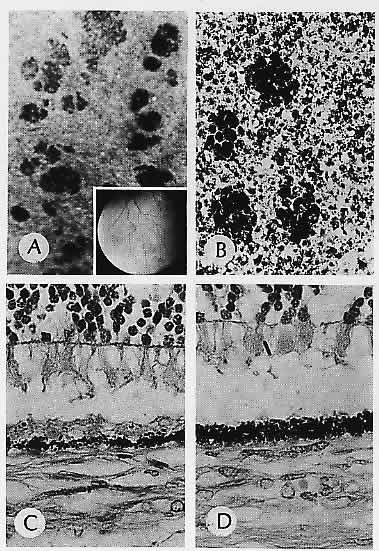
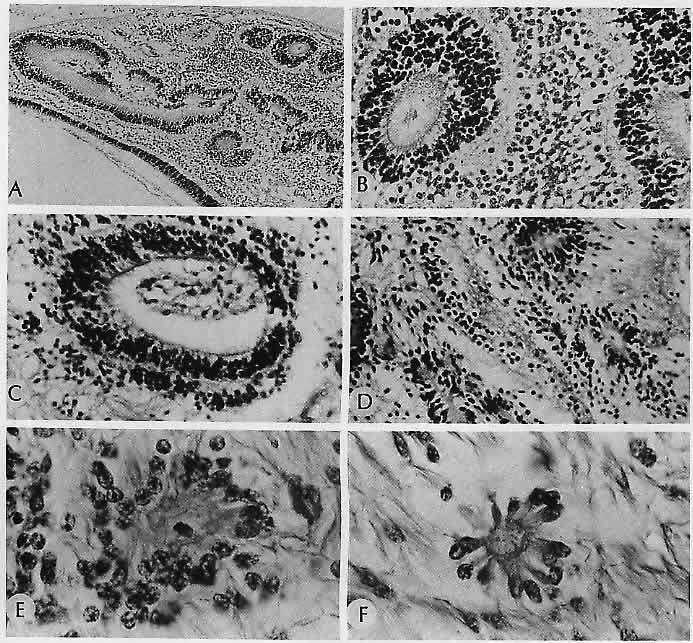
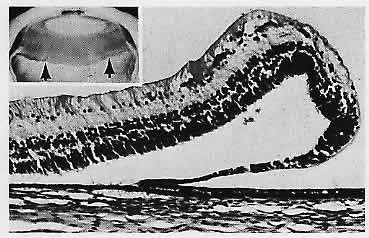
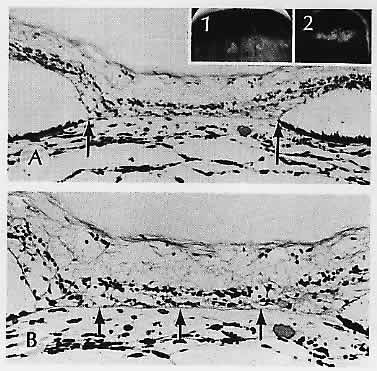
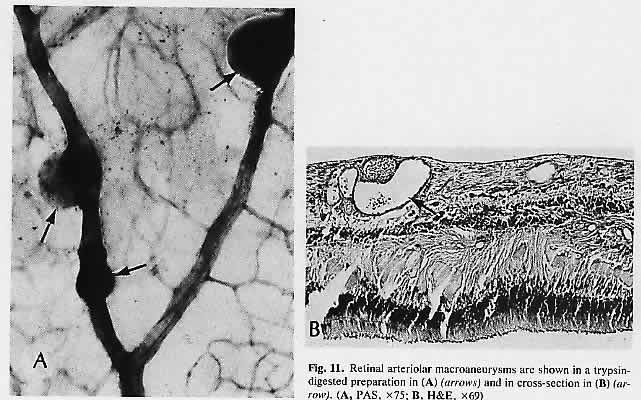
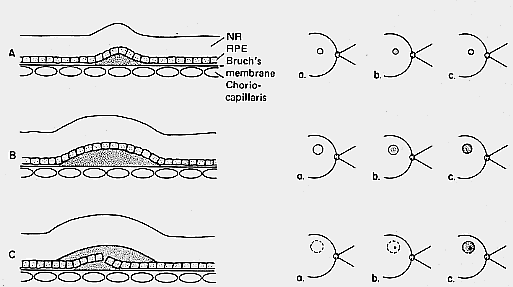
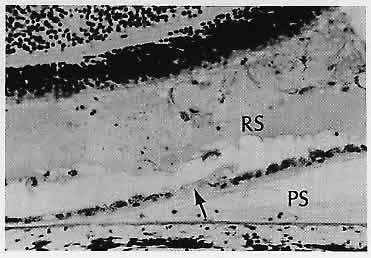
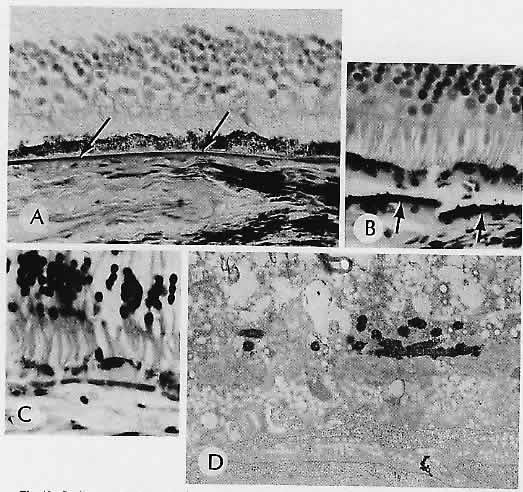
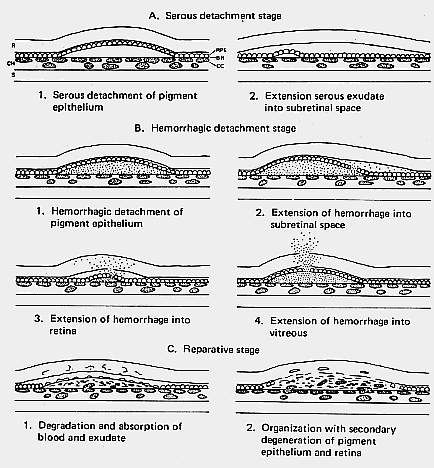
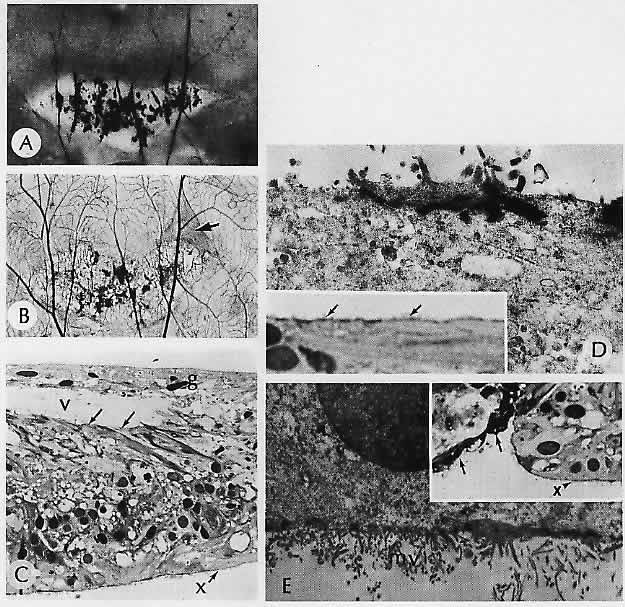
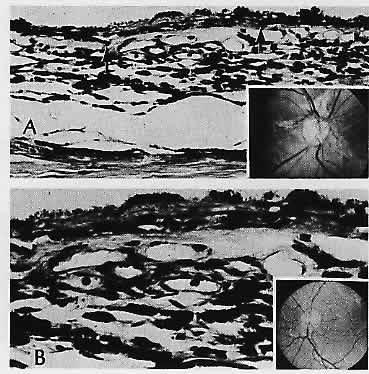
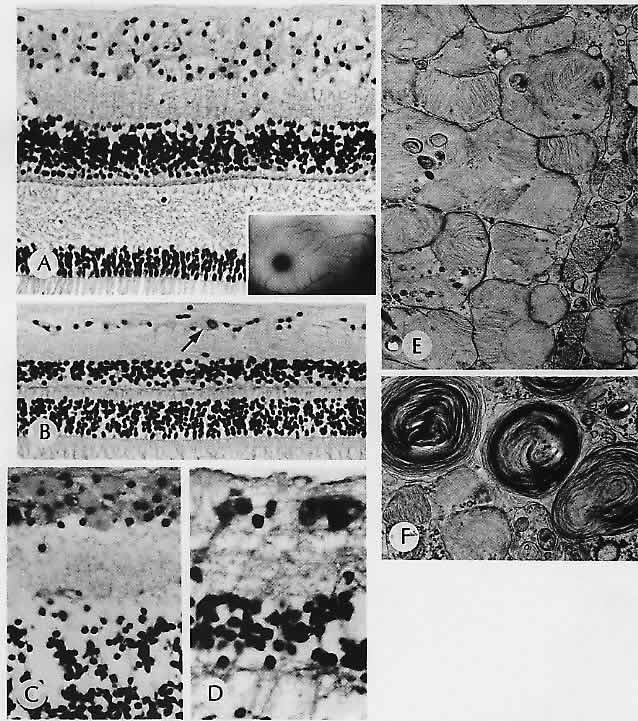
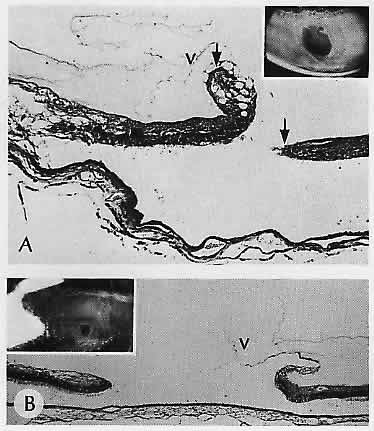
1. Castronuovo S, Simon JW, Kandel GL et al: Variable expression of albinism within a single kindred. Am J Ophthalmol 111:419, 1991 2. King RA, Lewis RA, Townsend D et al: Brown oculocutaneous albinism: Clinical, ophthalmological, and biochemical
characterization. Ophthalmology 92; 1496, 1985 3. Valenzuela R, Morningstar WA: The ocular pigmentary disturbance of human Chédiak-Higashi syndrome: A
comparative light- and electron-microscopic study and review of
the literature. Am J Clin Pathol 75:591, 1981 4. Wong L, O'Donnell FE Jr, Green WR: Giant pigment granules in the retinal pigment epithelium of a fetus with
X-linked ocular albinism. Ophthalmic Paediatr Genet 2:47, 1983 5. Weleber RG, Pillers DM, Powell BR et al: Åland Island eye disease (Forsius-Eriksson syndrome) associated with
contiguous deletion Syndrome at Xp21: Similarity to incomplete congenital
stationary night blindness. Arch Ophthalmol 107:1170, 1989 6. Hittner H, King R, Riccardi V et al: Oculocutaneous albinoidism as a manifestation of reduced neural crest derivatives
in the Prader-Willi syndrome. Am J Ophthalmol 94:328, 1982 7. Lloyd WC III, Eagle RC Jr, Shields JA et al: Congenital hypertrophy of the retinal pigment epithelium: Electron microscopic
and morphometric observations. Ophthalmology 97: 1052, 1990 8. Traboulsi El, Maumenee IH, Krush AJ et al: Pigmented ocular fundus lesions in the inherited gastrointestinal polyposis
syndromes and in hereditary nonpolyposis colorectal cancer. Ophthalmology 95:964, 1988 9. Munden PM, Sobol WM, Weingeist TA: Ocular findings in Turcot syndrome (glioma-polyposis). Ophthalmology 98:111, 1991 10. Traboulsi El, Maumenee IH, Krush AJ et al: Congenital hypertrophy of the retinal pigment epithelium predicts colorectal
polyposis in Gardner's syndrome. Arch Ophthalmol 108:525, 1990 11. Pagon RA: Ocular coloboma. Surv Ophthalmol 25:223, 1981 12. Fulton AB, Craft JL, Howard RO, Albert DM: Human retinal dysplasia. Am J Ophthalmol 85:690, 1978 13. Lahav M, Albert DM, Wyand S: Clinical and histopathologic classification of retinal dysplasia. Am J Ophthalmol 75:648, 1973 14. Kalina RE: A histopathologic postmortem and clinical study of peripheral retinal folds
in infant eyes. Am J Ophthalmol 71:446, 1971 15. Gartner S, Henkind P: Lange's folds: A meaningful ocular artifact. Ophthalmology 88: 1307, 1981 16. Levine RA, Gray DL, Gould N et al: Warburg syndrome. Ophthalmology 90: 1600, 1983 17. Straatsma BR, Foos RY, Heckenlively JR, Taylor GN: Myelinated retinal nerve fibers. Am J Ophthalmol 91:25, 1981 18. Baarsma GS: Acquired medullated nerve fibres. Br J Ophthalmol 64:651, 1980 19. Vaghefi HA, Green WR, Kelley JS et al: Correlation of clinicopathologic findings in a patient: Congenital night
blindness, branch retinal vein occlusion, cilioretinal artery, drusen
