X-LINKED JUVENILE RETINOSCHISIS
Synonyms: Congenital hereditary retinoschisis, congenital vascular veils, anterior dialysis of the young, cystic disease of the retina, inherited retinal detachment
Introduction
Initially described in two patients by Haas27 in 1898, X-linked juvenile retinoschisis is an X-linked recessive condition with 100% penetrance, but varying expressivity. The fundamental disorder in X-linked juvenile retinoschisis is a splitting of the superficial layers of the retina. The pathogenesis is unknown, but histopathologic and electrophysiologic studies have suggested an underlying defect in the Müller's cells.28,29 The gene locus for this condition is located on the distal short arm of the X chromosome localized to the p22 region.30 There is little heterogeneity with this disorder. X-linked juvenile retinoschisis has been described worldwide, and the large series have been reported from Finland, The Netherlands, Germany, and the United States.30–33 Most cases have been in Caucasians, but other races, including those of African and Asian descent, have also been reported.32,33 Only males are affected and female carriers have normal vision and are normal on clinical examination and electrophysiologic testing. A rare female homozygote has been reported.34
The characteristic histopathologic feature of juvenile retinoschisis is a split between the nerve fiber and ganglion cell layers. This is to be distinguished from the split between the outer plexiform and adjacent nuclear layers seen in senile or acquired retinoschisis.27,33 Juvenile retinoschisis also differs in that no acid mucopolysaccharide is present in the retinoschisis cavity. Some authors postulate that an inherited defect in the inner core of the cytoplasm of Müller's cells may result in the retinoschisis.27 Ultrastructural studies have demonstrated the presence of extracellular fine filaments 7 to 16 nm in diameter in the schisis cavity and adjacent retina. These filaments may be the product of the defective Müller's cells.28 Extracellular accumulation of these filaments within the retina is postulated to lead to further degeneration of Müller's cells and formation of the schisis cavity. When the retina splits, the inner aspect of Müller's cell is damaged and can no longer contribute to the formation of the inner limiting membrane, which is thinned. Vascular defects have been postulated in the pathogenesis of juvenile retinoschisis.36 Degenerative changes in the retinal pigment epithelium occur only where there is substantial photoreceptor cell degeneration and probably represent a secondary phenomenon.
Clinical Findings
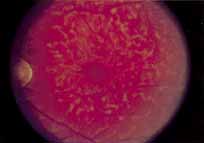
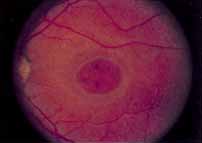
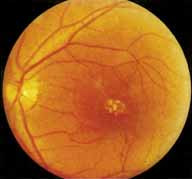
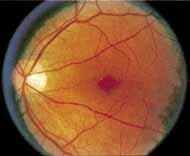
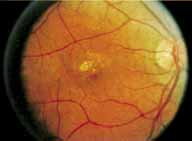
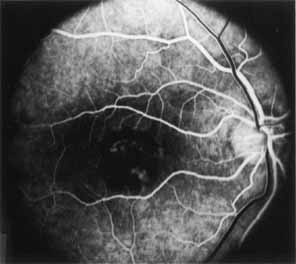
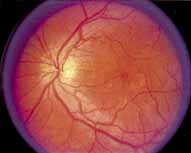
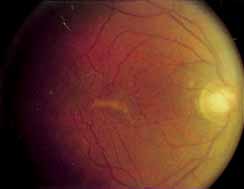
Although there is no reported genetic heterogeneity, there is wide phenotypic variation within the disorder. The major finding within the macula is a classic radial cystic maculopathy. Although retinal signs have been described in infants as young as 3 months, foveal schisis may be difficult to detect, leading to underdiagnosis. The diagnosis is usually not made until the affected male reaches school age (4 to 8 years of age) and encounters visual problems secondary to foveal involvement. Typical foveal schisis findings have been reported in 68% to 100% of eyes within various series.37,38 Foveal schisis is the only finding in about half the cases. It is characterized by the presence of radiate perifoveal microcysts located in the nerve fiber layer (Fig. 1) with radiate plications of the overlying internal limiting membrane that are seen especially well on monochromatic (red-free) photography (Fig. 2). The microcystoid change may slowly progress to form a macular cyst or hole. Foveal schisis has been reported in association with Goldmann-Favre vitreotapetoretinal dystrophy and rarely may be seen in rod-cone dystrophy or as an autosomal dominant or recessive condition.39–43
