|
IMMUNOPATHOLOGY OF LYMPHOMA
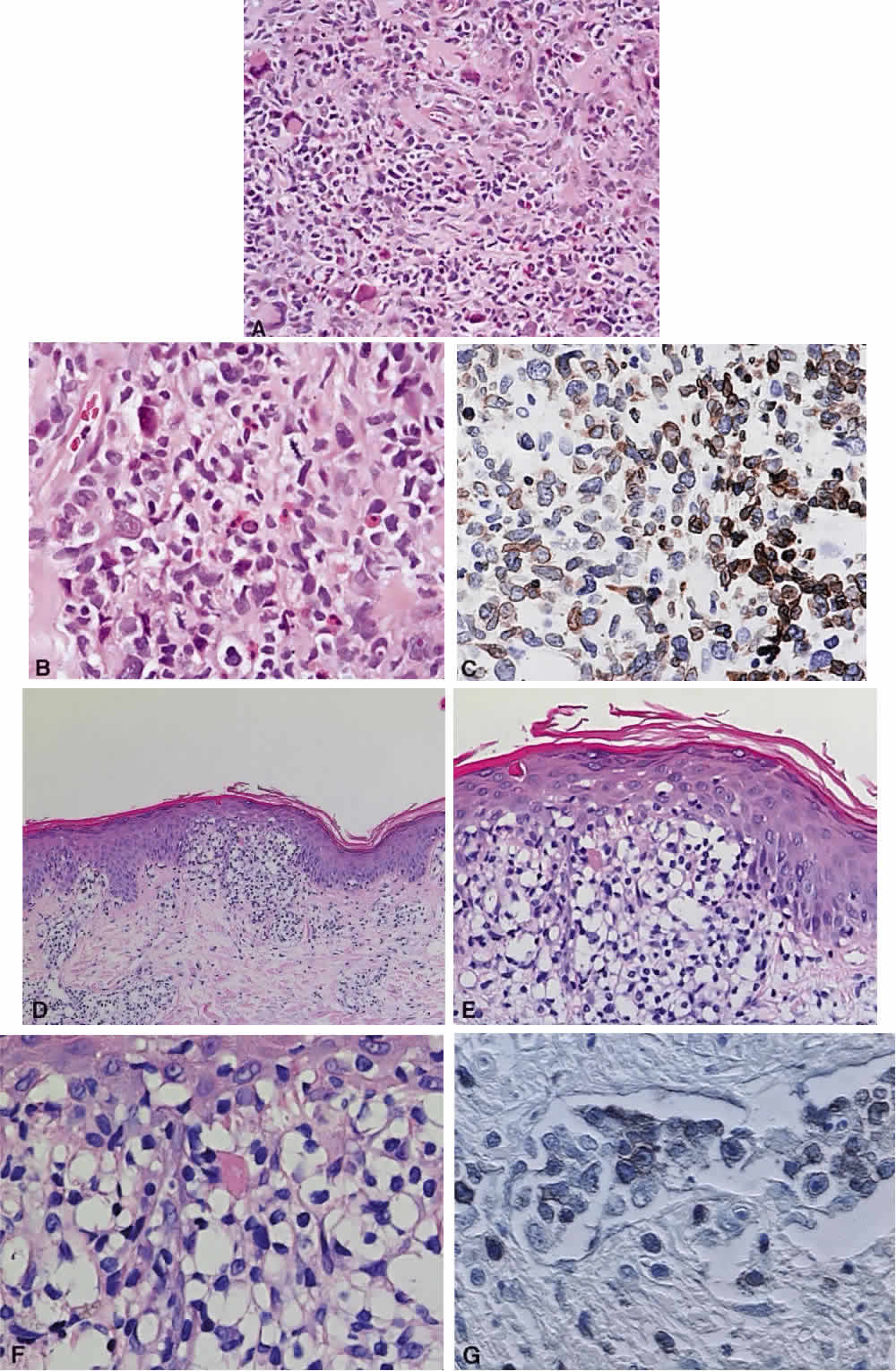
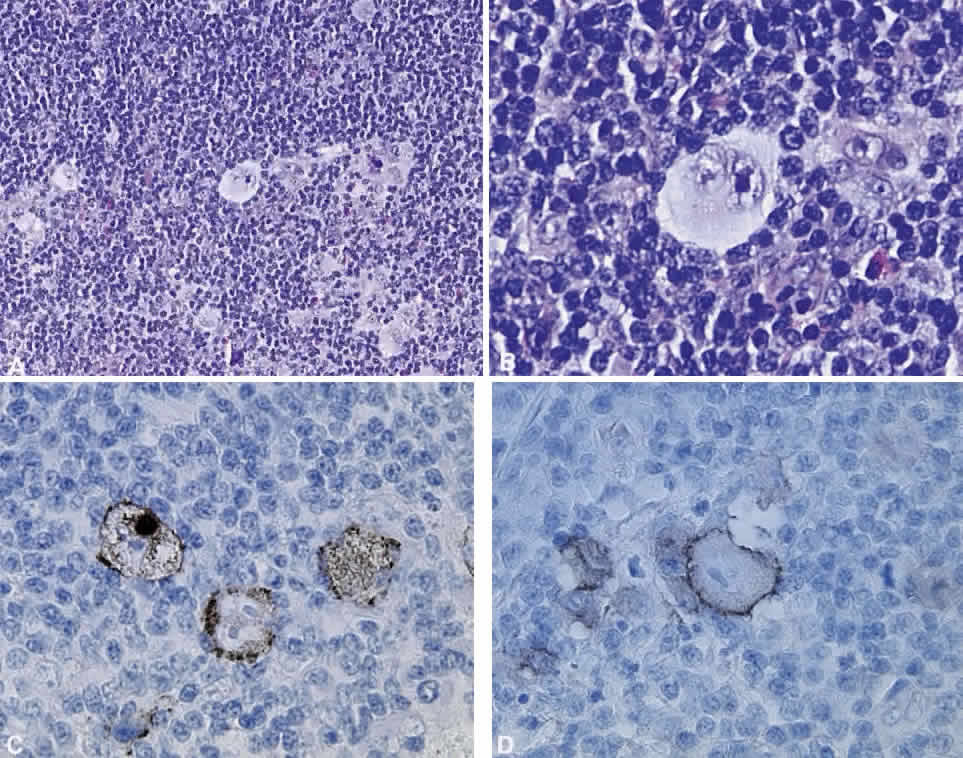
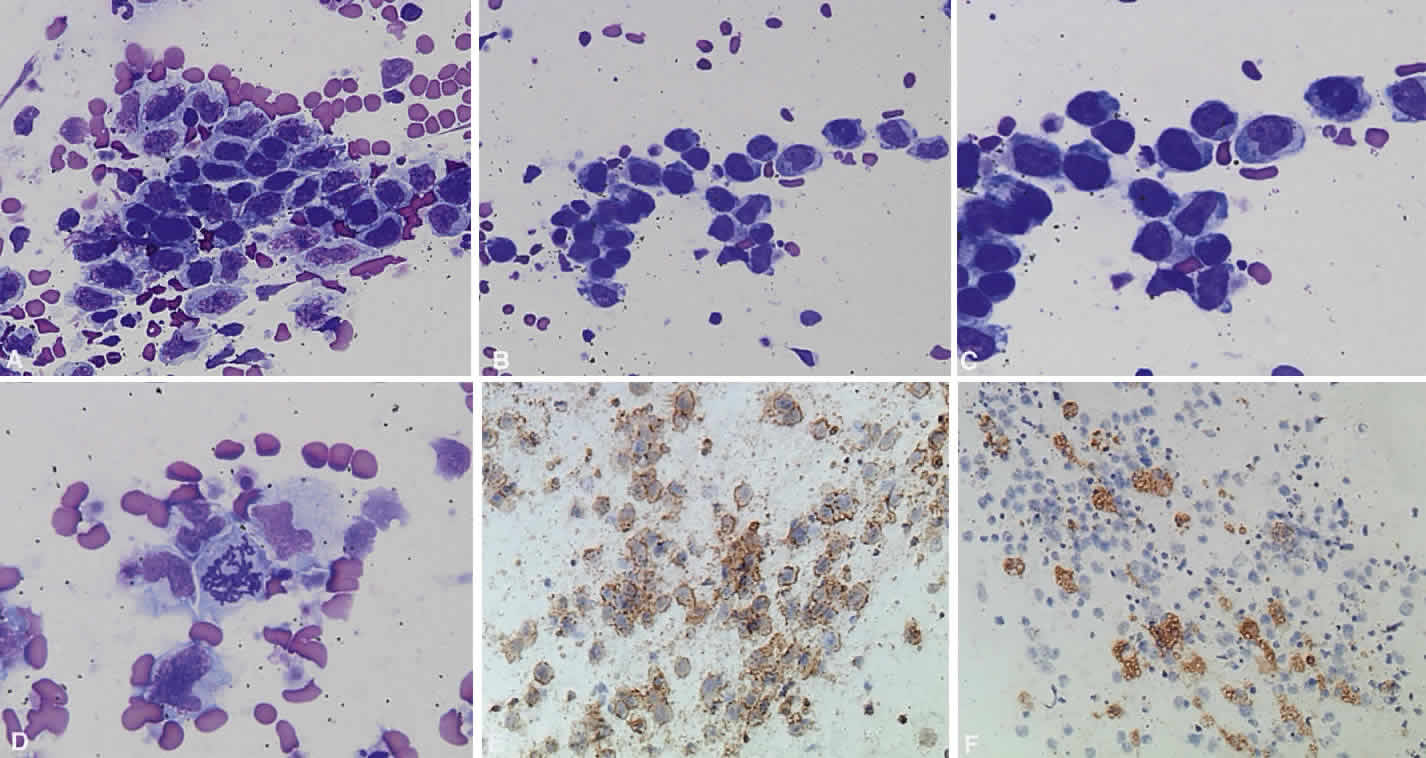
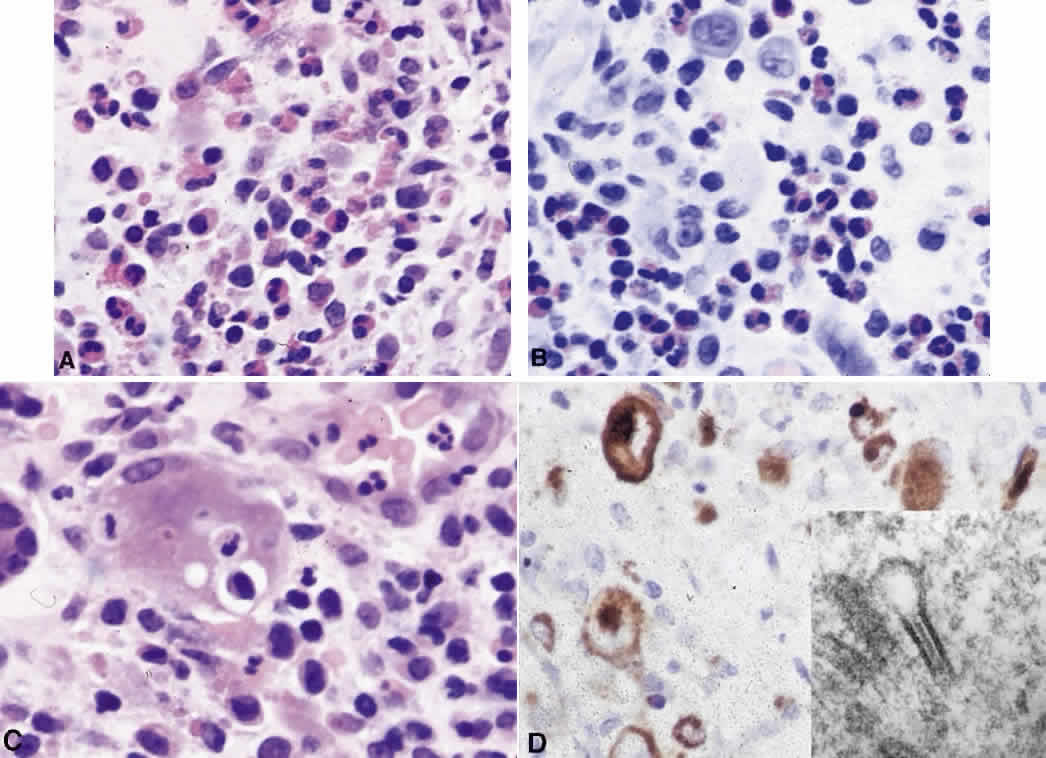
B cells arise from the bone marrow and produce immunoglobulin in a humoral response. T cells are thymus-derived and respond in a cell-mediated reaction via cells that are antigenically marked or produce lymphokines. Circulating blood and lymph nodes normally have a ratio of 70% T cells and 30% B cells, natural killer cells, or null cells. The normal lymph node is composed of follicles, each with germinal centers, mantle zones, and surrounding cortex (Fig. 2). The germinal center is composed of a framework of dendritic histiocytes, follicular dendritic cells, centroblasts, centrocytes, immunoblasts, small lymphocytes, and tingible body macrophages, whereas the surrounding cortex and paracortex consist mainly of B and T cells, respectively. Lymphomatous B cells were initially identified histologically using heterosera against immunoglobulin, which could then be stained with immunoperoxidase stain or rendered fluorescent with immunofluorescent markers.1b Plasma cells could be similarly identified by their cytoplasmic immunoglobulin. T cells were first identified cytologically by their rosette formation when combined with sheep erythrocytes.2 In addition, they were noted to uniquely contain cytoplasmic alphanaphthyl esterase, for which they could be stained histologically.3 Information regarding B-cell clonality could be obtained by staining for immunoglobulin, in particular, kappa and lambda light chains. In benign lesions, the kappa-lambda