of the optic nerve head, and intraretinal pigmented lesion. Arch Ophthalmol 96:2097, 1978 20. Orth DH, Fine BS, Fagman W, Quirk TC: Clarification of foveomacular nomenclature
and grid for quantization of macular disorders. Trans Am Acad
Ophthalmol Otolaryngol 83:OP506, 1977 21. Smith D, Oestreicher J, Musarella MA: Clinical spectrum of Leber's congenital amaurosis in the second to
fourth decades of life. Ophthalmology 97:1156, 1990 22. Noble KG, Cart RE: Leber's congenital amaurosis: A retrospective study of 33 cases and
a histopathological study of one case. Arch Ophthalmol 96:818, 1978 23. Hayasaka S, Hara S, Mizuno K et al: Leber's congenital amaurosis associated with hyperthreoninemia. Am J Ophthalmol 101:475, 1986 24. Mizuno K, Takei Y, Sears ML et al: Leber's congenital amaurosis. Am J Ophthalmol 83:32, 1977 25. Patrinely JR, Green WR, Randolph ME: Retinal phlebitis with chorioretinal emboli. Am J Ophthalmol 94:49, 1982 26. Kahn M, Green WR, Knox DL, Miller NR: Ocular features of carotid occlusive disease. Retina 6:239, 1986 27. Tso MOM, Jampol LM: Pathophysiology of hypertensive retinopathy. Ophthalmology 89:1132, 1982 28. Arruga J, Sanders MD: Ophthalmologic findings in 70 patients with evidence of retinal embolism. Ophthalmology 89:1336, 1982 29. Kresca LJ, Goldberg MF, Jampol LM: Talc emboli and retinal neovascularization in a drug abuser. Am J Ophthalmol 87:334, 1979 30. Brownstein S, Font RL, Alper MG: Atheromatous plaques of the retinal blood vessels: Histologic confirmation
of ophthalmoscopically visible lesions. Arch Ophthalmol 90:49, 1973 31. Zimmerman LE: Embolism of central retinal artery. Arch Ophthalmol 73:822, 1965 32. Gartner S, Henkind P: Neovascularization of the iris (rubeosis iridis). Surv Ophthalmol 22:291, 1978 33. Enzenauer RJ, Stock JG, Enzenauer RW et al: Retinal vasculopathy associated with systemic light chain deposition disease. Retina 10:115, 1990 34. Wolter JR: Axonal enlargements in the nerve-fiber layer of the human retina. Am J Ophthalmol 65: 1, 1968 35. Ashton N, Harry J: The pathology of cotton wool spots and cytoid bodies in hypertensive retinopathy
and other diseases. Trans Ophthalmol Soc UK 83:91, 1963 36. Kahn M, Knox DL, Green WR: Clinicopathologic studies of a case of aortic arch syndrome. Retina 6:228, 1986 37. McDonnell PJ, Moore GW, Miller NR et al: Temporal arteritis: A clinicopathologic study. Ophthalmology 93:518, 1986 38. Albert DM, Ruchman MC, Keltner JL: Skip areas in temporal arteritis. Arch Ophthalmol 94:2072, 1976 39. Hayreh SS: Classification of central retinal vein occlusion. Ophthalmology 90:458, 1983 40. Green WR, Chan CC, Hutchins GM, Terry JM: Central retinal vein occlusion: A prospective histopathologic study of 29 eyes
in 28 cases. Retina 1:27, 1981 41. Frangieh GT, Green WR, Barraquer-Somers E, Finkelstein D: Histopathologic study of nine branch retinal vein occlusions. Arch Ophthalmol 100:1132, 1982 42. Weinberg D, Dodwell DG, Fern SA: Anatomy of arteriovenous crossings in branch retinal vein occlusion. Am J Ophthalmol 109:298, 1990 43. Scheie HG: Evaluation of ophthalmoscopic changes of hypertension and arteriolar sclerosis. Arch Ophthalmol 49:117, 1953 44. Kishi S, Tso MOM, Hayreh SS: Fundus lesions in malignant hypertension: I. A pathologic study of experimental
