al: Autopsy report of acute necrotizing opticomyelopathy associated with thyroid
cancer. J Neurol Sci 120:29, 1993 166. Zucchini S, Ambrosetto P, Carla G et al: Primary empty sella: differences and similarities between children and
adults. Acta Paediatr 84:1382, 1995 167. McGrath P: Cysts of sellar and pharyngeal hypophyses. Pathology 3:123, 1971 168. Benes SL, Kansu T, Savino PJ et al: Ocular manifestations of arachnoid
cysts. In Glaser JS (ed): Neuro-Ophthalmology: Symposium of the University
of Miami, Vol 10, p 107. St. Louis, CV Mosby, 1980 169. Sumida M, Uozumi T, Mukada K et al: Rathke cleft cysts: correlation of enhanced MR and surgical findings. AJNR Am J Neuroradiol 15:525, 1994 170. Rao GP, Blyth CPJ, Jeffreys RV: Ophthalmic manifestations of Rathke's cleft cysts. Am J Ophthalmol 119:99, 1995 171. Baskin DS, Wilson CB: Transsphenoidal treatment of non-neoplastic intrasellar cysts: a report
of 38 cases. J Neurosurg 60:8, 1984 172. Appen RE, deVenecia G, Selliken JM, Giles LT: Meningeal carcinomatosis with blindness. Am J Ophthalmol 86:661, 1978 173. Takahashi T, Murase T, Isayama Y: Clinicopathological findings in the chiasmal
region with reference to carcinomatous optic neuropathy cases. In
Shimizu K, Oosterhuis JA (eds): Ophthalmology, Vol 2, p 1124. Amsterdam, Excerpta
Medica, 1979 174. Takahashi T, Yoshimasa I: Pathological findings in the chiasmal region with reference to malignant
lymphoma. Folia Ophthalmol Jpn 31:1118, 1980 175. Howard RS, Duncombe AS, Owens C et al: Compression of the optic chiasm due to a lymphoreticular malignancy. Postgrad Med J 63:1091, 1987 176. Tabarin A, Corcuff JB, Dautheribes M et al: Histiocytosis X of the hypothalamus. J Endocrinol Invest 14:139, 1991 177. Max MB, Deck F, Rottenberg DA: Pituitary metastases: incidence in cancer patients and clinical differentiation
from pituitary adenoma. Neurology 31:998, 1981 178. Woiciechowsky C, Vogel S, Meyer R et al: Magnetic resonance imaging of a glioblastoma of the optic chiasm: case
report. J Neurosurg 83:923, 1995 179. Liu GT, Galetta SL, Rorke LB et al: Gangliogliomas involving the optic chiasm. Neurology 46:1669, 1996 180. Morrison DA, Bibby K: Sellar and suprasellar hemangiopericytoma mimicking pituitary adenoma. Arch Ophthalmol 115: 1201, 1997 181. Balcer LJ, Galetta SL, Curtis M: von Hippel-Lindau disease manifesting as a chiasmal syndrome. Surv Ophthalmol 39: 302, 1995 182. Teixeira F, Penagos P, Lozano D et al: Medulloblastoma pre-senting as blindness of rapid evolution: a case report. J Clin Neuroophthalmol 11:250, 1991 183. Goodwin JA, Glaser JS: Chiasmal syndrome in sphenoid sinus mucocele. Ann Neurol 4:440, 1978 184. Valvassori GE, Putterman AM: Ophthalmologic and roentgenographic findings in sphenoidal mucoceles. Trans Am Acad Ophthalmol Otolaryngol 77:703, 1973 185. Abla AA, Maroon JC, Wilberger JE et al: Intrasellar mucocele simulating pituitary adenoma: case