ratio of B-cell production is 2:1, whereas in malignant lesions, the ratio increases to 10:1.3 Benign and atypical reactive lesions have 50% or greater T cells, whereas malignant B-cell lesions usually consist of only 10% to 20% T cells.3 Malignant T-cell lymphomas, on the other hand, consist of 90% T cells.3 In reactive lesions, the T-cell helper-to-suppressor ratio is increased from the normal 2:1 to 5:1, whereas the normal ratio is maintained in malignant B-cell lymphoma.3 Thus, information about cell lines and immunoglobulin can be used to determine the predominant cell type, the cell type ratio, and the monoclonality or polyclonality of the B-cell population from the immunoglobulin expressed. Reactive processes have been thought to result from polyclonal or oligoclonal populations wherein every distinct clonal population expresses a distinct gene rearrangement, whereas a malignant lymphoma is thought to arise from a monoclonal clone population, which would express only a single gene rearrangement. Polymerase chain reaction has been used to specifically amplify clonal immunoglobulin gene rearrangements. In addition, cell surface markers of the various cell lines have been identified, beginning with the immunoglobulins and interleukins, and, later, cell surface antigen receptors, termed clusters of differentiation (CD), which are used to characterize the degree of differentiation of the cell type in question.4,5 The list of identified cell surface markers subsequently has expanded to over 100 and has aided in the characterization of Hodgkin's lymphoma and different subsets of nonHodgkin's B- and T-cell lymphomas6 (Table 1).