hypertensive choroidopathy. Arch Ophthalmol 103:1189, 1985 45. Rosenthal AR: Ocular manifestations of leukemia: A review. Ophthalmology 90:899, 1983 46. Khouri GG, Murphy RP, Kuhajda FP, Green WR: Clinicopathologic features in two cases of multiple myeloma. Retina 6: 169, 1986 47. Ortiz JM, Yanoff M, Cameron JD, Schaffer D: Disseminated intravascular coagulation in infancy and in the neonate: Ocular
findings. Arch Ophthalmol 100:1413, 1982 48. Reeser F, Fleischman J, Williams GA, Goldman A: Efficacy of argon laser photocoagulation in the treatment of circinate
diabetic retinopathy. Am J Ophthalmol 92:762, 1981 49. Rabb MF, Gagliano DA, Teske MP: Retinal arterial macroaneurysms. Surv Ophthalmol 33:73, 1988 50. Fichte C, Streeten BW, Friedman AH: A histopathologic study of retinal arterial aneurysms. Am J Ophthalmol 85:509, 1978 51. Perry HD, Zimmerman LE, Benson WE: Hemorrhage from isolated aneurysm of a retinal artery. Arch Ophthalmol 95:281, 1977 52. Nagpal KC, Goldberg MF, Rabb MF: Ocular manifestations of sickle hemoglobinopathies. Surv Ophthalmol 21:391, 1977 53. Romayananda N, Goldberg MF, Green WR: Histopathology of sickle cell retinopathy. Trans
Am Acad Ophthalmol Otolaryngol 77:OP652, 1973 54. Jampol LM, Acheson R, Eagle RC Jr et al: Calcification of Bruch's membrane in angioid streaks with homozygous
sickle cell disease. Arch Ophthalmol 105:93, 1987 55. Renie WA, Murphy RP, Anderson KC et al: The evaluation of patients with Eales' disease. Retina 3:243, 1983 56. Whitson JT, Welch RB, Green WR: Von Hippel-Lindau disease: Case report of a patient with spontaneous regression
of a retinal angioma. Retina 6:253, 1986 57. Messmer E, Laqua H, Wessing A et al: Nine cases of cavernous hemangioma of the retina. Am J Ophthalmol 95:383, 1983 58. Pancurak J, Goldberg MF, Frenkel M, Crowell RM: Cavernous hemangioma of the retina: Genetic and central nervous system
involvement. Retina 5:215, 1985 59. Tilanus MD, Hoyng C, Deutman AF et al: Congenital arteriovenous communications and the development of two types
of leaking retinal macroaneurysms. Am J Ophthalmol 112:31, 1991 60. Kincaid MC, Green WR, Iliff WJ: Granulomatous reaction to Choyce-style intraocular lens. Ophthalmic Surg 13:292, 1982 61. Duane TD, Osher RH, Green WR: White centered hemorrhages: Their significance. Ophthalmology 87:66, 1980 62. Weiss JN, Hutchins RK, Balogh K: Simultaneous Aspergillus endophthalmitis and cytomegalovirus retinitis
after kidney transplantation. Retina 8: 193, 1988 63. Brod RD, Flynn HW Jr, Clarkson JG et al: Endogenous Candida endophthalmitis: Management without intravenous amphotericin B. Ophthalmology 97:666, 1990 64. Pepose JS, Kreiger AE, Tomiyasu U et al: Immunocytologic localization of herpes simplex type 1 viral antigens in
herpetic retinitis and encephalitis in an adult. Ophthalmology 92: 160, 1985 65. Partamian LG, Morse PH, Klein HZ: Herpes simplex type 1 retinitis in an adult with systemic herpes zoster. Am J Ophthalmol 92:215, 1981 66. Reynolds JD, Griebel M, Mallory S, Steele R: Congenital herpes simplex retinitis. Am J Ophthalmol 102:33, 1986 67. Hedges TR III, Albert DM: The progression of the ocular abnormalities of herpes zoster: Histopathologic
observations of nine cases. Ophthalmology 89: 165, 1982 68. Lambert SR, Taylor D, Kriss A et al: Ocular manifestations of the congenital varicella syndrome. Arch Ophthalmol 107:52, 1989 69. Bloom JN, Palestine AG: The diagnosis of cytomegalovirus retinitis. Ann Intern Med 109:963, 1988 70. Pepose JS, Newman C, Bach MC et al: Pathologic features of cytomegalovirus retinopathy after treatment with