|
|
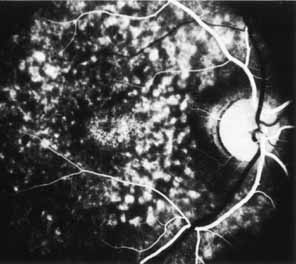
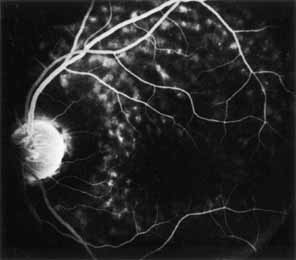
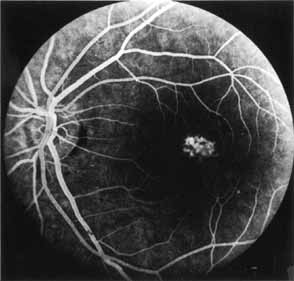
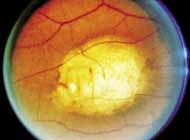
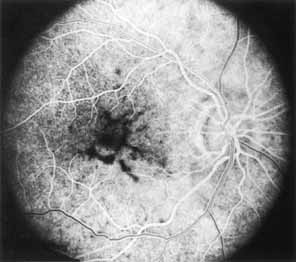
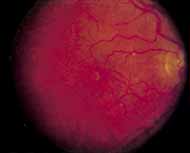
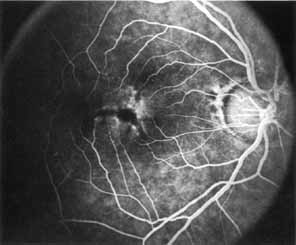
Peripheral features of this disorder include peripheral schisis, vitreous changes, perivascular sheathing, dendritiform patterns, retinal dragging, subretinal exudates, pigment lines, and a generalized tapetal-like reflex. Forsius and colleagues examined 268 eyes and classified severity of involvement into three cases, mild, moderate, and severe.31 Mild cases in 57% of eyes in this series demonstrated a typical schisis that was no longer readily distinguished except for pigment layer degeneration after 40 to 50 years of age. Moderate cases in 37% of patients in the series demonstrated similar macular findings with peripheral schisis or veils with retinal holes. Severe cases demonstrated schisis extending to the optic nervehead and severe anomalies involving retinal vessels and vitreous. In half of the cases, peripheral schisis is present which is usually bilateral, symmetric, and in the inferior temporal quadrant. The peripheral schisis only rarely extends to the ora serrata. The inner layer is usually elevated and may have multiple holes. Dendritic, opacified retinal vessels may be present within the retinal periphery, and fluorescein angiography in these cases reveals the presence of vascular leakage and capillary nonperfusion.36 Vitreous veils, which may contain branching retinal vessels, are sometimes prominent. Other vitreous changes include vitreous liquefaction, vitreous strands, traction bands, and vitreous detachment. Other changes associated with this disease include vitreous hemorrhage, optic atrophy, pseudopapilledema, ectopic maculae, iridocorneal angle anomalies, cataracts, disc and peripheral retinal neovascularization, neovascular glaucoma, and hemorrhagic retinal cyst.44–48 In end-stage disease, the inner wall of the schisis cavity may be a focus for glial proliferation and vitreous condensation, forming a retrolental membrane that results in leukocoria as a possible clinical presentation.35
Vision in this disease is variable and cannot be predicted based on the fundus findings alone. On occasion, it has been shown to be normal even in the presence of a foveal schisis. Visual acuity is usually in the 20/60 range in the affected young adult and may remain stationary for many years, with gradual deterioration to about 20/100 by the sixth decade. Most affected patients are legally blind by the seventh decade.31
Laser photocoagulation, cryopexy, and scleral buckling have been used with varying degrees of success to treat peripheral schisis that is rapidly advancing and threatening the posterior pole.49–53 The incidence of retinal detachment has varied from 0% to more than 20%.54 For these patients, scleral buckling may be used to treat retinal detachment that may arise in areas of peripheral retinoschisis with inner and outer layer holes. The redetachment rate may be as high as 40% regardless of scleral buckling versus vitrectomy.45 Vitrectomy may also be needed if significant vitreous traction or vitreous hemorrhage is present.54
Ancillary Tests
The posterior pole most often appears normal on fluorescein angiography and this may be helpful in the clinical differentiation of this entity from cystoid macular edema. In some cases there is evidence of pigment epithelial atrophy at the site of the macula. Kellner reported macular focal hyperfluorescence in the early phase, more distinct in older patients, paralleling pigment derangements found on ophthalmoscopy.44 Early in the course of the disease, color testing using the Farnsworth-Munsell 100-hue test shows a tritan defect.55,56 Later, a progressive deutan defect is found, correlating well with the visual acuity. Absolute visual field defects corresponding to zones of peripheral retinoschisis are present, as are relative central scotomas.