|
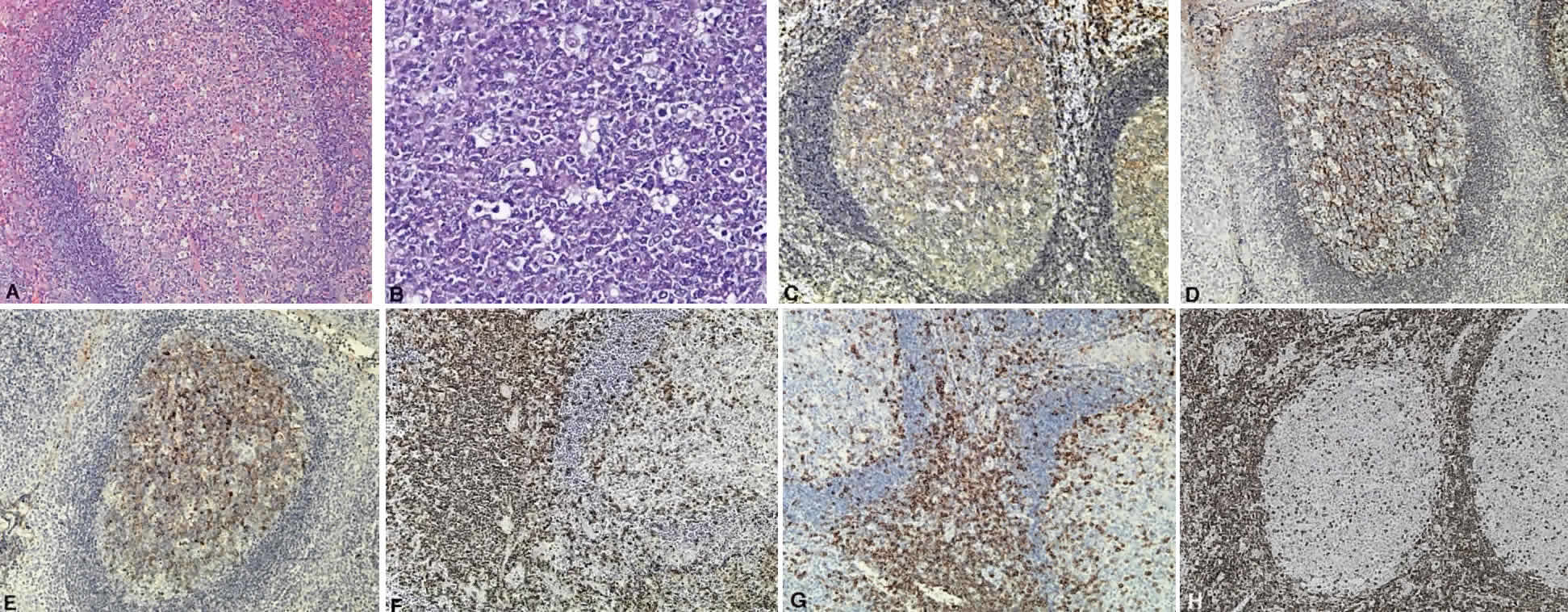
TABLE 1. List of Lymphoid Lesions and Their Characteristic Cell Markers
| B-Cell Lymphomas | |
| Precursor B-cell neoplasm | TdT+ CD19+ CD79a+ CD22+ CD20- /+ CD10+ /- HLA-DR+ SIg- cMu- /+ CD34+ /- |
| B-cell CLL | BCAA+ SIgM+ CD5+ CD23+ CD43+ CD11c- /+ CD10- |
| Lymphoplasmacytoid | BCAA+ IgM+ CD22+ CD5- CD10- CD43+ /- CD25+ CD11c+ |
| Mantle cell lymphoma | BCAA+ SigM+ IgD+ CD5+ CD10- /+ CD23- CD43+ CD11c- |
| Follicle center lymphoma | BCAA+ SIg+ CD10+ /- CD5- CD23- /+ CD43- CD11c- |
| Marginal zone lymphoma | BCAA+ SIgM+ IgD- CIg+ CD5- CD10- CD23- CD43- /+ CD11c+ /- |
| Plasmacytoma/myeloma | Sig- CIg+ CD19- CD20- CD22- CD79a+ /- CD45+ /- HLA-DR-/+ CD38+ EMA-/+ CD43+ /- CD56+ /- |
| Large cell lymphoma | BCAA+ SIg+ /- CIg-/+ CD45+ /- CD5-/+ CD10-/+ |
| Burkitt's lymphoma | BCAA+ SIgM+ CD22+ CD10+ CD5- CD38+ CD23- |
| T-Cell Lymphomas | |
| Precursor T-cell lymphoma | CD7+ CD3+ TdT+ Ig- BCAA- |
| Peripheral T-cell CLL | CD7+ CD2+ CD3+ CD5+ |
| Mycosis fungoides/Sézary syndrome | CD2+ CD3+ CD5+ CD7+ CD4+ |
| Anaplastic large T-cell | CD30+ CD45+ /- CD25+ /- EMA+ /- CD15-/+ CD3-/+ CD43-/+ CD45RO-/+ CD68- |
| Hodgkin's Disease | |
| Lymphocyte predominant | BCAA+ CD45+ CDw75+ EMA-/+ CD15- CD30-/+ Ig- |
| Nodular sclerosis | CD15+ /- CD30+ CD45- |
| Mixed cellularity | CD15+ /- CD30+ CD45- |
| Lymphocyte depletion | CD15+/- CD30+ CD45- BCAA- EMA- |
CLL, chronic lymphocytic leukemia; LGL, lymphocytic granular leukemia; TdT, terminal deoxynucleotidyl transferase; IgH-R, immunoglobulin heavy chain rearrangement; IgL-R, immunoglobulin light chain rearrangement; TCR-R, T-cell receptor rearrangement; SIg, surface immunoglobulin; CIg, cytoplasmic immunoglobulin; EMA, epithelial membrane antigen; CD, cluster of differentiation; BCAA, CD19, CD20, CD79a (B-cell antigens); + , 90% incidence; + /—, over 50%; —/+ , less than 50%; —, 10% or less.