report. Neurosurgery 18: 197, 1986 186. Heinz GW, Nunery WR, Grossman CB: Traumatic chiasmal syndrome associated with midline basilar skull fractures. Am J Ophthalmol 117:90, 1994 187. Elisevich KV, Ford RM, Anderson DP et al: Visual abnormalities with multiple trauma. Surg Neurol 22:565, 1984 188. Savino PJ, Glaser JS, Schatz NJ: Traumatic chiasmal syndrome. Neurology 30:963, 1980 189. Ess T, Weiler G: Histomorphologische Befunde and Chiasma opticum bei Schadel-Hirntrauma. Z Rechtsmed 82:257, 1979 190. Crompton MR: Hypothalamic lesions following closed head injury. Brain 94:165, 1971 191. Arkin MS, Rubin AD, Bilyk JR et al: Anterior chiasmal optic nerve avulsion. AJNR Am J Neuroradiol 17:1777, 1996 192. Hayman A, Carter K, Schiffman JS et al: A sellar misadventure: imaging considerations. Surv Ophthalmol 41:252, 1996 193. Warman R, Glaser JS: Radionecrosis of optico-hypothalamic glioma. Neuroophthalmology 9:219, 1989 194. Kline LB, Kim JV, Ceballos R: Radiation optic neuropathy. Ophthalmology 92:1118, 1985 195. Ebner R, Slamovits TL, Friedland S et al: Visual loss following treatment of sphenoid sinus carcinoma. Surv Ophthalmol 40: 62, 1995 196. Atkinson AB, Allen IV, Gordon DS et al: Progressive visual failure in acromegaly following external pituitary irradiation. Clin Endocrinol 10:469, 1979 197. Hammer HM: Optic chiasmal radionecrosis. Trans Ophthalmol Soc UK 103:208, 1983 198. Girkin CA, Comey CH, Lunsford LD et al: Radiation optic neuropathy after stereotactic radiosurgery. Ophthalmology 104: 1634, 1997 199. Zimmerman CF, Schatz NJ, Glaser JS: Magnetic resonance imaging of radiation optic neuropathy. Am J Ophthalmol 110: 389, 1990 200. Hudgins PA, Newman NJ, Dillong WP et al: Radiation-induced optic neuropathy: characteristics on gadolinium-enhanced
MR. AJNR Am J Neuroradiol 13:235, 1992 201. Hufnagel TJ, Kim JH, Lesser R et al: Malignant glioma of the optic chiasm eight years after radiotherapy for
prolactinoma. Arch Ophthalmol 106:1701, 1988 202. Aristizabal S, Caldwell WL, Avila J: The relationship of time dose fractionation factors to complication in
the treatment of pituitary tumors by irradiation. Int J Radiat Oncol Biol Phys 2:667, 1977 203. Parsons JT, Bova FJ, Fitzgerald CR et al: Radiation optic neuropathy after megavoltage external-beam irradiation: analysis
of time-dose factors. Int J Radiation Oncol Biol Phys 30:755, 1994 204. Wilson WB, Perez GM, Kleinschmidt-Demasters BK: Sudden onset of blindness in patients treated with oral CCNU and low-dose
cranial irradiation. Cancer 59:901, 1987 205. Glantz MJ, Burger PC, Friedman AH et al: Treatment of radiation-induced nervous system injury with heparin and warfarin. Neurology 44:2020, 1994 206. Borruat FX, Schatz NJ, Glaser JS et al: Visual recovery from radiation-induced optic neuropathy: the role of hyperbaric
oxygen therapy. J Clin Neuroophthalmol 13:98, 1993 207. Borruat FX, Schatz NJ, Glaser JS: Radiation optic neuropathy: report of cases, role of hyperbaric oxygen