the antiviral agent gancyclovir. Ophthalmology 94:414, 1987 71. Culbertson WW, Blumenkranz MS, Pepose JS et al: Varicella zoster virus is a cause of the acute retinal necrosis syndrome. Ophthalmology 93:559, 1986 72. Freeman WR, Thomas EL, Rao NA et al: Demonstration of herpes group virus in acute retinal necrosis syndrome. Am J Ophthalmol 102:701, 1986 73. Duker JS, Nielsen JC, Eagle RC Jr et al: Rapidly progressive acute retinal necrosis secondary to herpes simplex
virus, type 1. Ophthalmology 97: 1638, 1990 74. Pepose JS, Holland GN, Nestor MS et al: Acquired immune deficiency syndrome: Pathogenic mechanisms of ocular disease. Ophthalmology 92:472, 1985 75. Jabs DA, Green WR, Fox R et al: Ocular manifestations of acquired immune deficiency syndrome. Ophthalmology 96:1092, 1989 76. Tanenbaum M, Russell S, Richmond P, Gass JDM: Calcified cytoid bodies in acquired immunodeficiency syndrome. Retina 7:84, 1987 77. Jabs DA, Enger C, Bartlett JG: Cytomegalovirus retinitis and acquired immunodeficiency syndrome. Arch Ophthalmol 107:75, 1989 78. Forster DJ, Dugel PU, Frangieh GT et al: Rapidly progressive outer retinal necrosis in the acquired immunodeficiency
syndrome. Am J Ophthalmol 110:341, 1990 79. Heinemann R-H, Gold JMW, Maisel J: Bilateral Toxoplasma retinochoroiditis in a patient with acquired immune deficiency syndrome. Retina 6:224, 1986 80. Parke DW II, Font RL: Diffuse toxoplasmic retinochoroiditis in a patient with AIDS. Arch Ophthalmol 104:571, 1986 81. Macher A, Rodrigues MM, Kaplan W et al: Disseminated bilateral chorioretinitis due to Histoplasma capsulatum in a patient with the acquired immunodeficiency syndrome. Ophthalmology 92:1159, 1985 82. Levy JH, Liss RA, Maguire AM: Neurosyphilis and ocular syphilis in patients with concurrent human immunodeficiency
virus infection. Retina 9: 175, 1989 83. Shami MJ, Freeman W, Friedberg D et al: A multicenter study of Pneumocystis choroidopathy. Am J Ophthalmol 112:15, 1991 84. Rao NA, Zimmerman PL, Boyer D et al: A clinical, histopathologic, and electron microscopic study of Pneumocystis carinii choroiditis. Am J Ophthalmol 107:218, 1989 85. Laatikainen LT, Immonen I JR: Acute posterior multifocal placoid pigment epitheliopathy in connection
with acute nephritis. Retina 8: 122, 1988 86. O'Brien DM, Farmer SG, Kalina RE, Leon JA: Acute macular neuroretinopathy following intravenous sympathomimetics. Retina 9:281, 1989 87. Foos RY: Senile retinoschisis: Relationship to cystoid degeneration. Trans Am Acad Ophthalmol Otolaryngol 74:33, 1970 88. Laatikainen L, Tarkkanen A, Saksela T: Hereditary X-linked retinoschisis and bilateral congenital retinal detachment. Retina 7:24, 1987 89. Schlernitzauer DA, Green WR: Peripheral retinal albinotic spots. Am J Ophthalmol 72:729, 1971 90. Curtin B J: The posterior staphyloma of pathologic myopia. Trans Am Ophthalmol Soc 75:67, 1977 91. Hotchkiss ML, Fine SL: Pathologic myopia and choroidal neovascularization. Am J Ophthalmol 91:177, 1981 92. Levy JH, Pollock HM, Curtin BJ: The Fuchs' spot: An ophthalmoscopic and fluorescein angiographic study. Ann Ophthalmol 9: 1433, 1977 93. Hampton GR, Kohen D, Bird AC: Visual prognosis of disciform degeneration in myopia. Ophthalmology 90:923, 1983 94. Yannuzzi LA, Shakin JL, Fisher YL, Altomonte MA: Peripheral retinal detachments and retinal pigment epithelial atrophic
