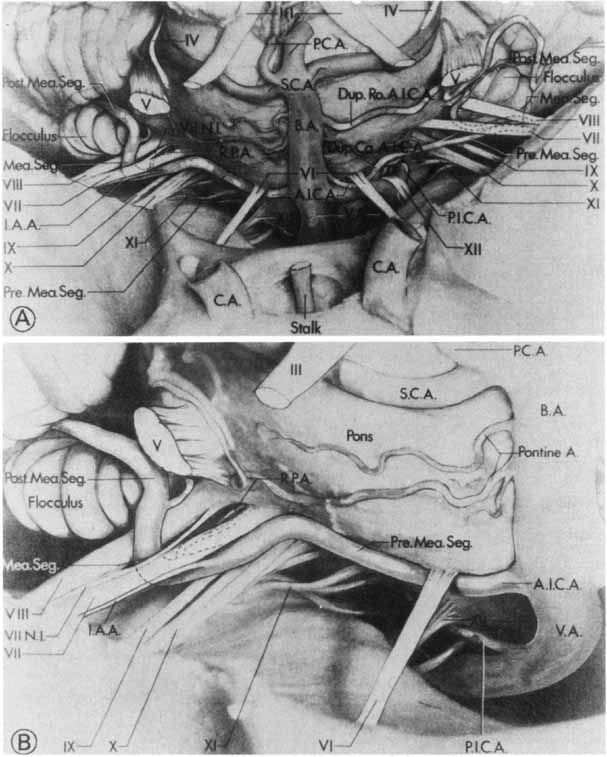
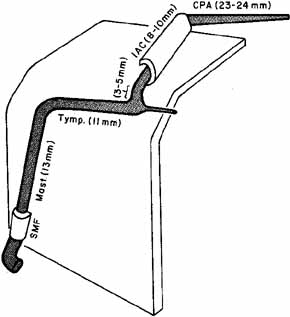
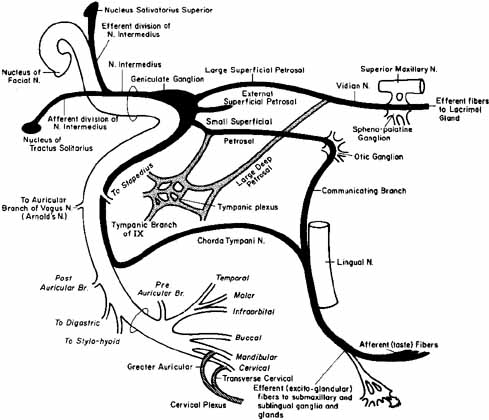
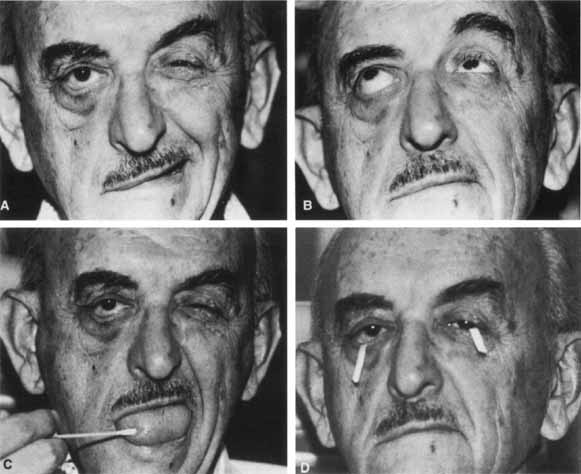
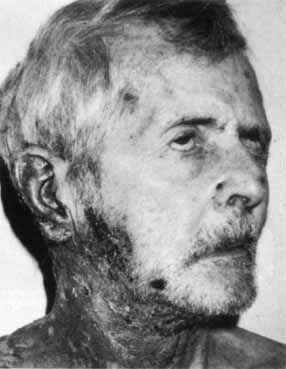
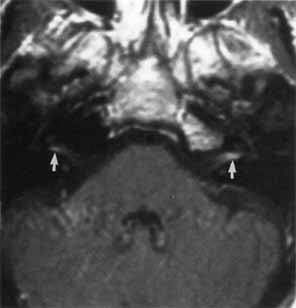
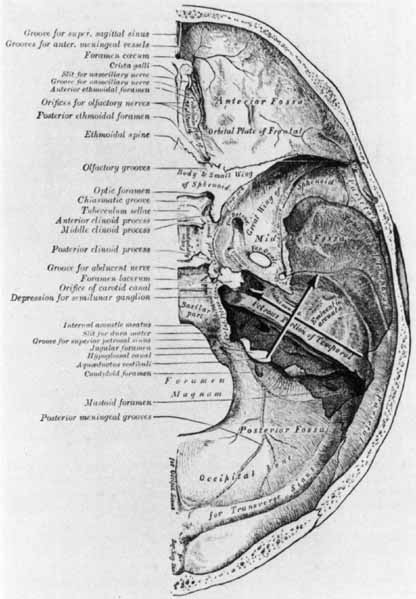
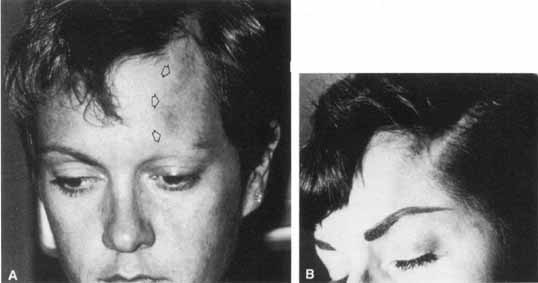
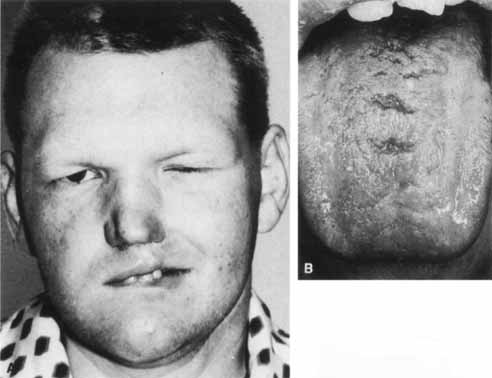
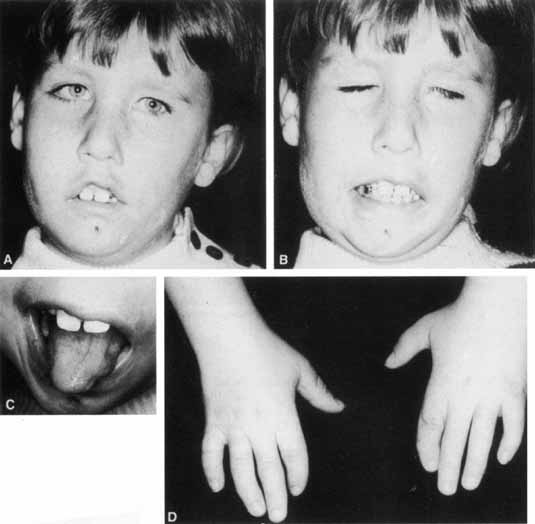
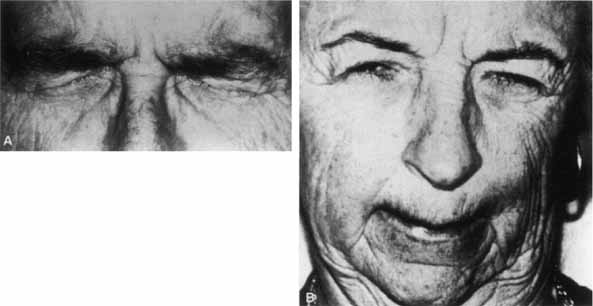
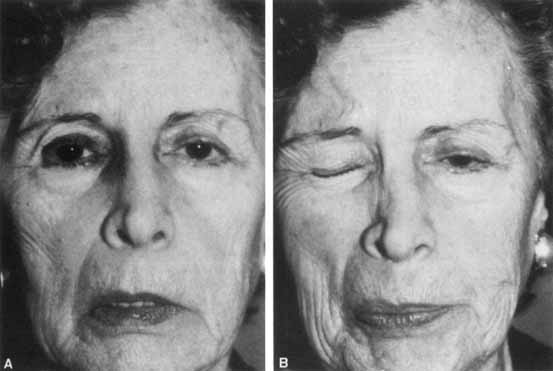
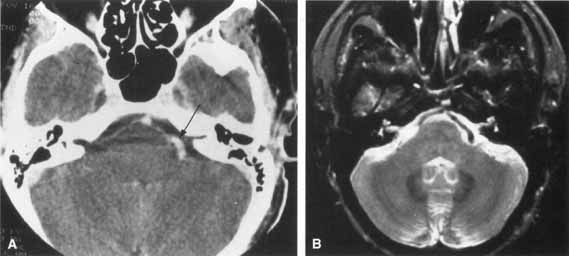
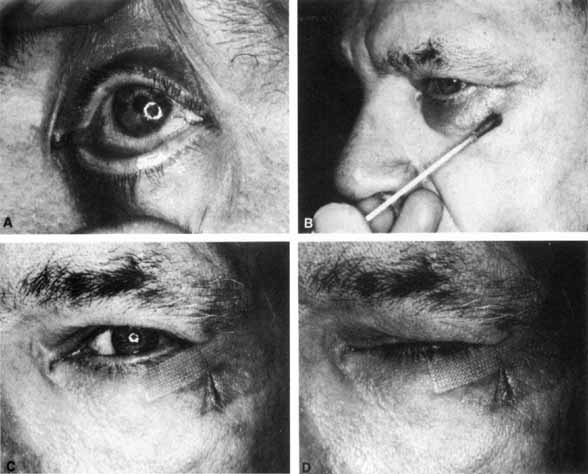
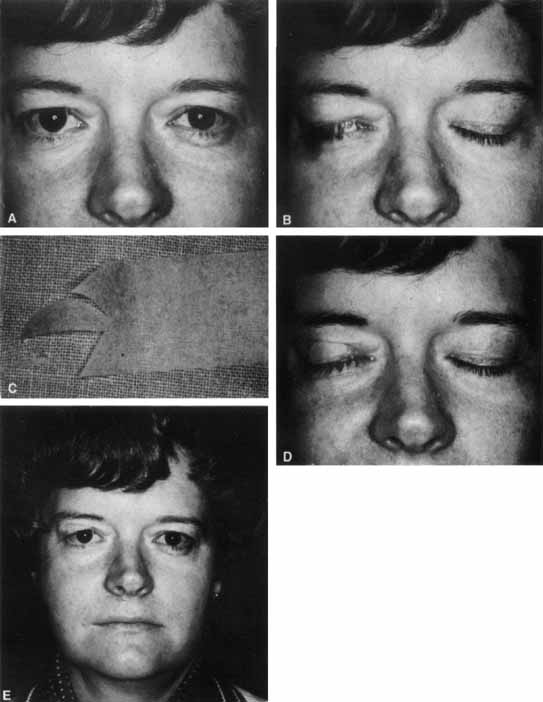
1. Gasser RF: The development of the facial nerve in man. Ann Otol Rhinol Laryngol 76:37, 1967 2. Gasser RF: The development of the facial muscles in man. Am J Anat 120:357, 1967 3. Goycoolea MV, Paparell MM, Carpenter AM: Ganglia and ganglion cells in the middle ear. Arch Otolaryngol 108:276, 1982 4. Bischoff EPE: Microscopic analysis of anastomoses between the cranial nerves. In Sachs E, Valtin EW (eds): The facial nerve. Hanover, NH: University Press of New England, 1977: 43–76 5. Jahrsdoerfer RA: The facial nerve in congenital middle ear malformations. Laryngoscope 91:1217, 1981 6. Terao S, Miura N, Takeda A, et al: Course and distribution of facial corticobulbar tract fibers in the lower
brainstem. J Neurol Neurosurg Psych 69:262, 2000 7. Crosby EC, DeJonge BR: Experimental and clinical studies of the central connections and the central
relations of the facial nerve. Ann Otol 72:735, 1963 8. Van Buskirk C: The seventh nerve complex. J Comp Neurol 82:303, 1945 9. Carpenter M: Core Text of Neuro-Anatomy, Baltimore: Williams & Wilkins, 1985: 151 10. Martin RG, Grant JL, Peace D: Microsurgical relationship of the anterior inferior cerebellar artery and
the facial vestibulocochlear nerve complex. Neurosurgery 6:483, 1980. 11. Guerrier Y: Surgical anatomy, particularly vascular supply of the facial nerve. In Fisch U (ed): Facial Nerve Surgery. Birmingham: Aesculapius Publishing Co., 1977: 12–23 12. Davis RA, Anson BJ, Puddinger JM, Kurth RE: Surgical anatomy of the facial nerve and parotid gland based upon a study
of 350 cervical facial halves. Surg Gynecol Obstet 102:385, 1956 13. Nager GT, Nager N: The arteries of the human middle ear, with particular regard to the blood
supply of the auditory ossicle. Ann Otol Rhinol Laryngol 62:923, 1953 14. Blunt MJ: The possible role of vascular changes in the etiology of Bell's palsy. J Laryngol 70:701, 1956 15. Donath T, Lengyel I: The vascular structure of the intrapetrosal section of the facial nerve, with
special reference to peripheral facial palsy. Acta Med Acad Sci Hung 10:249, 1957 16. May M, Hardin WB: Facial palsy: interpretation of neurologic findings. Trans Am Acad Ophthalmol Otolaryngol 84:710, 1977 17. Schmidtke K, Buttner-Ennever JA: Nervous control of eyelid function. A review of clinical, experimental