therapy, and literature review. Neuroophthalmology 16:255, 1996 208. Sinclair AHH, Dott NM: Hydrocephalus simulating tumour in the production of chiasmal and other
parahypophysial lesions. Trans Ophthalmol Soc U K 51:232, 1931 209. Hughes EBC: Some observations on the visual fields in hydrocephalus. J Neurol Neurosurg Psychiatry 9:30, 1946 210. Corbett JJ: Neuro-ophthalmologic complications of hydrocephalus and shunting procedures. Semin Neurol 6:111, 1986 211. Calogero JA, Alexander E: Unilateral amaurosis in a hydro-cephalic child with an obstructed shunt: case
report. J Neurosurg 34:236, 1971 212. Kupersmith MJ, Rosenberg C, Kleinberg D: Visual loss in pregnant women with pituitary adenomas. Ann Intern Med 121: 473, 1994 213. Roelvink NCA, Kamphorst W, van Alphen HAM et al: Pregnancy-related primary brain and spinal tumors. Arch Neurol 44:209, 1987 214. Abramsky O: Pregnancy and multiple sclerosis. Ann Neurol 36:38, 1994 215. Cosman F, Post KD, Holub DA et al: Lymphocytic hypophysitis: report of three new cases and review of the literature. Medicine 68:240, 1989 216. Sunness JS: The pregnant woman's eye. Surv Ophthalmol 32: 219, 1988 217. Kaufman B: The “empty” sella turcica, a manifestation of the intrasellar
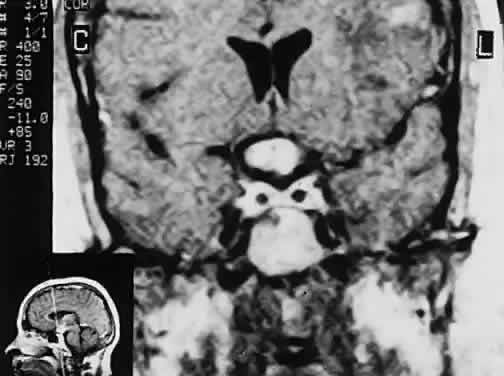
subarachnoid space. Radiology 90:931, 1968 218. Bergland RM, Ray BS, Torack RM: Anatomical variations in the pituitary gland and adjacent structures in 225 human
autopsy cases. J Neurosurg 28:93, 1968 219. Zagardo MT, Cail WS, Kelman SE et al: Reversible empty sella in idiopathic intracranial hypertension: an indicator
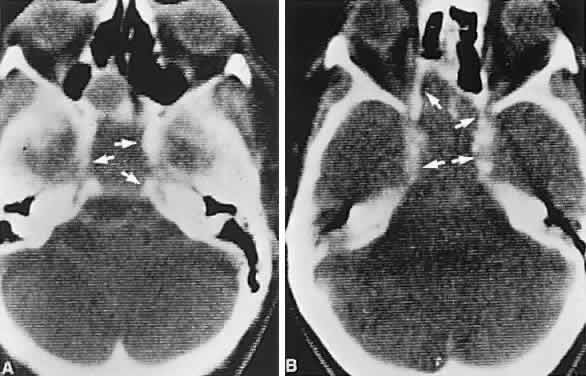
of successful therapy. AJNR Am J Neuroradiol 17:1953, 1996 220. Neelon FA, Goree JA, Lebovitz HE: The primary empty sella: clinical and radiographic characteristics and
endocrine function. Medicine 52:73, 1973 221. Brismar K: Prolactin secretion in the empty sella syndrome in prolactinomas and in
acromegaly. Acta Med Scand 209:397, 1981 222. Tremoulet M, Petrus M, Bonafe A, Rochiccioli P: La selle turcique vide de l'enfant. Rev Otoneuroophthalmol 54: 405, 1982 223. Wilkinson IA, Duck SC, Gager WE, Daniels DL: Empty-sella syndrome: occurrence in childhood. Am J Dis Child 136:245, 1982 224. Berke JP, Buxton LF, Kokmen E: The “empty sella.” Neurology 25:1137, 1975 225. Olson DR, Guiot G, Derome P: The symptomatic empty sella: prevention and correction via the transsphenoidal
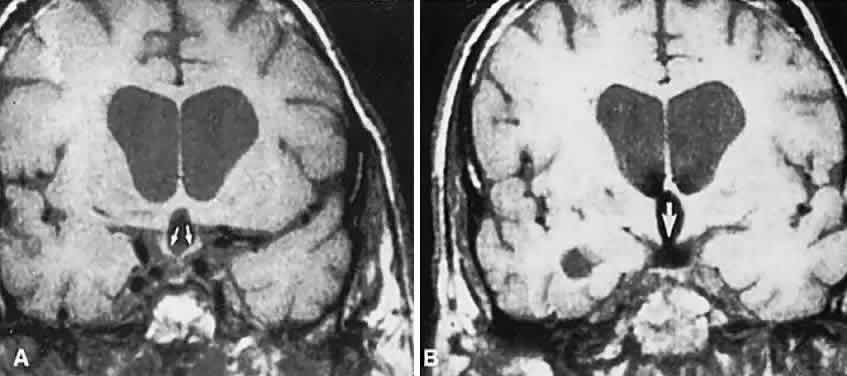
approach. J Neurosurg 37:553, 1972 226. Bursztyn EM, Lavyne MH, Aisen M: Empty sella syndrome with intrasellar herniation of the optic chiasm. AJNR Am J Neuroradiol 4:167, 1983 227. Kaufman B, Tomsak RL, Kaufman BA et al: Herniation of the suprasellar visual system and third ventricle into empty
sellae: morphologic and clinical consideration. AJNR Am J Neuroradiol 10:65, 1989 |