tracts secondary to central serous pigment epitheliopathy. Ophthalmology 91:1554, 1984 95. Kincaid MC, Green WR, Kelley JS: Acute ocular leukemia. Am J Ophthalmol 87:698, 1979 96. Green WR, Key SN III: Senile macular degeneration: A histopathologic study. Trans Am Ophthalmol Soc 75: 180, 1977 97. Weiter J J, Delori FC, Wing GL, Fitch KA: Retinal pigment epithelial lipofuscin and melanin and choroidal melanin
in human eyes. Invest Ophthalmol Vis Sci 27:145, 1986 98. Feeney-Burns L: The pigments of the retinal pigment epithelium. Curr Topics Eye Res 2:119, 1980 99. Feeney-Burns L, Burns RP, Gao C-L: Age-related macular changes in humans over 90 years old. Am J Ophthalmol 109:265, 1990 100. Eagle RC Jr: Mechanisms of maculopathy. Ophthalmology 91:613, 1984 101. Feeney-Burns L, Ellersieck MR: Age-related changes in the ultrastructure of Bruch's membrane. Am J Ophthalmol 100:686, 1985 102. Sarks SH: Drusen and their relationship to senile macular degeneration. Aust J Ophthalmol 8:117, 1980 103. Green WR, McDonnell PJ, Yeo JH: Pathologic features of senile macular degeneration. Ophthalmology 92:615, 1985 104. van der Schaft TL, de Bruijn WC, Mooy CM et al: Is basal laminar deposit unique for age-related macular degeneration? Arch Ophthalmol 109:420, 1991 105. Coffey AJH, Brownstein S: The prevalence of macular drusen in postmortem eyes. Am J Ophthalmol 102:164, 1986 106. Ulshafer RJ, Allen CB, Nicolaissen B Jr, Rubin ML: Scanning electron microscopy of human drusen. Invest Ophthalmol Vis Sci 28:683, 1987 107. Burns R, Feeney-Burns L: Clinico-morphologic correlations of drusen of Bruch's membrane. Trans Am Ophthalmol Soc 78:206, 1980 108. Green WR: Clinicopathologic studies of senile macular degeneration. in
Nicholson DH (ed): Ocular Pathology Update. New York, Masson, 1980 109. Kenyon KR, Maumenee AE, Ryan SJ et al: Diffuse drusen and associated complications. Am J Ophthalmol 100:119, 1985 110. El Baba F, Green WR, Fleischmann J et al: Clinicopathologic correlation of lipidization and detachment of the retinal
pigment epithelium. Am J Ophthalmol 101:576, 1986 111. Keno DD, Green WR: Retinal pigment epithelial window defect, Arch Ophthalmol 96:854, 1978 112. Henkind P, Gartner S: The relationship between retinal pigment epithelium and the choriocapillaris. Trans Ophthalmol Soc UK 103:444, 1983 113. Yeo JH, Marcus S, Murphy RP: Retinal pigment epithelial tears: Patterns
and prognosis, Ophthalmology 95;8, 1988 114. Heriot WJ, Henkind P, Bellhorn RW: Choroidal neovascularization can digest Bruch's membrane: A prior
break is not essential. Ophthalmology 91:1603, 1984 115. Green WR, Gass JDM: Senile disciform degeneration of the macula: Retinal
arterialization of the fibrous plaque demonstrated clinically and histopathologically. Arch
Ophthalmol 86: 487, 197 1 116. Guyer DR, Green WR, de Bustros S, Fine SL: Histopathologic features of idiopathic macular holes and cysts. Ophthalmology 97: 1045, 1990 117. Wood WJ, Smith TR: Senile disciform macular degeneration complicated by massive hemorrhagic
retinal detachment and angle closure glaucoma. Retina 3:296, 1983 118. Wolter JR, McWilliams JR: Rupture of disciform macular degeneration: Causing massive retroretinal
hemorrhage. Am J Ophthalmol 59: 1044, 1965 119. El Baba F, Jarrett WH II, Harbin TS Jr et al: Massive hemorrhage complicating age-related macular degeneration: Clinicopathologic