and pathologic data. Brain 115:227, 1992 18. Evinger C, Manning KA, Sibony PA: Eyelid movements. Mechanisms and normal data. Invest Ophthalmol Vis Sci 32:387, 1991 19. Francis IL, Loughhead JA: Bell's phenomenon. A study of 508 patients. Aust J Ophthalmol 12:15, 1984 20. Hauser WA, Karnes WE, Annis J: Incidence and prognosis of Bell's palsy in the population of Rochester, MN: Mayo Clin Proc 46:258, 1971 21. Adour KK, Byl FM, Hilsinger RL: The true nature of Bell's palsy: Analysis of 1000 consecutive patients. Laryngoscope 88:787, 1978 22. Peitersen E: The natural history of Bell's palsy. Am J Otol 4:107, 1982 23. Katusic SK, Beard CM, Wiederholt WC: Incidence, clinical features and prognosis in Bell's palsy. Ann Neurol 20:622, 1986 24. Kohler A, Chofflon M, Sztajzel R, et al: Cerebrospinal fluid in acute peripheral facial palsy. J Neurol 246:165, 1999 25. Park HW, Watkins AL: Facial paralysis: Analysis of 500 cases. Arch Phys Med 30:749, 1949 26. Paolino E, Granieri E, Tola MR: Predisposing factor in Bell's palsy: A case control study. J Neurol 232:363, 1985 27. Pechet P, Schattner A: Concurrent Bell's palsy and diabetes mellitus: A diabetic mononeuropathy? J Neurol 232:363, 1985 28. Hilsinger RL, Aduor KK, Doty HE: Idiopathic facial paralysis, pregnancy and the menstrual cycle. Ann Otol Rhinol Laryngol 84:433, 1975 29. Alter M: Familial aggregation of Bell's palsy. Arch Neurol 8:557, 1963 30. Takahash A, Fujiwara R: Familial Bell's palsy. Report of seven families. Clin Neurol (Tokyo) 11:454, 1971 31. May M: Muscle transposition for facial reanimation. Indications and results. Arch Otolaryngol 110:184, 1984 32. Dumitru D, Walsh NE, Porter LD: Electrophysiologic evaluation of the facial nerve in Bell's palsy: A
review. Am J Phys Med Rehabil 67:137, 1988 33. Morgan M, Nathwani D: Facial palsy and infection: The unfolding story. Clin Infect Dis 14:263, 1992 34. Adour KK: Medical management of idiopathic Bell's palsy. Otolaryngol Clin North Am 24:666, 1991 35. Hyden D, Roberg M, Forsberg P: Acute idiopathic peripheral facial palsy: Clinical, serological, and cerebrospinal
fluid findings and effects of corticosteroids. Am J Otolaryngol 14:179, 1993 36. Murakami S, Mizobuchi M, Nakashiro Y, et al: Bell palsy and herpes simplex virus: Identification of viral DNA in endoneural
fluid and muscle. Ann Intern Med 124:27, 1996 37. Kohsyu H, Aoyagi M, Tojima H: Facial nerve enhancement in Gd-MRI in patients with Bell's palsy. Acta Otolaryngol 511(suppl):165, 1994 38. Knox G: Treatment controversies in Bell's palsy. Arch Otolaryngol Head Neck Surg 124:821, 1998 39. May M, Klein SR, Taylor FH: Idiopathic (Bell's) facial palsy: Natural history defies
