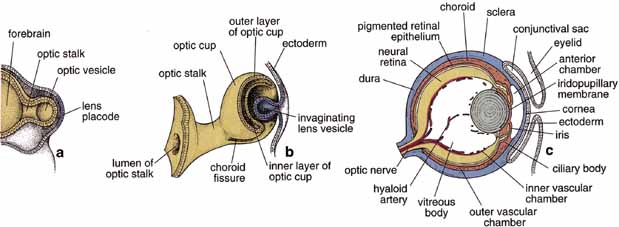
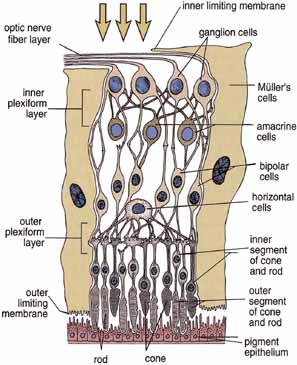
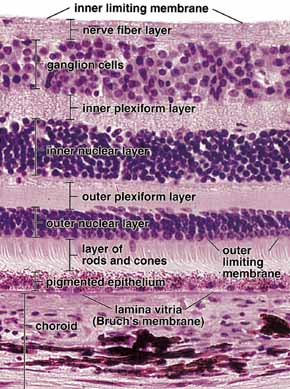
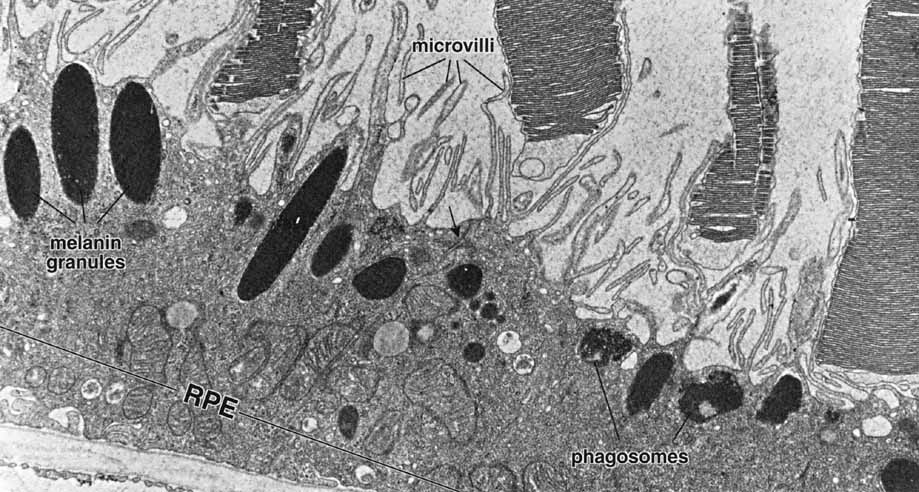
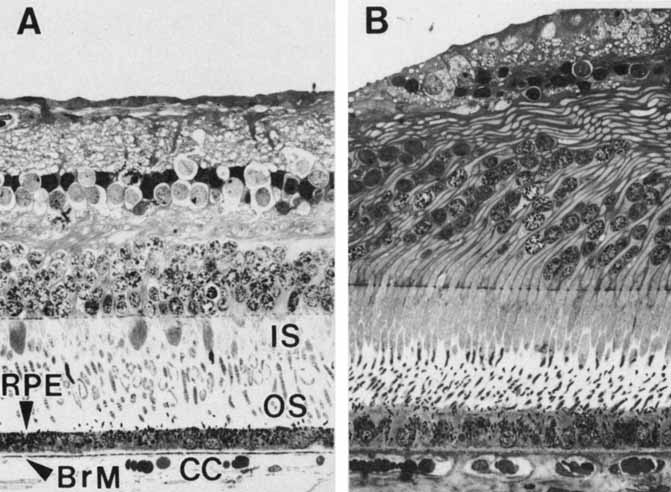
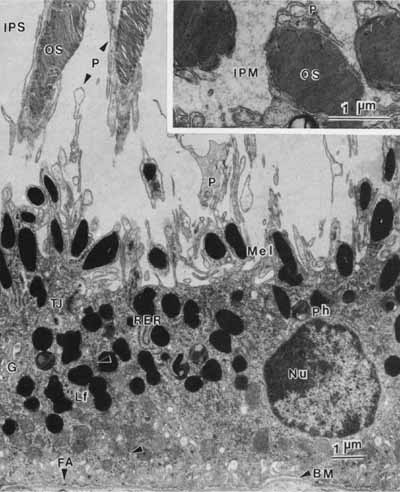
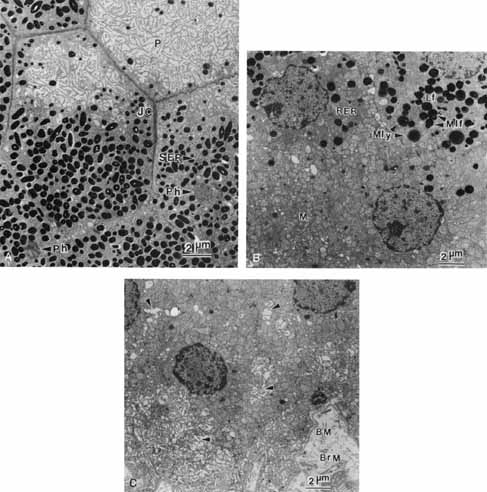
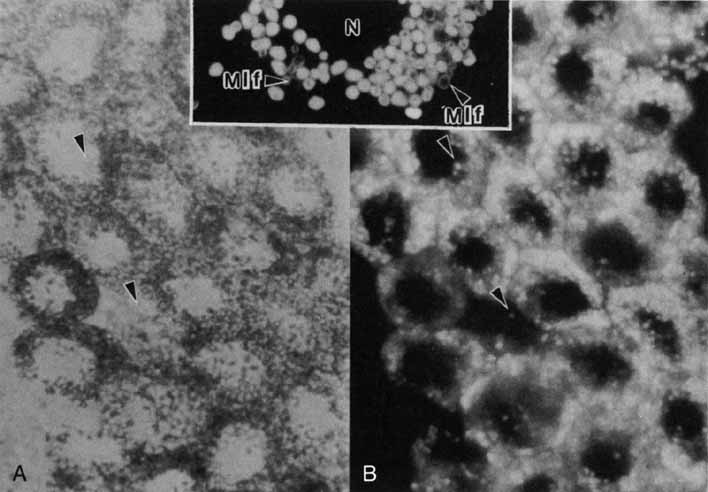
1. Feeney-Burns L, and Katz M: Retinal pigment epithelium. In: Tasman W, Jaeger EA, eds., Duane's Foundations of Clinical Ophthalmology, Vol. 1, Ch. 21. Philadelphia: Lippincott Williams & Wilkins, 1992:1–20. 2. Wolfensberger TJ: The historical discovery of the retinal pigment epithelium. In Marmor MF, Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:13–22. 3. Marmorstein AD, Finneman SC, Bonilha VL, and Rodriguez-Boulan E: Morphogenesis of the retinal pigment epithelium: toward understanding retinal
degenerative diseases. Ann NY Acad Sci 857:1–12, 1998 4. Hageman GS, and Kuehn MH: Biology of the interphotoreceptor matrix-retinal pigment epithelium-retinal
interface. In Marmor MF and Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:361–391 5. Thumann G: Development and cellular functions of the iris pigment epithelium. Surv Ophthalmol 45:345–354, 2001 6. Fuhrmann S, Levine EM, and Reh TA: Extraocular mesenchyme patterns the optic vesicle during early eye development
in the embryonic chick. Development 127:4599–4609, 2000 7. Tachibana M: MITF: a stream flowing for pigment cells. Pigment Cell Res 13:230–240, 2000 8. Martinez-Morales JR, Dolez V, Rodrigo I, Zaccarini R, Leconte L, Bovolenta P, and Saule S: OTX2 activates the molecular network underlying retinal pigment epithelium
differentiation. J Biol Chem 278:21721–21731, 2003 9. Baumer N, Marquardt T, Stoykova A, Spieler D, Treichel D, Ashery-Padan R, Gruss P: Retinal pigment epithelium determination requires the redundant activation
of Pax2 and Pax6. Development 130:2903–2915, 2003 10. Shields CL, Shields JA, Marr BP, Sperber DE, Gass JD: Congenital simple hamartoma of the retinal pigment epithelium: a study
of five cases. Ophthalmology 110:1005–1011, 2003 11. Santos A, Morales L, Hernandez-Quintela E, Jimenez-Sierra JM, Villalobos JJ, Panduro A: Congenital hypertrophy of the retinal pigment epithelium associated with
familial adenomatous polyposis. Retina 14:6–9, 1994 12. Shields JA, Shields CL, Eagle RC Jr , Singh AD: Adenocarcinoma arising from congenital hypertrophy of retinal pigment epithelium. Arch Ophthalmol 119:597–602, 2001 13. Shields CL, Mashayekhi A, Ho T, Cater J, Shields JA: Solitary congenital hypertrophy of the retinal pigment epithelium: clinical