correlation and role of anticoagulants. Ophthalmology 93: 1581, 1986 120. Sheffer A, Green WR, Fine SL, Kincaid M: Presumed ocular histoplasmosis syndrome. Arch Ophthalmol 98:335, 1980 121. Khalil MK: Histopathology of presumed ocular histoplasmosis. Am J Ophthalmol 94:369, 1982 122. Pulido JS, Folberg R, Carter KD, Coonan P: Histoplasma capsulatum endophthalmitis after cataract extraction. Ophthalmology 97:217, 1990 123. Tso MOM: Pathology of Cystoid macular edema. Ophthalmology 89:902, 1982 124. Fine BS, Brucker AJ: Macular edema and cystoid macular edema. Am J Ophthalmol 92:466, 1981 125. Yanoff M, Fine BS, Brucker AJ, Eagle RC Jr: Pathology of human cystoid macular edema. Surv Ophthalmol 28(suppl):505, 1984 126. Gass JDM, Anderson DR, Davis EB: A clinical, fluorescein angiographic, and electron microscopic correlation
of cystoid macular edema. Am J Ophthalmol 100:82, 1985 127. Frangieh GT, Green WR, Engel HM: A histopathologic study of macular cysts and holes. Retina 1:311, 1981 128. Smiddy WE, Michels RG, de Bustros S et al: Histopathology of tissue removed during vitrectomy for impending idiopathic
macular holes. Am J Ophthalmol 108:360, 1989 129. Craythorn JM, Swartz M, Creel DJ: Clofazimine-induced bull's eye retinopathy. Retina 6:50, 1986 130. Ramsey MS, Fine BS: Chloroquine toxicity in the human eye: Histopathologic observations by
electron microscopy. Am J Ophthalmol 73:229, 1972 131. Miller FS III, Bunt-Milam AH, Kalina RE: Clinical-ultrastructural study of thioridazine retinopathy. Ophthalmology 89:1478, 1982 132. Kaiser-Kupfer MI, Kupfer C, Rodrigues MM: Tamoxifen retinopathy: A clinicopathologic report. Ophthalmology 88:89, 1981 133. Brown GC, Shields JA, Sanborn G et al: Radiation retinopathy. Ophthalmology 89: 1494, 1982 134. Midena E, Segato T, Piermarocchi S et al: Retinopathy following radiation therapy of paranasal sinus and nasopharyngeal
carcinoma. Retina 7: 142, 1987 135. Boozalis GT, Schachat AP, Green WR: Subretinal neovascularization from the retina in radiation retinopathy. Retina 7:156, 1987 136. Tso MOM, Wallow IHL, Powell JO, Zimmerman LE: Recovery of the rod and cone cells after photic injury. Trans Am Acad Ophthalmol Otolaryngol 76: 1247, 1972 137. McDonald HR, Irvine AR: Light-induced maculopathy from the operating microscope in extracapsular
cataract extraction and intraocular lens implantation. Ophthalmology 90:945, 1983 138. Kuhn F, Morris R, Massey M: Photic retinal injury from endoillumination during vitrectomy. Am J Ophthalmol 111:42, 1991 139. Michels M, Sternberg P Jr: Operating microscope-induced retinal phototoxicity: Pathophysiology, clinical
manifestations and prevention. Surv Ophthalmol 34:237, 1990 140. Rinkoff J, Machemer R, Hida T, Chandler D: Temperature-dependent light damage to the retina. Am J Ophthalmol 102:452, 1986 141. Bastek JV, Fops RY, Heckenlively J: Traumatic pigmentary retinopathy. Am J Ophthalmol 92:621, 1981 142. Aguilar JP, Green WR: Choroidal rupture: A histopathologic study of 47 cases. Retina 4:269, 1984 143. Gaynon MW, Koh K, Marmor MF, Frankel LR: Retinal folds in the shaken baby syndrome. Am J Ophthalmol 106:423, 1988 144. Elner SG, Elner VM, Arnall M, Albert DM: Ocular and associated systemic findings in suspected child abuse: A necropsy
study. Arch Ophthalmol 108:1094, 1990 145. Massicotte SJ, Folberg R, Torczynski E et al: Vitreoretinal traction and perimacular retinal folds in the eyes of deliberately