steroid or surgical treatment. Laryngoscope 95:406, 1985 40. Stankiewicz JA: A review of the published data on steroids and idiopathic facial paralysis. Otolaryngol Head Neck Surg 97:481, 1987 41. Grogan PM, Gronseth GS: Practice parameter: Steroids, acyclovir, and surgery for Bell's palsy (an
evidence-based review). Report of the Quality
Standards Subcommittee of the American Academy of Neurology. Neurology 56:830, 2001 42. Adour KK, Ruboyianes JM, Von Doersten PG, et al: Bell's treatment with acyclovir and prednisone compared with prednisone
alone: A double-blind randomized controlled trial. Ann Otol Rhinol Laryngol 105:37, 1996 43. Hunt JR: On herpetic inflammations of the geniculate ganglion. A new syndrome and
its implications. J Nerve Ment Dis 34:73, 1907 44. Hunt JR: A further contribution to herpetic inflammations of the geniculate ganglion. Am J Med Sci 136:226, 1916 45. Portenoy RK, Duma C, Foley KM: Acute herpetic and postherpetic neuralgia. Clinical review and current
management. Ann Neurol 20:651, 1986 46. Stafford FW, Welch AR: The use of acyclovir in Ramsay Hunt syndrome. J Laryngol Otol 100:337, 1986 47. Inamura H, Aoyagi M, Tojima H, Koike Y: Effects of acyclovir in Ramsay Hunt syndrome. Acta Otolaryngol 446(suppl):111, 1988 48. Kinishi M, Amatsu M, Mohri M, et al: Acyclovir improves recovery rate of facial nerve palsy in Ramsay Hunt syndrome. Ausis, Nasus, Larynx 28(3):223, 2001 49. Abramsky O, Webb C, Tietelbaum D: Cellular immune response to peripheral nerve basic protein in idiopathic
facial paralysis (Bell's palsy). J Neurol Sci 26:13, 1975 50. Asbury AK: Diagnostic considerations in Guillain-Barré syndrome. Ann Neurol 9(suppl):1, 1981 51. Ohtsuka K, Nakamura Y, Hashimoto M: et al.: Fisher syndrome associated with IgG antiGQ1b antibody following infection
by a specific serotype of Campylobacter jejuni. Ophthalmology 105: 1281, 1998. 52. Charous DI, Saxe BI: Landry-Guillain-Barré syndrome: Report of an unusual case with a
comment on Bell's palsy. N Engl J Med 267:1334, 1962 53. Clark JR, Carlson RD, Sasak CT: Facial paralysis in Lyme disease. Laryngoscope 95:1341, 1985 54. Peltomaa M, Pyykko I, Seppala I, et al: Lyme borreliosis and facial paralysis-a prospective analysis of
risk factors and outcome. Am J Otolaryngol 23(3), 125, 2002. 55. Markby DP: Lyme disease facial palsy: Differentiation from Bell's palsy. Br Med J 299:605, 1989 56. Rawlings JA, Fournier PU, Teltow GJ: Isolation of Borrelia spirochetes from patients in Texas. J Clin Microbiol 25:1148. 1987 57. Hunter EF, Russel H, Farstly CE: Evaluation of sera from patients with Lyme disease in the fluorescent treponemal
antibody-absorption test for syphilis. Sex Transm Dis 13:232, 1986 58. Kamitsuka M, Feldman R, Richardson M: Facial paralysis associated with otitis media. Pediatr Infect Dis 6:682, 1985 59. Yetiser S, Tosun F, Kazkayasi M, et al: Facial nerve paralysis due to chronic otitis media. Otology and Neurotology 23:580, 2002 60. Gradenigo G: A special syndrome of endocranial otitic complications. Ann Otol Rhinol Laryngol 13:637, 1904 61. Kohut RF, Lindsay JR: Necrotizing (malignant) external otitis histopathologic processes. Ann Otol 88:714, 1979 62. Naldol JB: Histopathology of Pseudomonas osteomyelitis of the temporal bone starting