features and frequency of enlargement in 330 patients. Ophthalmology 110:1968–1976, 2003 14. Santos A, and Traboulsi EI: Congenital abnormalities of the retinal pigment epithelium. In Marmor MF and Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:307–325 15. Ross MH, Kaye GI, Pawlina W: Histology, a Text and Atlas. 4th ed. Philadelphia: Lippincott Williams & Wilkins, 2003 16. Streeten BW: Development of the human retinal pigment epithelium and the posterior segment. Arch Ophthalmol 81:383–394, 1969 17. Feeney-Burns L, Burns RP, Gao C-L: Age-related macular changes in humans over 90 years old. Am J Ophthalmol 109:265, 1990 18. Zhao S, Rizolo LJ, Barnstable CJ: Differentiation and transdifferentiation of the retinal pigment epithelium. Int Rev Cytol 171:225–266, 1997 19. Burke JM: Determinants of retinal pigment epithelial cell phenotype and polarity. In Marmor MF and Wolfensberger TJ, (eds):: The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:86–102 20. Korte GE, Perlman JI, Pollack A: Regeneration of mammalian retinal pigment epithelium. Int. Rev. Cytol. 152:223–263, 1994 21. Wang H, Ninomiya Y, Sugino IK, Zarbin M: Retinal pigment epithelium wound healing in human Bruch's membrane
explants. Invest Ophthalmol Vis Sci 44:2199–2210, 2003 22. Hiscott P, and Sheridan CM: The retinal pigment epithelium, epiretinal membrane formation, and proliferative
vitreoretinopathy. In Marmor MF and Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:478–491. 23. Pastor C: Proliferative vitreoretinopathy: an overview. Survey Ophthalmol. 43:3–18, 1998 24. Nelson WJ: Adaptation of core mechanisms to generate cell polarity. Nature 422:766–774, 2003 25. Marmorstein AD: The polarity of the retinal pigment epithelium. Traffic 2:867–872, 2001 26. Philp NJ, Ochrietor JD, Rudoy C, Muramatsu T, and Linser PJ: Loss of MCT1, MCT3 and MCT4 expression in the retinal pigment epithelium
and neural retina of the 5A11/basigin-null mouse. Invest Ophthalmol Vis Sci 44:1305–1311, 2003 27. Rizzolo LJ: Polarity and the development of the outer blood-retinal barrier. Histol Histopathol 12:1057–1067, 1997 28. Marrs JA, Anderson-Fisone C, Jeong MC, Cohen-Gould L, Zurzolo C, Nabi IR, Rodriguez-Boulan E, Nelson WJ: Plasticity in epithelial cell phenotype: modulation by expression of different
cadherin cell adhesion molecules. J Cell Biol 129:507–519, 1995 29. Burke JM, Cao F, Irving PE: High levels of E-/P-cadherin: correlation with decreased
apical polarity of Na/K ATPase in bovine RPE cells in situ. Invest Ophthalmol Vis Sci 41:1945–1952, 2000 30. Hughes BA, Gallemore RP, Miller SS: Transport mechanisms in the retinal pigment epithelium. In Marmor MF and Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:103–134. 31. Yang D, Pan A, Swaminathan A, Kumar G, Hughes BA: Expression and localization of the inwardly rectifying potassium channel
Kir7.1 in native bovine retinal pigment epithelium. Invest. Ophthalmol. Vis Sci 44:3178–3185, 2003 32. Stamer WD, Bok D, Hu J, Jaffe GJ, McKay BS: Aquaporin–1 channels in human retinal pigment epithelium: role in