(Harris NL, Jaffe ES, Stein H et al: A revised European—American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 84: 1361, 1994)
GENETICS OF LYMPHOMA
Lymphomas are thought to occur as the result of an immunoregulatory defect, caused by somatic mutations resulting either in loss of a tumor suppressor gene or in the upregulation of a protooncogene or factor regulating mitosis and differentiation of naive lymphocytes to memory cells and plasma cells. As a result, patients who are immunosuppressed because of congenital immunodeficiencies, autoimmune diseases, cytotoxic agents after organ transplantation, or acquired immunodeficiency syndrome are thought to be at increased risk of developing nonHodgkin's lymphomas, and, in patients with acquired immunodeficiency syndrome in particular, lymphomas tend to be more aggressive with bone marrow, bowel, and central nervous system involvement and are associated with a poor prognosis despite treatment.7–12
Immunoglobulins have been studied at the molecular level in lymphoma. Monoclonal lymphoid lesions express gene rearrangements that can be detected with restriction endonuclease cleavage and Southern blot.13–25 The gene rearrangement appears as a unique band on the blot, known as a DNA fingerprint, and a polyclonal lesion, which expresses the immunoglobulins made by several different lymphocyte populations in undetectable amounts, then would demonstrate only the control band (Fig. 3). Many cytogenetic abnormalities that are characteristic of certain lymphoma subtypes have been isolated, mostly in the form of chromosomal translocations, some of which have been recognized to cause upregulation of protooncogenes, upregulation of transcription factors, and downregulation of factors involved in apoptosis. Chromosomal translocations are thought to occur so frequently in lymphoma because they take place during the normal process of B-cell differentiation. Examples of mutations and the affected gene include the c-myc oncogene (8q24) in Burkitt's Lymphoma, the bcl-1 gene (11q13) in mantle cell lymphoma, the bcl-2 (18q21) in follicular lymphoma, the bcl-2 t(14;18) gene in 70% to 90% of follicular lymphomas and 10% to 35% of nodal large cell lymphomas, the bcl-6 gene (3q27) in diffuse large cell lymphoma, and the PAX-5 gene t(9;14)(p13;q32) identified in 50% of lymphoplasmacytoid lymphomas25–37 (Table 2). The genes that are overexpressed or underexpressed, as a result of the mutations or translocations, have begun to be elucidated. The bcl-2 gene is a regulator of normal cell apoptosis.34 The t(14:18) translocation of bcl-2 is thought to allow unregulated cell longevity of the centrocytes in follicular lymphoma. The c-myc gene is known to code for a protooncogene, which, when upregulated, allows for unmitigated cell division.26,27 The PAX-5 gene belongs to a family of PAX genes, which have been identified as critical regulators of the cell cycle and differentiation.27,31,35 In particular, PAX-5 (BSAP) codes for the B-cell-specific transcription factor expressed in all stages of B-cell differentiation except in plasma cells.35 BSAP factor is suspected of enhancing B-cell lymphopoiesis, but the exact mechanism is not known. A mutation in bcl-1 allows for upregulation of the PRAD-1 gene, which codes for cyclin D1, a cell cycle regulator.26,27
|
TABLE 2. Lymphoid Lesions and Their Characteristic Gene Rearrangements
and Mutations
| B-Cell Lymphomas | |
| Precursor B-cell/B-ALL | IgH, IgL gene rearrangement |
| Peripheral B-cell B-CLL | IgH, IgL gene rearrangement, trisomy 12 |
| Lymphoplasmacytoid lymphoma/immunocytoma | IgH, IgL gene rearrangement, 50% t(9;14) (p13;q32) rearrangement, PAX-5 on chr 9 to IgH on chr 14 |
| Mantle cell lymphoma | t(11:14) involving IgH locus and bcl-1 on chr 11, overexpression of PRAD-1, codes for cyclinD1, p53 gene mutation (tumor suppressor gene) |
| Follicle center lymphoma grade I-III | 70–95% t(14;18) of bcl-2 gene, expresses antiapoptosis gene |
| Marginal zone lymphoma | 35% t(11;18) of bcl-2 gene, 60% trisomy 3, trisomy 18 |
| Monocytoid and MALT-type | p53 gene mutation |
| plasmacytoma, multiple myeloma | IgH and IgL genes rearrangement or deletion, t(9;14)(p13;q32), PAX-5 rearrangement |
| Diffuse large cell B-lymphoma | 45% bcl-6 (3q27) rearrangement |
| 30% bcl-2 rearrangement, 23% REL gene amplification (progression-associated marker), c-myc rearrangement | |
| Burkitt's lymphoma | t(8;14) c-myc translocation from chr 8 to IgH region chr 14 |
| t(2;8) c-myc to IgL chr 2, t(8;22) c-myc to IgL chr 22 | |
| 25–40% African Burkitt's, some nonendemic EBV genome+ | |
| T-Cell Lymphomas | |
| Precursor T-cell/NK/T-ALL | TCR gene rearrangement, IgH rearrangement |
| Peripheral T-cell lymphoma/T-CLL | TCR gene rearrangement, trisomy 8q, 75% inv 14(q11;q32) |
| Mycosis fungoides/Sézary syndrome | TCR gene rearrangement |
| Anaplastic large cell T-lymphoma | 50–60% TCR gene rearrangement, t(2;5) (p23;q35) |
| Hodgkin's Disease | |
| Lymphocyte predominant | IgH or TCR gene rearrangement |
| Nodular sclerosis | IgH or TCR gene rearrangement |
| Mixed cellularity | IgH or TCR gene rearrangement |
| 60–70% EBV genome+ | |
| Lymphocyte depletion | IgH or TR gene rearrangement |
ALL, acute lymphocytic leukemia; CLL, chronic lymphocytic leukemia; IgH, immunoglobulin heavy chain; IgL, immunoglobulin light chain; TCR, T-cell receptor; EBV, Epstein-Barr virus; inv, inversion; MALT, mucosal-associated lymphoid tissue; chr, chromosome; NK, natural killer.