as malignant external otitis. Am J Otolaryngol 115:359, 1980 63. McGove FH: Bilateral Bell's palsy. Laryngoscope 75:1070, 1965 64. Dreifus FE, Martin JD, Green RC: Brainstem encephalitis. Va Med Month 91:15, 1964 65. Yasui I, Miyasaki T: Case of poliomyelitis due to virus type I manifested only by right facial
paralysis. J Jpn Assn Infect Dis 36:427, 1962 66. Sklar VEF, Patriarca PA, Onorato IM: Clinical findings and results of treatment in an outbreak of acute hemorrhagic
conjunctivitis is southern Florida. Ain J Ophthalmol 95:45, 1983 67. Bosher SK: Leprosy presenting as facial palsy. J Laryngol 76:827, 1962 68. Lucente FE, Tobias GW, Parisier SC: Tuberculous otitis media. Laryngoscope 88:1107, 1978 69. Bergstrom L, Hemenway WG, Barnhardt RA: Rhinocerebral and otologic mucormycosis. Ann Otol 79:70, 1970 70. Gussen R, Canalis RF: Mucormycosis of the temporal bone. Ann Otol 91:27, 1982 71. Verduijn PG, Bleeker JD: Secondary syphilis of the facial nerve. Arch Otolaryngol 48:675, 1939 72. Dastur FD, Shahani MT, Dastoor DH, et al: Cephalic tetanus: demonstration of a dual lesion. J Neurol Neurosurg Psychiatry 40:782, 1977 73. Harrison TR. Principles of internal medicine. 12th ed. New York: McGraw-Hill, 1987 74. Thompson PK, Vaphiades MS, Saccente M, et al: Cat scratch disease presenting as neuroretinitis and peripheral facial
palsy. J Neuro-Ophthalmology 19:240, 1999 75. Grant AC, Hunter S, Partin WC: A case of acute monocytic ehrlichiosis with prominent neurologic signs. Neurology 48:1619, 1997 76. Sahludovich S: Accidents due to antirabies vaccine: pseudoperitoneal syndrome followed
by bilateral paralysis: Case. Ed Dia Medico (Buenos Aires) 18:1454, 1946 77. Jappich G: Effects and side effects of oral poliomyelitis vaccinations. Monatsschr Kinderheilkd 112:112, 1964 78. Danforth HB: Familial Bell's palsy. Ann Otol 73:179, 1964 79. Lerond J: Ascending paralysis after tetanus antiserum's rapid regression in
member lingering facial paralysis. Bull Mem Soc Med Hop Paris 50:1695, 1926 80. Wechsler AF, Ho DD: Bilateral Bell's palsy at the time of HIV seroconversion. Neurology 39:747, 1989 81. Wright RE: Blocking the main trunk of the facial nerve in cataract operations. Arch Ophthalmol 55:555, 1926 82. Lambert PR, Brackman DE: Facial paralysis in longitudinal temporal bone fractures: A review of 26 cases. Laryngoscope 94:1022, 1984 83. Cannon CR, Jahrsdoerfer RA: Temporal bone fractures: Review of 90 cases. Arch Otolaryngol 109:285, 1983 84. Hitselberger WE, House WF: Tumors of the cerebellopontine angle. Arch Otolaryngol 80:720, 1964 85. Kettel K: Peripheral facial palsies due to tumors: Pathology and clinical picture: Review