traumatized children. Ophthalmology 98:1124, 1991 146. Maumenee IH: Vitreoretinal degeneration as a sign of generalized connective tissue diseases. Am J Ophthalmol 88:432, 1979 147. Nasr YG, Cherfan GM, Michels RG, Wilkinson CP: Goldmann-Favre Maculopathy. Retina 10:178, 1990 148. Brockhurst RJ, Albert DM, Zakov ZN: Pathologic findings in familial exudative vitreoretinopathy. Arch Ophthalmol 99:2143, 1981 149. Boldrey EE, Egbert P, Gass JDM, Friberg T: The histopathology of familial exudative vitreoretinopathy: A report of
two cases. Arch Ophthalmol 103:238, 1985 150. Blair NP, Albert DM, Liberfarb RM, Hirose T: Hereditary progressive arthro-ophthalmopathy of Stickler. Am J Ophthalmol 88:876, 1979 151. Straatsma BR, Zeegen PE, Fops RY et al: Lattice degeneration of the retina: XXX Edward Jackson memorial lecture. Am J Ophthalmol 77:619, 1974 152. Robinson MR, Streeten BW: The surface morphology of retinal breaks and lattice retinal degeneration: A
scanning electron microscopic study. Ophthalmology 93:237, 1986 153. Parelhoff ES, Wood WJ, Green WR, Kenyon KR: Radial perivascular lattice degeneration of the retina. Ann Ophthalmol 12:25, 1980 154. Wilson DJ, Weleber RC, Green WR: Ocular clinicopathologic study of gyrate atrophy. Am J Ophthalmol 111:24, 1991 155. Lopez PF, Maumenee IH, de la Cruz Z, Green WR: Autosomal-dominant fundus flavimaculatus: Clinicopathologic correlation. Ophthalmology 97:798, 1990 156. Eagle RC Jr, Lucier AC, Bernardino VB Jr, Yanoff M: Retinal pigment epithelial abnormalities in fundus flavimaculatus: A light
and electron microscopic study. Ophthalmology 87: 1189, 1980 157. Steinmetz RL, Garner A, Maguire JI, Bird AC: Histopathology of incipient
fundus flavimaculatus, Ophthalmology 98:953, 1991 158. McDonnell PJ, Kivlin JD, Maumenee IH, Green WR: Fundus flavimaculatus without maculopathy: A clinicopathologic study. Ophthalmology 93:116, 1986 159. Mohler CW, Fine SL: Long-term evaluation of patients with Best's vitelliform dystrophy. Ophthalmology 88:688, 1981 160. Frangieh GT, Green WR, Fine SL: A histopathologic study of Best's macular dystrophy. Arch Ophthalmol 100:1115, 1982 161. O'Gorman S, Flaherty WA, Fishman GA, Berson EL: Histopathologic findings in Best's vitelliform macular dystrophy. Arch Ophthalmol 106: 1261, 1988 162. Weingeist TA, Kobrin JL, Watzke RC: Histopathology of Best's macular dystrophy. Arch Ophthalmol 100:1108, 1982 163. Patrinely JR, Lewis RA, Font RL: Foveomacular vitelliform dystrophy, adult type: A clinicopathologic study
including electron microscopic observations. Ophthalmology 92:1712, 1985 164. Jaffe G J, Schatz H: Histopathologic features of adult-on-set foveomacular pigment epithelial
dystrophy. Arch Ophthalmol 106:958, 1988 165. Sneed SR, Sieving PA: Fenestrated sheen macular dystrophy. Am J Ophthalmol 112:1, 1991 166. Pagon RA: Retinitis pigmentosa. Surv Ophthalmol 33:137, 1988 167. Bastek JV, Siegel EB, Straatsma BR, Fops RY: Chorioretinal juncture: Pigmentary patterns of the peripheral fundus. Ophthalmology 89: 1455, 1982 168. Szamier RB, Betson EL: Retinal histopathology of a carrier of X-chromosome-linked retinitis pigmentosa. Ophthalmology 92:271, 1985 169. Bunt-Milam AH, Kalina RE, Pagon RA: Clinical-ultra-structural study of a retinal dystrophy. Invest Ophthalmol Vis Sci 24:458, 1983 170. Meyer KT, Heckenlively JR, Spitznas M, Fops RY: Dominant retinitis pigmentosa: A clinicopathologic correlation. Ophthalmology 89:1414, 1982 171. Gartner S, Henkind P: Pathology of retinitis pigmentosa. Ophthalmology 89: 1425, 1982 172. Rodrigues MM, Bardenstein D, Wiggert B et al: Retinitis pigmentosa with segmental massive retinal gliosis: An immunohistochemical, biochemical, and ultrastructural study. Ophthalmology 94: 180, 1987 173. Albert DM, Pruett RC, Craft JL: Transmission electron microscopic observations of vitreous abnormalities