(Harris NL, Jaffe ES, Stein H et al: A revised European—Americcan classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 84: 1361, 1994)
CLASSIFICATION OF LYMPHOMA
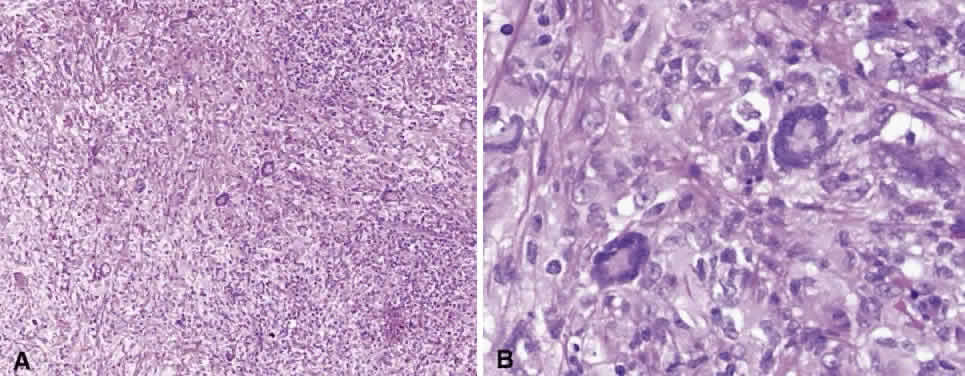
Numerous attempts have been made to classify lymphoid processes for clinical management and prediction of prognosis. Early classifications included only disease localized to the lymph node, which made classification of extranodal disease difficult and inaccurate. The Rappaport classification, first developed in 1956 and then modified in 1978, attempted to categorize lymphomas in two ways, first, using cytologic characteristics identified by conventional stains, and second, distinguishing between the follicular and diffuse growth pattern histologically38,39 (Table 3). The distinction of nodular, or follicular, and diffuse growth was considered useful because of the generally indolent nature of follicular growth, in which the tumor cell aggregates resemble germinal centers and disrupt the normal architecture of the node, compared with the appearance of diffuse growth, in which the lymph node is completely obliterated by a dense monotonous sheet of lymphocytes. In subsequent years, however, it was found that the descriptive growth pattern and cytogenetic characteristics of the Rappaport system did not predict prognosis reliably and were biologically inaccurate. The complexity of correlating degrees of differentiation, mitotic activity, and cytologic characteristics to prognosis have made lymphomas difficult to classify and have led to subsequent systems. The second system, proposed by Lukes-Collins in 1974, classifies lymphoma histologically according to its normal counterpart B-cell, T-cell, or null cell origin40,41 (see Table 3). Histologically, cells may appear small cleaved, large cleaved, small noncleaved, or large noncleaved, depending on the stage of B-cell arrest during normal transformation to immunoblast. Ninety percent of lymphomas are of B-cell origin, and the null cell also usually is of B-cell origin, although 10% may originate from T cells or histiocytes.42,43 Burkitt's lymphoma, the only lymphoma common in children, is a B-cell variant with a background of reactive histiocytes. As a result of histologic classification by Lukes-Collins, 76% of histiocytic lymphomas according to the Rappaport system were found to be not of histiocytic origin but of lymphocytic origin.40,44 The third system, the Working Formulation devised by the National Cancer Institute in 1982, attempted to predict prognosis by grouping lymphoma according to natural history, response to therapy, and overall survival.45 Three broad categories were established in terms of 5-year survival rates, the low-grade with a 50% to 70% survival rate, intermediate with 35% to 45%, and high grade with 23% to 32% (see Table 3). Orbital reactive hyperplasia, a relatively low-grade lesion, can be associated with systemic disease, whereas malignant or high-grade orbital lymphomas may be isolated findings. The Ann Arbor Staging Classification for Hodgkin's and non-Hodgkin's lymphomas was developed to stage disease based on systemic areas of involvement as a means of establishing a baseline for treating disease and following clinical progression46 (Table 4). Histologic classification, however, has been recognized as more useful than localization in the clinical management of nonHodgkin's lymphoma.47
TABLE 3. Rappaport, Lukes-Collins, and Working Formulation Classifications
| 5-y Survival Rate (%) Low Grade | Working Formulation | Rappaport Classification | Lukes-Collins Classification |
| 50–70 | Small lymphocytic | Lymphocytic, well-differentiated | Small lymphocyte and plasma-cytoid lymphocytic |
| Follicular, predominantly small cleaved cell | Nodular, poorly differentiated lymphocytic | FCC, small cleaved | |
| Follicular, mixed small and large cleaved | Nodular, mixed lymphocytic-histiocytic | FCC, small and large cleaved | |
| Intermediate Grade | |||
| 35–40 | Follicular, predominantly large cell | Nodular, histiocytic | FCC, large cleaved and/or noncleaved |
| Diffuse, small cleaved | Diffuse, poorly differentiated lymphocytic | FCC, small cleaved, diffuse | |
| Diffuse, mixed large and small cell | Diffuse, mixed lymphocytic and histiocytic | FCC, small cleaved, large cleaved, or large noncleaved | |
| Diffuse, large cell | Diffuse histiocytic | FCC, large cleaved or large noncleaved | |
| High Grade | |||
| 23–32 | Large cell, immunoblastic | Diffuse histiocytic | Immunoblastic B- or T-cell type |
| Lymphoblastic | Lymphoblastic lymphoma | Convoluted T-cell lymphoma | |
| Small noncleaved cell | Undifferentiated Burkitt's and non-Burkitt's | FCC, small noncleaved |
FCC, follicle center cell.