of the literature and a report of three cases of intratemporal
tumors of the facial nerve. Arch Otolaryngol 69:276, 1959 86. Cawthorne T, Griffith A: Primary cholesteatoma of the temporal bone. Arch Otolaryngol 73:252, 1961 87. Koide C, Imai A, Nagaba A: Pathological findings of the facial nerve in a case of facial palsy associated
with benign parotid tumor. Arch Otolaryngol Head Neck Surg 120:410, 1994 88. Pulec JL, House WF: Facial nerve involvement and testing in acoustic neuromas. Arch Otolaryngol 80:685, 1964 89. Lee SH, Rao K. Cranial computed tomography and MRI. 2nd ed. New York: McGraw-Hill, 1987 90. Harner SG, Daube JR, Beatty CW, Ebersold MJ: Intraoperative monitoring of the facial nerve. Laryngoscope 98:209, 1988 91. Uziel A, Benezech J, Frerebeau P: Intraoperative facial nerve monitoring in posterior fossa acoustic neuroma
surgery. Otolaryngol Head Neck Surg 108:126, 1993 92. Kunihiro T, Kanzaki J, Shiobara R, et al: Long-term prognosis of profound facial nerve facial nerve paralysis
secondary to acoustic neuroma resection. J Oto-Rhinolaryngol and Related Specialties 61:98, 1999 93. Pillsbury HE, Price HC, Gardner LH: Primary tumors of the facial nerve. Laryngoscope 93:1045, 1983 94. Sherman JD, Dagnew E, Pensak ML, et al: Facial nerve neuromas: Report of 10 cases and review of the literature. Neurosurgery 50:450, 2002 95. Wasserstorm WR, Glass PT, Posner JB: Diagnosis and treatment of leptomeningeal metastasis from solid tumors. Cancer 49:759, 1982 96. Olson ME, Chernik NC, Posner JB: Infiltration of the leptomeninges by systemic cancer. Arch Neurol 30:122, 1974 97. Cohen JP, Lachman LJ, Hammerschlag PE: Reversible facial paralysis in sarcoidosis. Confirmation by serum angiotensin-converting
enzyme assay. Arch Otolaryngol 109:832, 1983 98. Stern BT, Krumholz A, Johns C: Sarcoidosis and its neurological manifestations. Arch Neurol 42:909, 1985 99. Facial palsy in Heerfordt's syndrome: Electrophysiological localization
of the lesion. Muscle and Nerve 22(9): 1279, 1999 100. Lewis M, Kallenbach J, Hockman M: Otolaryngologic complications of acute porphyria. Laryngoscope 93:483, 1983 101. Suarez JI, Cohen ML, Larkin J, et al: Acute intermittent porphyria: Clinicopathologic correlation: report of
a case and review of the literature. Neurology 48:1678, 1997 102. Ridley A: The neuropathy of acute intermittent porphyria. Q J Med 151:301, 1969 103. Osher RH, Griggs RC: Orbicularis fatigue: The “peek” sign of myasthenia gravis. Arch Ophthalmol 97:677, 1979 104. Milone M, Monaco ML, Evoli A: Ocular myasthenia: Diagnostic value of single fiber EMG in the orbicularis