in retinitis pigmentosa. Am J Ophthalmol 101:665, 1986 174. Hsieh RC, Fine BS, Lyons JS: Patterned dystrophies of the retinal pigment epithelium. Arch Ophthalmol 95:429, 1977 175. de Jong PTVM, Delleman JW: Pigment epithelial pattern dystrophy: Four different
manifestations in a family, Arch Ophthalmol 100:1416, 1982 176. Wilson DJ, Weleber RG, Klein ML et al: Bietti's crystalline dystrophy: A clinicopathologic correlative study. Arch Ophthalmol 107:213, 1989 177. Harrison RJ, Acheson RR, Dean-Hart JC: Bietti's tapetoretinal degeneration with marginal corneal dystrophy (crystalline
retinopathy): Case report. Br J Ophthalmol 71:220, 1987 178. Small KW, Letson R, Scheinman J: Ocular findings in primary hyperoxaluria. Arch Ophthalmol 108:89, 1990 179. Novak MA, Roth AS, Levine MR: Calcium oxalate retinopathy associated with methoxyflurane abuse. Retina 8:230, 1988 180. Wells CG, Johnson RJ, Qingli Let al: Retinal oxalosis: A clinicopathologic report. Arch Ophthalmol 107: 1638, 1989 181. Dreyer R, Green WR: The pathology of angioid streaks: A study of twenty-one cases. Trans Pa Acad Ophthalmol Otolaryngol 31: 158, 1978 182. Cibis GW, Harris DJ, Chapman AL, Tripathi RC: Mucolipidosis I. Arch Ophthalmol 101:933, 1983 183. Matthews JD, Weiter JJ, Kolodny EH: Macular halos associated with Niemann-Pick type B disease. Ophthalmology 93:933, 1986 184. Arnold AC, Pepose JS, Hepler RS, Foos RY: Retinal periphlebitis and retinitis in multiple sclerosis: k Pathologic
characteristics. Ophthalmology 91:255, 1984 185. Baker RH, Trautman JC, Younge BR et al: Late juvenile-onset Krabbe's disease. Ophthalmology 97:1176, 1990 186. Nork TM, Ghobrial MW, Peyman GA, Tso MOM: Massive retinal gliosis: A reactive proliferation of Müller cells. Arch Ophthalmol 104: 1383, 1986 187. Leys AM, Van Eyck LM, Nuttin BJ et al: Metastatic carcinoma to the retina. Arch Ophthalmol 108:1448, 1990 188. Kincaid MC, Green WR: Ocular and orbital involvement in leukemia. Surv Ophthalmol 27:211, 1983 189. Gass JDM, Weleber RG, Johnson DR: Non-Hodgkin's lymphoma causing fundus picture simulating fundus flavimaculatus. Retina 7:209, 1987 190. Foos RY, Simons KB: Vitreous in lattice degeneration of retina. Ophthalmology 91:452, 1984 191. Meyer E, Kurz GH: Retinal pits: A study of pathologic findings in two cases. Arch Ophthalmol 70:640, 1963 192. Smiddy WE, Green WR: Retinal dialysis: Pathology and pathogenesis. Retina 2:94, 1982 193. Kampik A, Kenyon KR, Michels RG et al: Epiretinal and vitreous membranes: Comparative study of 56 cases. Arch Ophthalmol 99: 1445, 1981 194. Schwartz D, de la Cruz Z, Green WR, Michels RG: Proliferative vitreoretinopathy: Ultrastructural study of 20 retroretinal
membranes removed by vitreous surgery. Retina 8:275, 1988 195. Wilson DJ, Green WR: Histopathologic study of the effect of retinal detachment surgery on 49 eyes
obtained post mortem. Am J Ophthalmol 103:167, 1987 196. Barr CC: The histopathology of successful retinal reattachment. Retina 10: 189, 1990 |