TABLE 4. Ann Arbor Staging of non-Hodgkin's Lymphoma
| Clinical Staging (CS) | |
| Stage I: | Involvement of a single lymph node region (I) or of a single lymphatic organ or site (IE) |
| Stage II: | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or localized involvement of an extralymphatic origin or site of one or more lymph node regions on the same side of the diaphragm (IIE) |
| Stage III: | Involvement of lymph node regions on both sides of the diaphragm (III), which may also be accompanied by localized involvement of thespleen (IIIS), extralymphatic site (IIIE), or both (IIISE) |
| Stage IV: | Diffuse or disseminated involvement of one or more extralymphatic organs or tissue with or without associated lymph node enlargement |
| Pathologic Staging (PS) | |
| Involvement found at primary laparotomy or by any further removal of tissue for histologic examination other than that taken for the original diagnosis. Include annotations for specific biopsy sites. | |
| N+ or N- | For other lymph node positive or negative by biopsy |
| H+ or H- | For liver positive or negative by biopsy |
| S+ or S- | For spleen positive or negative after splenectomy |
| L+ or L- | For lung positive or negative by biopsy |
| M+ or M- | For bone marrow positive or negative by biopsy or smear |
| P+ or P- | For pleura or pleural fluid positive or negative by biopsy or by cytologic examination |
| O+ or O- | For bone positive or negative by biopsy |
| D+ or D- | For skin positive or negative by biopsy |
While the Lukes-Collins and Working Formulation classifications were in wide use in the United States, the European literature made references to the Kiel and updated Kiel classifications, which led to disparities in classifying lymphoma. Another classification proposed by Jakobiec and coworkers was the most comprehensive classification available for orbital disease but failed to integrate systemic lymphoma, which is known to be associated in approximately half of cases.1 The most recent classification has made the system universal, comprehensive, and useful to interdisciplinary teams that characteristically manage patients with lymphoma. The International Lymphoma Study Group in 1994 developed the Revised European-American Lymphoma (REAL) classification (Table 5), which classifies lymphoid disease by the cell of origin into B-cell, T-cell, and natural killer cell lymphomas, leukemias, myeloma, and variants of Hodgkin's disease26 (Fig. 4). The identification of the putative benign progenitor cells has been inferred through the use of cell marker studies. The results of molecular genetic studies to identify immunoglobulin gene rearrangements and cytogenetic studies to detect chromosomal translocations in monoclonal proliferations have also been incorporated. A significant contribution of the REAL classification has been to incorporate primary extranodal lymphomas as recognizable and classifiable entities. As a result, new variants in this list include lymphoplasmacytic lymphoma, mantle cell lymphoma, marginal zone B-cell lymphoma, particularly mucosal-associated lymphoid tissue (MALT) lymphoma, subclasses of large cell lymphoma, and the natural killer cell lymphomas. Comparisons of the REAL classification to the Working Formulation and the Kiel classification are shown in Tables 6 and 7, respectively.48 The first series of 112 orbital lymphomas using the REAL classification reported the accuracy and utility of the system in classifying orbital lesions and predicting prognosis in combination with currently available immunophenotyping and immunocytogenetic studies.49 The REAL classification does not classify disease based on the degree of differentiation or clinical prognosis. However, a proposed prognostic scheme has been developed in accordance with the REAL classification28 (Table 8). In this text, tumor nomenclature adheres as strictly as possible to the REAL classification.
TABLE 5. Revised European—American Lymphoma (REAL) Classification
B-Cell Neoplasms
- Precursor B-cell neoplasm: Precursor B-lymphoblastic leukemia/lymphoma
- Peripheral B-cell neoplasms:
- B-cell chronic lymphocytic leukemia/prolymphocytic leukemia/small lymphocytic
lymphoma
- Lymphoplasmacytoid lymphoma/immunocytoma
- Mantle cell lymphoma
- Follicle center lymphoma, follicular
Provisional cytologic grades: I (small cell), II (mixed small and large cell), III (large cell) - Marginal zone B-cell lymphoma
Extranodal (MALT-type + /— monocytoid B cells)
Provisional subtype: diffuse, predominantly small cell type - Provisional entity: Splenic marginal zone lymphoma (+ /— villous
lymphocytes)
- Hairy cell leukemia
- Plasmacytoma/plasma cell myeloma
- Diffuse large B-cell lymphoma*
- Burkitt's lymphoma
- Provisional entity: high-grade B-cell lymphoma, Burkitt's-like
- B-cell chronic lymphocytic leukemia/prolymphocytic leukemia/small lymphocytic
lymphoma
T-Cell and Putative NK Cell Neoplasms
- Precursor T-cell neoplasm: Precursor T-cell lymphoblastic lymphoma/leukemia
- Peripheral T-cell and NK-cell neoplasms
- T-cell chronic lymphocytic leukemia/promyelocytic leukemia
- Large granular lymphocytic leukemia (LGL), T-cell and NK-cell types
- Mycosis fungoides/Sézary syndrome
- Peripheral T-cell lymphomas, unspecified.*
Provisional cytologic catgories: Medium-sized cell, mixed medium and large cell, large cell, lymphoepithelioid cell
Provisional subtype: Hepatosplenic γδ T-cell lymphoma
Provisional subtype: Subcutaneous panniculitic T-cell lymphoma - Angioimmunoblastic T-cell lymphoma (AILD)
- Angiocentric lymphoma
- Intestinal T-cell lymphoma (+ /— enteropathy associated)
- Adult T-cell lymphoma/leukemia (ATLL)
- Anaplastic large cell lymphoma (ALCL), CD30+ , T- and null-cell
- Provisional entity: Anaplastic large-cell lymphoma. Hodgkin's-like
- T-cell chronic lymphocytic leukemia/promyelocytic leukemia
Hodgkin's Disease
- Lymphocyte predominance
- Nodular sclerosis
- Mixed cellularity
- Lymphocyte depletion
- Provisional entity: Lymphocyte-right classic
MALT, mucosal-associated lymphoid tissue; NK, natural killer.
*These categories are thought likely to include more than one disease entity.
(Harris NL, Jaffe ES, Stein H et al: A revised European—American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 84:1361, 1994)
|
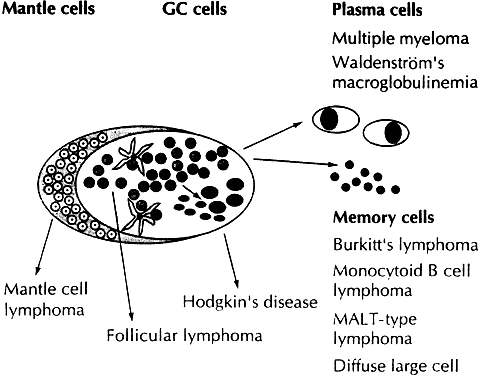
TABLE 6. Comparison of REAL Classification with Working Formulation
| REAL Classification | Working Formulation |
| B-Cell | |
| Low-Grade Lymphoma | |
| CLL | Small lymphocytic consistent with CLL |
| Small lymphocytic/B-cell prolymphocytic leukemia | Lymphoplasmacytoid/Immunocytoma |
| Lymphoplasmacytoid | Small lymphocytic, plasmacytoid |
| Extranodal marginal zone | Small lymphocytic |
| Nodal marginal zone | Small lymphocytic |
| Splenic marginal zone/hairy cell leukemia | Small lymphocytic |
| Plasmacytoma/myeloma | Plasmacytoma |
| Follicle center, follicular, grade I | Follicular, predominantly small cleaved |
| Follicle center, follicular, grade II | Follicular, mixed small and large cell |
| Intermediate-Grade Lymphoma | |
| Follicle center, follicular, grade III | Follicular, predominantly large cell |
| Follicle center, diffuse, small cell | Diffuse, small cleaved cell/diffuse, mixed small and large cell |
| Mantle cell | Diffuse small cleaved cell |
| Diffuse large cell | Diffuse, large cell |
| High-Grade Lymphoma | |
| Diffuse large cell | Diffuse large immunoblastic |
| Burkitt's | Small noncleaved, Burkitt's |
| High-grade, Burkitt's-like | Small noncleaved, non-Burkitt's |
| Precursor lymphoblastic | Lymphoblastic |
| T-Cell | |
| Low-Grade Lymphoma | |
| CL, prolymphocytic leukemia | Small lymphocytic |
| Large granulocytic leukemia, T-Cell or NK cell | Small lymphocytic |
| Mycosis fungoides/Sézary syndrome | Mycosis fungoides |
| Peripheral | Diffuse, small, cleaved |
| Diffuse, mixed small and large | |
| Diffuse, large cell | |
| Large cell, immunoblastic | |
| Angioimmunoblastic | Diffuse mixed small and large, large cell immunoblastic |
| Angiocentric | Diffuse mixed small and large, large cell immunoblastic |
| Intestinal | Large cell immunoblastic |
| Adult lymphoma/leukemia | Diffuse small cleaved, diffuse mixed small and large |
| Anaplastic large T- and null cell | Large cell immunoblastic |
CLL, chronic lymphocytic leukemia; NK, natural killer; REAL, revised European—American Lymphoma classification.