oculi muscle. J Neurol Neurosurg Psychiatry 56:720, 1993 105. Shugar MA, Granich MS, Reardon EJ: The otolaryngologic presentation of botulism. Laryngoscope 91:121, 1981 106. Hanson PA, Rowland LP: Möbius' syndrome and facioscapulohumeral muscular dystrophy. Arch Neurol 24:31, 1971 107. Bandello F, Rosa N, Ghisolfi F: New Findings in the Parry Romberg syndrome: A case report. Eur J Ophthalmol 12: 556, 2002 108. Miller MT, Sloane H, Goldberg MF: Progressive hemifacial atrophy (Parry-Romberg disease). J Pediatr Ophthalmol Strabismus 24:27, 1987 109. Stone J: Parry-Romberg syndrome: A global survey of 205 patients using the Internet. Neurology 61:674, 2003 110. Chung MH, Sum J, Morrell MJ, Horoupian DS: Intracerebral involvement in scleroderma en coup de sabre: Report of a
case with neuropathologic findings. Ann Neurol 37:679, 1995 111. Saberman MN, Tenta LT: The Melkersson-Rosenthal syndrome. Arch Otolaryngol 84:292, 1966 112. Chen C, Huigol SC, James C, et al: Melkersson-Rosenthal syndrome presenting with upper lid edema and facial
palsy. Can J Ophthalmol 37(6):361, 2002 113. Minor MW, Fox RW, Bukantz SL: Melkersson-Rosenthal syndrome. J Allergy Clin Immunol 80:64, 1987 114. May M, Fria TJ, Blumenthal F: Facial paralysis in children: Differential diagnosis. Otolaryngol Head Neck Surg 89:841, 1981 115. Harris JP, Davidson TM, May M: Evaluation and treatment of congenital facial paralysis. Arch Otolaryngol 109:145, 1983 116. McHugh HE: Facial paralysis in birth injury and skull fractures. Arch Otol 78:443, 1963. 117. Hepner WR: Some observations on facial paresis in the newborn infant: Etiology and
incidence. Pediatrics 8:494, 1951 118. Falco NA, Eriksson E: Facial nerve palsy in the newborn: Incidence and outcome. Plast Reconstr Surg 85:1, 1990 119. Pape KE, Pickering B: Asymmetric crying facies: An index of other congenital anomalies. J Pediatr 81:21, 1972 120. Towfighi T, Marks K, Palmer E: Möbius' syndrome: neuropath observations. Acta Neuropathol 48:11, 1979 121. Keane JR, Young JA: Blepharospasm with bilateral basal ganglia infarction. Arch Neurol 42:1206, 1985 122. Hallet M: Blepharospasm: recent advances. Neurology 59:1306, 2002 123. Schmidt KE, Linden DEJ, Goebel R, et al: Striatal activation during blepharospasm revealed by fMRI. Neurology 60:1738, 2003 124. Evinger C, Perlmutter JS: Blind men and blinking elephants. Neurology 60:1732, 2003 125. Jankovic J, Havins WE, Wilkins RB: Blinking and blepharospasm: Mechanism, diagnosis and management. JAMA 284:3160, 1982 126. Reynolds DH, Smith JL, Walsh TH: Differential sections of the facial nerve for blepharospasm. Trans Am Acad Ophthalmol Otolaryngol 71:656, 1967 127. Fox SA: Essential blepharospasm. Arch Ophthalmol 76:318, 1966 128. Biglan A, May M: Treatment of facial spasm with Oculinum. J Pediatr Ophthalmol Strabismus 23:216, 1986 129. Engstrom P, Arnoult J, Malow M: Effectiveness of botulinum toxin therapy for essential blepharospasm. Ophthalmology 94:971, 1987 130. Jordan DR, Patrinely JR, Anderson RL: Essential blepharospasm and related dystonias. Surv Ophthalmol 34:123, 1989 131. Fukuda H, Ishikawa M, Okumura R: Demonstration of neurovascular compression in trigeminal neuralgia and