transepithelial water movement. Invest Ophthalmol Vis Sci 44:2803–2808, 2003 33. Philp NJ, Yoon H, Grollman EF: Monocarboxylate transporter MCT1 is located in the apical membrane and
MCT3 in the basal membrane of rat RPE. Am J Physiol 274:R1824–R1828, 1998 34. Hudspeth AJ, Yee AG: The intercellular junctional complexes of retinal pigment epithelia. Invest Ophthalmol Vis Sci 12:354–365, 1973 35. Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P: Molecular Biology of the Cell. 4th ed. New York: Garland, 2002 36. Philp NJ, Nachmias V: Components of the cytoskeleton in retinal pigment epithelium of the chick. J Cell Biol 101:358–362, 1985 37. Kojima S, Rahner C, Peng S, Rizzolo LJ: Claudin 5 is transiently expressed during the development of the retinal
pigment epithelium. J Membr Biol 186:81–88, 2002 38. Lagunowich LA, Grunwald GB: Expression of calcium-dependent cell adhesion during ocular development: a
biochemical, histochemical and functional analysis. Dev Biol 135:158–171, 1989 39. Grunwald GB: Cadherin cell adhesion molecules in retinal development and pathology. Prog. in Retinal and Eye Res 15(2) 363–392, 1996a 40. Grunwald GB: Discovery and analysis of the classical cadherins. In Colman DR (ed). Adhesion Molecules. A volume in the series. Advances in Molecular and Cell
Biology. Greenwich, CT: JAI Press, 1996b:16:63–112 41. Kaida M, Cao F, Skumatz CMB, Irving PE, Burke JM: Time at confluence for human RPE cells: effects on the adherens junction
and in vitro wound closure. Invest Ophthalmol Vis Sci 41:3215–3224, 2000 42. Malfait M, Gomez P, van Vreen TA, Parys JB, De Smedt H, Vereecke J, Himpens B: Effects of hyperglycemia and protein kinase C on conexin43 expression in
cultured rat retinal pigment epithelial cells. J Membr Biol 181:31–40, 2001 43. Turksen K, Aubin JE, Sodek J, Kalnins VI: Localization of laminin, type IV collagen, fibronectin, and heparan sulfate
proteoglycan in chick retinal pigment epithelium basement membrane
during embryonic development. J Histochem Cytochem 33:665–671, 1985 44. Chu P, Grunwald GB: Functional inhibition of retinal pigment epithelial cell-substrate
adhesion with a monoclonal antibody against the β1-subunit
of integrin. Invest Ophthalmol Vis Sci, 32:1763–1769, 1991 45. Zarbin M: Analysis of retinal pigment epithelium integrin expression and adhesion
to aged submacular Bruch's membrane. Trans Am Ophthalmol Soc 101:499–520, 2003 46. Opas M, and Kalnins VI: Light-microscopical analysis of focal adhesions of retinal pigmented