(Harris NL, Jaffe ES, Stein H et al: revised European—American classification of lymphoid neoplasms: a proposal from the International Lymphoma Study Group. Blood 84:1361, 1994)
TABLE 7. Comparison of REAL Classification With Updated Kiel Classification
| REAL Classification | Updated Kiel Classification |
| Precursor B-lymphoblastic leukemia/lymphoma | B-lymphoblastic |
| B-cell chronic lymphocytic leukemia/promyelocytic leukemia/small lymphocytic lymphoma | B-lymphocytic, CLL |
| B-lymphocytic, prolymphocytic leukemia | |
| Lymphoplasmacytoid immunocytoma | |
| Lymphoploasmacytoid lymphoma | Lymphoplasmacytic immunocytoma |
| Mantle cell lymphoma | Centrocytic (mantle cell) |
| Centroblastic, centrocytoid subtype | |
| Follicle center lymphoma, follicular | Centroblastic-centrocytic, follicular |
| Grade I | |
| Grade II | |
| Grade III | Centroblastic, follicular |
| Follicle center lymphoma, diffuse, predominantly small cell (provisional) | Centroblastic, follicular |
| Centroblastic-centrocytic, diffuse | |
| Extranodal marginal zone B-cell lymphoma (provisional) | - |
| Nodal marginal zone B-cell lymphoma | Monocytoid, including marginal zone |
| Immunocytoma | |
| Splenic marginal zone B-cell lymphoma (provisional) | - |
| Hairy cell leukemia | Hairy cell leukemia |
| Plasmacytoma/myeloma | Plasmacytic |
| Diffuse large cell | Centroblastic (monomorphic, polymorphic, and multilobated subtypes) |
| B-immunoblastic | |
| B-large cell anaplastic | |
| Primary mediastinal large B-cell lymphoma | - |
| Burkitt's lymphoma | Burkitt's lymphoma |
| High-grade B-cell lymphoma, Burkitt-like (Provisional) | ? |
| Precursor T-lymphoblastic lymphoma/leukemia | T-lymphoblastic |
| T-cell chronic lymphocytic leukemia/prolymphocytic leukemia | T-lymphocytic, CLL type |
| T-lymphocytic, prolymphocytic leukemia | |
| Large granular lymphocytic leukemia, | |
| T-cell type | T-lymphocytic, CLL type |
| NK-cell type | - |
| Mycosis fungoides/Sézary syndrome | Small cell cerebriform (mycosis fungoides/Sézary syndrome) |
| Peripheral T-cell lymphomas, unspecified | T-zone |
| Lymphoepithelioid | |
| Including subtype: subcutaneous panniculytic T-cell lymphoma (provisional) | Pleomorphic, small T-cell |
| Pleomorphic, medium-sized and large T-cell | |
| T-immunoblastic | |
| Hepatosplenic gd T-cell lymphoma (provisional) | - |
| Angioimmunoblastic T-cell lymphoma with dysproteinemia) | Angioimmunoblastic (angioimmuno-blastic lymphadenopathy |
| Angiocentric T-cell lymphoma | - |
| Intestinal T-cell lymphoma | - |
| Adult T-cell Lymphoma/Leukemia | Pleomorphic small T-cell, HTLV1+ |
| Pleomorphic medium-sized and large T-cell, HTLV1+ | |
| Anaplastic large T- and null cell | T-large cell anaplastic |
AILD, ; CLL, chronic lymphocytic leukemia; HTLV1, human T-lymphotropic virus type 1; LgX, ; NK, natural killer; REAL, revised European—American Lymphoma classification.
(Chan JKC, Banks PM, Cleary ML et al: A proposal for classification of lymphoid neoplasms (by the international Lymphoma Study Group). Histopathology 25:517, 1994)
TABLE 8. Proposed Clinical Scheme for Malignancies of the Lymphoid System
| B-Cell Lineage | T-Cell Lineage |
| I. Indolent lymphomas (low risk) | I. Indolent lymphomas (low risk) |
| Chronic lymphocytic leukemia/small lymphocytic lymphoma | Large granular lymphocytic leukemia, T-cell and NK-cell |
| Mycosis fungoides/Sézary syndrome | |
| Lymphoplasmacytic lymphoma/immunocytoma/ Waldenstrom's macroglobulinemia | Smoldering and chronic adult T-cell leukemia/lymphoma, HTLV-1 |
| Hairy cell leukemia | |
| Splenic marginal zone lymphoma | |
| Marginal zone B-cell lymphoma | |
| Extranodal (MALT B-cell lymphoma) | |
| Nodal (monocytoid) | |
| Follicle center lymphoma/follicular (small cell); grade I | |
| Follicle center lymphoma/follicular (mixed small and large cell); grade II | |
| II. Aggressive lymphomas (intermediate risk) | II. Aggressive lymphomas (intermediate risk) |
| Prolymphocytic leukemia | Prolymphocytic leukemia |
| Plasmacytoma/multiple myeloma | Peripheral T-cell lymphoma, unspecified |
| Mantle cell lymphoma | Angioimmunoblastic lymphoma |
| Follicle center lymphoma/follicular (large cell); grade III | Angiocentric lymphoma |
| Diffuse large B-cell lymphoma (includes immunoblastic and diffuse large and centroblastic lymphoma) | Intestinal T-cell lymphoma |
| Primary mediastinal (thymic) large B-cell lymphoma | Anaplastic large cell lymphoma |
| High-grade B-cell lymphoma, Burkitt-like | |
| III. Very aggressive lymphomas (high risk) | III. Very aggressive lymphomas (high risk) |
| Precursor B-lymphoblastic lymphoma/leukemia | Precursor T-lymphoblastic |
| Burkitt's lymphoma/B-cell acute leukemia | Adult T-cell lymphoma/leukemia |
| Plasma cell leukemia | |
| IV. Hodkin's disease |
HTLV-1, human T-lymphotropic virus type 1; MALT, mucosal-associated lymphoid tissue. NK, natural killer.