hemifacial spasm with magnetic resonance imaging: Comparison with surgical
findings in 60 consecutive cases. Surg Neurol 59(2):93, 2003 132. Jannetta PT, Abbasy M, Marion JC: Etiology and definitive microsurgical treatment of hemifacial spasm: Operative
techniques and results in 47 patients. J Neurosurg 47:321, 1977 133. Gardner WJ, Sava GA: Hemifacial spasm: A reversible pathophysiologic state. J Neurosurg 19:240, 1962 134. Nielsen VK: Pathophysiology of hemifacial spasm: A reversible pathophysiologic state. Neurology 34:418, 1984 135. Ferguson JH: Hemifacial spasm and the facial nucleus. Ann Neurol 4:97, 1978 136. Davis WE, Luterman BF, Pulliam MW: Hemifacial spasm caused by cholesteatoma: Am J Otol 2:272, 1981 137. Adler CH, Zimmerman RA, Savino PJ: Hemifacial spasm: Evaluation by magnetic resonance imaging and magnetic
resonance tomographic angiography. Ann Neurol 32:502, 1992 138. Auger R, Piepras D, Laws E: Hemifacial spasm: Results in microvascular decompressions of the facial
nerve in 54 patients. Mayo Clin Proc 61:650, 1986 139. Huang CI, Chen IH, Lee LS: Microvascular decompression of hemifacial spasm: Analysis of operative
findings and results in 310 patients. Neurosurgery 30:53, 1992 140. Garland PE, Patrinely JR, Andreson RI: Hemifacial spasm: Results of unilateral myectomy. Ophthalmology 94:288, 1987 141. Alexander GE, Moses H: Carbamazepine for hemifacial spasm. Neurology 32:286, 1982 142. Sandyk R: Facial myokymia. J Neurosurg 59:1108, 1983 143. Frueh BR: Associated facial contracting after seventh nerve palsy mimicking jaw-winking. Ophthalmology 90:1105, 1983 144. Lubkin V: The inverse Marcus Gunn phenomenon. Arch Neurol 35:249, 1978 145. Chorobski J: Syndrome of crocodile tears. Arch Neurol Psychiatry 65:299, 1951 146. Axelsson A, Laage-Hellman JE: The gustolachrymal reflex: The syndrome of crocodile tears. Acta Otolaryngol 54:239, 1962 147. Frey L: Syndrome of auriculotemporal nerve. Rev Neurol 2:97, 1923 148. Laage-Hellman JE: Gustatory sweating and flushing. Aetiological implications of latent period
and mode of development after parotidectomy. Acta Otolaryngol 49:366, 1958 149. Guntinas-Luchius O: Increased botulinum toxin type A dosage is more effective in patients with
Frey's syndrome. Laryngoscope 112:746, 2002 150. Andermann F, Cosgrove JBR, Lloyd-Smith DL: Facial myokymia in multiple sclerosis. Brain 84:31, 1961 151. Tenser RB, Corbett JJ: Myokymia and facial contraction in brainstem glioma: An electrographic