epithelial cells. Invest Ophthalmol Vis Sci 27:1622–1633, 1986 47. Marshall J, Hussain AA, Starita C, Moore DJ, Patmore AL: Aging and Bruch's membrane. In Marmor MF, Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:669–692 48. Chader GJ, Pepperberg DR, Crouch R, Wiggert B: Retinoids and the retinal pigment epithelium. In Marmor MF, Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:135–151 49. Thompson DA, Gal A: Genetic defects in vitamin A metabolism of the retinal pigment epithelium. Dev Ophthalmol 37:141–154, 2003 50. Gonzalez-Fernandez F: Interphotoreceptor retinoid-binding protein--an old
gene for new eyes. Vision Res 43:3021–3036, 2003 51. Edwards RB, Adler J: IRBP enhances removal of 11-cis-retinaldehyde from isolated
RPE membranes. Exp Eye Res 70:235–245, 2000 52. Hooks JJ, Detrick B, Percopo C, et al: Development and characterization of monoclonal antibodies directed against
the retinal pigment epithelial cell. Invest Ophthalmol Vis Sci 30:2106–2113, 1989 53. Gu SM, Thompson DA, Srikumari CR, Lorenz B, Finckh U, Nicoletti A, Murthy KR, Rathmann M, Kumaramanickavel G, Denton MJ, Gal A: Mutations in RPE65 cause autosomal recessive childhood-onset severe
retinal dystrophy. Nat Genet 17:194–197, 1997 54. Gollapalli DR, Maiti P, Rando RR: RPE65 operates in the vertebrate visual cycle by stereospecifically binding
all-trans-retinyl esters. Biochem. 42:11824–1830, 2003 55. Young RW: Shedding of discs from rod outer segments in the rhesus monkey. J Ultrastruct Res 34:190–203, 1971 56. La Vail MM: Rod outer segment disc shedding in relation to cyclic lighting. Exp Eye Res 23:277–280, 1976 57. Hollyfield JG: Phagocytic capacities of the pigment epithelium. Exp Eye Res 22:457–468, 1976 58. Besharse JC, DeFoe DM: Role of the retinal pigment epithelium in photoreceptor membrane turnover. In Marmor MF, Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press; 1998:152–172 59. Katz ML: Incomplete proteolysis may contribute to lipofuscin accumulation in the
retinal pigment epithelium. In Porta EA (ed): Lipofuscin and Ceroid Pigments. New York: Plenum Press, 1990:109–118 60. Young RW: The renewal of rod and cone outer segments in the rhesus monkey. J Cell Biol 49:303–318, 1971 61. Lutz DA, Guo Y, McLaughlin BJ: Natural, high-mannose glycoproteins inhibit ROS binding and ingestion
by RPE cell cultures. Exp. Eye Res 61:487–493, 1995 62. Finneman SC, Bonilha VL, Marmorstein AD, Rodriguez-Boulan E: Phagocytosis of rod outer segments by retinal pigment epithelial cells
requires alpha(v)beta5 integrin for binding but not for internalization. Proc Natl Acad Sci USA 94:12932–12937, 1997 63. Finneman SC, Silverstein RL: Differential roles of CD36 and alphavbeta5 integrin in photoreceptor phagpcytosis
by the retinal pigment epithelium. J Exp Med 194:1289–1298, 2001 64. Schraermeyer U, Heiman K: Current understanding on the role of retinal pigment epithelium and its
pigmentation. Pigment Cell Res 12:219–236, 1999 65. Schraermeyer U, Peters S, Thumann G, Mociok N, Heiman K: Melanin granules of the retinal pigment epithelium are connected with the