(Harris NL, Jaffe ES, Stein H et al: A revised European—American classification of lymphoid neoplasms: A proposal from the International Lymphoma Study Group. Blood 84:1361, 1994)
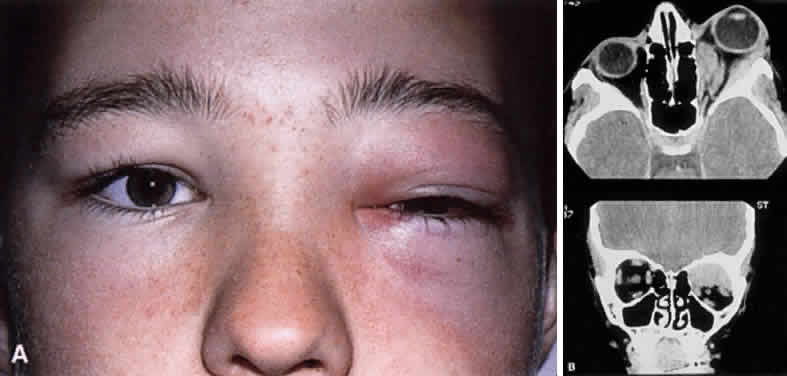
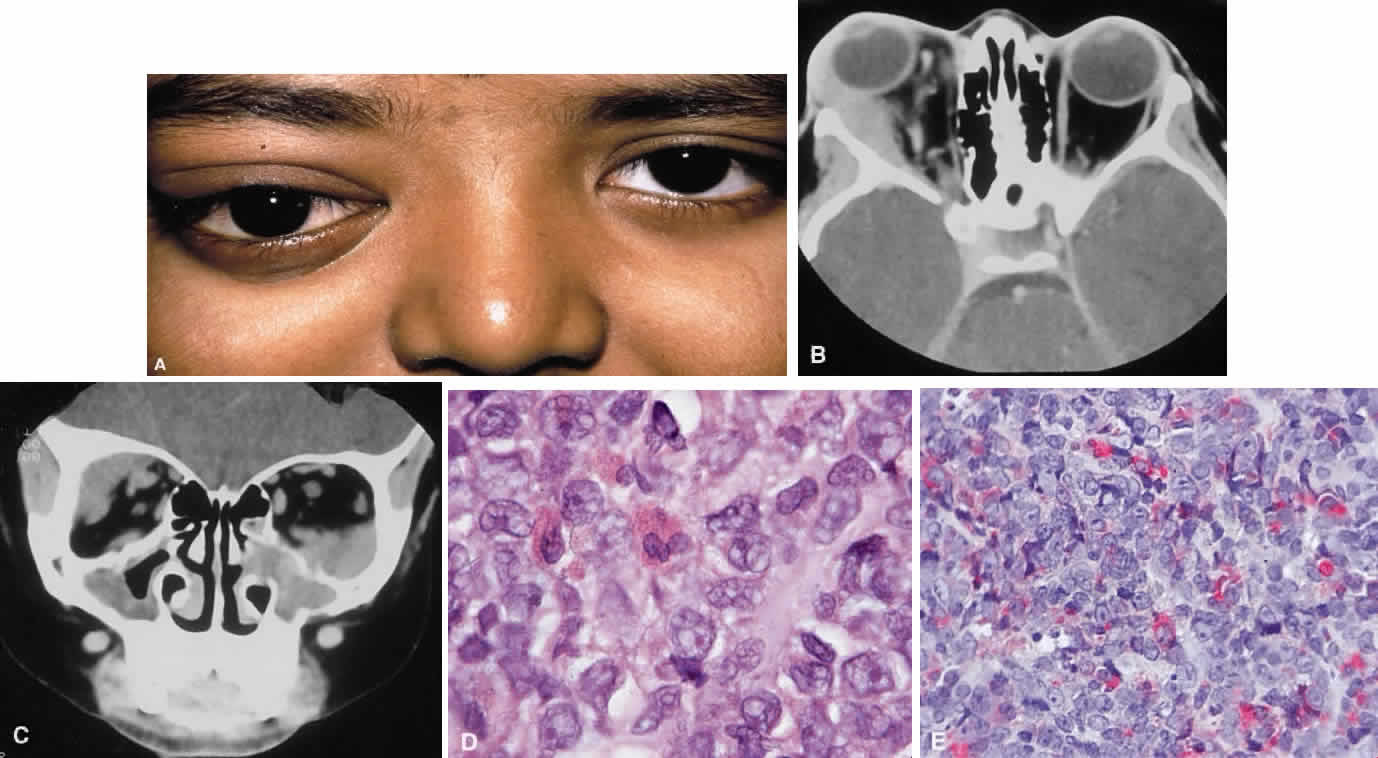
CLINICAL PRESENTATION OF SYSTEMIC NON-HODGKIN'S LYMPHOMA
Systemic non-Hodgkin's lymphomas usually present with painless lymph node enlargement in one or more nodes. Adults usually are in their fifth or sixth decade, and presentation in childhood is extremely rare. The fever, night sweats, and weight loss characteristic of Hodgkin's disease generally are absent. Splenomegaly may develop in 20% of patients. Patients usually have normal blood counts, although they may develop lymphocytosis, especially with well-differentiated lymphocytic lymphomas, or pancytopenia with anemia, hemorrhage, petechiae, ecchymosis, and infection—the pancytopenia occurring in one third of patients as a result of bone marrow involvement or chemotherapy and radiation. Patients with B-cell lymphomas tend to develop difficulty with bacterial infection, whereas those with T-cell lymphoma may develop difficulty with delayed-type hypersensitivity and viral infections. Leukemic conversion is rare in adults, although it occurs in 25% of children. In children, an acute leukemic phase may be the initial presentation. Furthermore, children are more likely to develop extranodal and aggressive disease, although they may respond well to therapy.50 In a series of 1269 patients, of whom only 3 (0.42%) presented initially with proptosis, one third of patients presented with extranodal disease and a few with bone marrow invasion.51 There was a 1.3% incidence rate of orbital disease secondary to systemic lymphoma.