study. Arch Neurol 30:425, 1974 152. Espinosa RE, Lambert EH, Klass DW: Facial myokymia affecting the electroencephalogram. Mayo Clin Proc 42:258, 1967 153. Radu EW, Skorpil V, Raeser HE: Facial myokymia. Eur Neurol 13:499, 1975 154. Wasserstrom WR, Starr A: Facial myokymia in the Guillain-Barré syndrome. Arch Neurol 35:576, 1977 155. Feldman RG, Lessell S, Travers PM: Neuro-ophthalmologic and neuropsychological effect of trichloroethylene
intoxication: 18 year follow-up. Neurology 34:242, 1984 156. Morris HH, Esters MC: Bilateral facial myokymia following cardiopulmonary arrest. Arch Neurol 38:393, 1981 157. Waybright EA, Gutman L, Chou SM: Facial myokymia. Pathological features. Arch Neurol 36:244, 1979 158. Jacobs L, Kaba S, Pullicino P: The lesion causing continuous facial myokymia in multiple sclerosis. Arch Neurol 51:1115, 1994 159. Tozolovanu V, Forget R, Iancu A, et al: Prolonged orbicularis oculi activity: A major factor in apraxia of lid
opening. Neurology 57(6):1013, 2001 160. Goldstein JE, Cogan DG: Apraxia of lid opening. Arch Ophthalmol 73:155, 1965 161. Keane JR: Lid opening apraxia in Wilson's disease. J Clin Neuro-Ophthalmol 8:31, 1988 162. Lepore F, Duvoisin R: “Apraxia” of eyelid opening: An involuntary levator inhibition. Neurology 35:423, 1985 163. Boghen D: Apraxia of lid opening: a review. Neurology 48:1491, 1997 164. Johnston J, Rosenbaum DM, Picone CM: Apraxia of eyelid opening secondary to right hemisphere infarction. Ann Neurol 25:622, 1989 165. Dewey RB, Maragonore DM: Isolated eyelid opening apraxia: Report of a new levodopa-responsive
syndrome. Neurology 44:752, 1994 166. Ross Russell RW: Supranuclear palsy of eyelid closure. Brain 103:71, 1980 167. Lessell S: Supranuclear paralysis of voluntary lid closure. Arch Ophthalmol 88:241, 1972 168. Lepore F: Bilateral cerebral ptosis. Neurology 37:1043, 1987 169. Barton JS: Bilateral central ptosis in acquired immunodeficiency syndrome. Can J Neurol Sci 22:52, 1995 170. Galetta SL, Gray LG, Raps EC, Schatz NJ: Pretectal eyelid retraction and lag. Ann Neurol 33:554, 1993 171. Adams GG, Kirkness JP: Botulinum toxin A induced protective ptosis. Eye 1:603, 1987 172. Morel-Fatio D, Lalardrie JP: Palliative surgical treatment of facial paralysis. The palpebral spring. Plast Reconstr Surg 33:446, 1964 173. Morel-Fatio D, Lalardrie JP: Le ressort palpebral: contribution a l'etude de la chirurgie plastique
de la paralysie faciale. Neurochirurgia 11:303, 1965 174. Levine RE, House WF, Hitselberger WE: Ocular complications of seventh nerve paralysis and management with the
palpebral spring. Am J Ophthalmol 73:219, 1972 175. Jobe RP: A technique for lid-loading in the management of lagophthalmos in
facial palsy. Plast Reconstr Surg 53:29, 1974 176. Sansone V, Boyton J, Palenski C: Use of gold weights to correct lagophthalmos in neuromuscular disease. Neurology 48:1500, 1997 177. Tankere F, Bernat I, Vitte E, et al: Hypoglossal-facial nerve anastomosis: Dynamic insight into the cross-innervation
phenomenon. Neurology 61:693, 2003 |