lysosomal degradation pathway. Exp Eye Res 68:237–245, 1999 66. Eldred GE: Lipofuchsin and other lysosomal storage deposits in the retinal pigment
epithelium. In Marmor MF, Wolfensberger TJ, (eds): The Retinal Pigment Epithelium. New York: Oxford University Press, 1998:651–668 67. Feeney-Burns L, Hilderbrand ES, Eldridge S: Aging human RPE: Morphometric analysis of macular, equatorial, and peripheral
cells. Invest Ophthalmol Vis Sci 25:195–280, 1984 68. Wolf G: Lipofuchsin and macular degeneration. Nutr Rev 61:342–346, 2003 69. Boulton M, Dayhew-Barker P: The roles of the retinal pigment epithelium: topographical variation and
ageing changes. Eye 15:384–389, 2001 70. Katz ML, Robison WG: What is lipofuchsin? Defining characteristics and differentiation
from other autofluorescent lysosomal storage bodies. Arch Gerontol Geriatr 34:169–184, 2002 71. Zarbin MA: Age-related macular degeneration: review of pathogenesis. Eur J Ophthalmol 8:199–206, 1998 72. Campochiaro PA, Soloway P, Ryan SJ, Miller JW: The pathogenesis of choroidal neovascularization in patients with age-related
macular degeneration. Mol Vis 5:34, 1999 73. Glaser LC, Dryja TP: Understanding the etiology of Stargardt's disease. Ophthalmol Clin North Am 15:93–100, 2002 74. Nagasaki H, Shinagawa K, Moxhizuki M: Risk factors for proliferative vitreoretinopathy. Prog Retina Eye Res 17:77–98, 1998 75. Campochiaro PA: Gene therapy for retinal and choroidal diseases. Expert Opin Biol Ther 2:537–544, 2002 76. Grierson I, Hiscott P, Hogg P, Robey H, Mazure A, Larkin G: Development, repair and regeneration of the retinal pigment epithelium. Eye 8:255–262, 1994 77. Lund RD, Kwan AS, Keegan DJ, Sauve Y, Coffey PJ, Lawrence UK: Cell transplantation as a treatment for retinal diseases. Prog Retina Eye Res 20:415–449, 2001 78. Aramant RB, Seiler MJ: Retinal transplantation--advantage on intact fetal sheets. Prog Retina Eye Res 21:57–73, 2002 79. Stanga PE, Kychenthal A, Fitzke FW, Halfyard AS, Chan R, Bird AC, Aylward GW: Retinal pigment epithelium translocation and central visual function in
age related macular degeneration: preliminary results. Int Ophthalmol 23:297–307, 2001 80. Hadlock T, Singh S, Vacanti JP, McLaughlin BJ: Ocular cell monolayers cultured on biodegradable substrates. Tissue Eng 5:187–196, 1999 81. Lu L, Yaszemski MJ, Mikos AG: Retinal pigment epithelium engineering using synthetic biodegradable polymers. Biomaterials 22:3345–3355, 2001 82. Davis AA, Bernstein PS, Bok D, Turner J, Nachtigal M, Hunt RC: A human retinal pigment epithelial cell line that retains epithelial characteristics
after prolonged culture. Invest Ophthalmol Vis Sci 36:955–964, 1995 83. Dunn KC, Aotaki-Keen AE, Putkey FR, Hjelmeland LM: ARPE–19, a human retinal pigment epithelial cell line with differentiated
properties. Exp Eye Res 62:155–169, 1996 84. Kanuga N, Winton HL, Beachene L, Koman A, Zerbib A, Halford S, Couraud PO, Keegan D, Coffey P, Lund RD, Adamson P, Greenwood J: Characterization of genetically modified human retinal pigment epithelial
cells developed for in vitro and transplantation studies. Invest Ophthalmol Vis Sci 43:546–555, 2002 85. West KA, Yan L, Mitagi M, Crabb JS, Marmorstein AD, Marmorstein L, Crabb JW: Proteome survey of proliferating and differentiating rat RPE-J cells. Exp Eye Res 73:479–491, 2001 86. Hergott GJ, Sandig M, Kalnins VI: Cytoskeletal organization of migrating retinal pigment epithelial cells
during wound healing in organ culture. Cell Motil Cytoskeleton 13:83–93, 1989 87. Sugino IK, Wang H, Zarbin MA: Age-related macular degeneration and retinal pigment epithelium
wound healing. Mol Neurobiol 28:177–194, 2003 88. Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR: Retinal degeneration mutants in the mouse. Vision Res. 42:517–525, 2002 89. Gal A, Li Y, Thompson DA, Weir J, Orth U, Jacobson SG, Apfelstedt-Sylla E, Vollrath D: Mutations in MERTK, the human orthologue of the RCS rat retinal dystrophy
gene, cause retinitis pigmentosa. Nat Genet 26:270–271, 2000 90. D'Cruz PM, Yasumura D, Weir J, Matthes MT, Abderrahim H, LaVail MM, Vollrath D: Mutation of the receptor tyrosine kinase gene Mertk in the retinal dystrophic
RCS rat. Hum Mol Genet 9:645–651, 2000 91. Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J: Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 28:92–95, 2001 92. Alge CS, Suppman S, Priglinger SG, Neubauer AS, May CA, Hauck S, Welge-Lussen U, Ueffing M, Kampik A: Comparative proteome analysis of native differentiated and cultured dedifferentiated
human RPE cells. Invest Ophthalmol Vis Sci 44:3629–3641, 2003 93. Honda S, Farboud B, Hjelmeland LM, Handa JT: Induction of an aging mRNA retinal pigment epithelial phenotype by matrix-containing
advanced glycation end products in vitro. Invest Ophthalmol Vis Sci 42:2419–2425, 2001 94. Weigel AL, Handa JT, Hjelmeland LM: Microarray analysis of H2O2-, HNE- or tBH-treated
ARPE–19 cells. Free Rad Biol Med 33:1419–1432, 2002 95. Dufour EM, Nandrot E, Marchant D, Van Den Berghe L, Gadin S, Issilame M, Dufier JL, Marsac C, Carper D, Menasche M, Abitbol M: Identification of novel genes and altered signaling pathways in the retinal
pigment epithelium during the Royal College of Surgeons rat retinal
degeneration. Neurobiol. Dis 14:166–180, 2003 96. Wistow G, Bernstein SL, Wyatt MK, Fariss RN, Behal A, Touchman JW, Bouffard G, Smith D, Peterson K: Expressed sequence tag analysis of human RPE/choroid for the NEIBank Project: over 6000 non-redundant transcripts, novel genes and slice
variants. Mol Vis 8:205–220, 2002 |