Electrophysiologic Tests
There is an overlapping of the fundus, electrophysiologic, and psychophysical findings among X-linked juvenile retinoschisis and the dominant (Wagner's) and recessive (Goldmann-Favre) types of vitreotapetoretinal dystrophies.55 In X-linked retinoschisis, the electroretinogram (ERG) is often abnormal. Both the photopic and scotopic b-wave, which arise from the bipolar cell layer, are usually reduced, whereas the a-wave, which arises from the inner segments of the photoreceptors, is intact, suggesting an inner retinal localization for these diseases. A demonstrated decrease in amplitude over several years indicates progressive disease, with nonrecordable ERG responses in advanced disease. The electrooculogram (EOG) is normal in mild cases, but may become subnormal in advanced cases. Dark adaptometry generally shows normal-to-subnormal cone and rod segments.
Differential Diagnosis
The diagnosis of the female carrier has been problematic because these females have normal visual function and electrophysiologic testing reveals no abnormalities. The fundus examination is also normal, although one case of a female carrier with radial wrinkling of the inner limiting membrane around the fovea, but no retinoschisis, has been reported.57 Recent studies of rod-mediated cone flicker thresholds and genetic linkage studies using restriction fragment length polymorphisms show promise in identifying the fundus-copically normal female carrier.57–59
X-linked juvenile retinoschisis must be differentiated from Goldmann-Favre disease and the other forms of inherited juvenile retinoschisis described in the next section, senile or acquired retinoschisis, cystoid macular edema, idiopathic macular hole formation, Wagner's disease, Stickler's syndrome, and other types of peripheral vitreoretinal abnormality.
FAMILIAL FOVEAL RETINOSCHISIS
Introduction
Lewis and Lee40 have described foveal retinoschisis in three daughters of funduscopically normal parents in a nonconsanguineous marriage. A male sibling was not affected. The condition is believed to be autosomal recessive. All three patients exhibited a mild degree of visual loss in the 20/30 to 20/50 range. Fundus changes were limited to the fovea. The pathology was identical to that seen in the early stages of X-linked retinoschisis. Fluorescein angiography revealed a barely discernible area of hyperfluorescence within the fovea of the oldest girl and the fovea was normal in the other two girls. Each patient showed a relative central scotoma on visual field testing.
Electrophysiologic testing (ERG and EOG) was normal in all girls tested. Dark adaptometry showed normal-to-subnormal cone and rod segments, and color testing revealed a mild tritan defect. The authors suggested that a deficiency of macular xanthophyll or a reduced sensitivity of foveal blue cones may exist in these patients.40 The complete natural history of this disorder has not yet been elucidated.
Subsequently, two families who demonstrated autosomal dominant transmission of juvenile retinoschisis have been reported. Yassur and investigators41 described eight affected members (five males and three females) in a three-generation pedigree with peripheral retinoschisis observed in all affected members. Of these individuals, only three had evidence of foveal schisis.41 The ERG was normal in mild cases, and subnormal b-waves were reported in the more severely affected individuals. Shimazaki and co-workers42 described a female with foveal and peripheral schisis whose mother also had foveal schisis. They proposed that an autosomal dominant pattern of inheritance was probably present in their limited pedigree.
Autosomal juvenile peripheral retinoschisis without foveal retinoschisis and autosomal juvenile retinoschisis associated with generalized rod and cone dysfunction probably represent yet other entities that are distinct from those described above.43,60,61
CONE DYSTROPHIES
Introduction
St. Albertus Magnus has been credited with the first description of cone dystrophy in the thirteenth centrury.62 The cone dystrophies represent a heterogenous group of disorders that can be inherited as an autosomal recessive, autosomal dominant, or X-linked recessive trait. The stationary cone dystrophies present as congenital disorders with with various degrees of cone dysfunction but normal rod function.63 In contrast, the progressive cone dystrophies often present in childhood or even early adulthood. These patients often develop rod dysfunction in later life and therefore overlap in clinical features with cone-rod dystrophies64,65
Based on an analysis of the cone dysfunction syndromes by Goodman and collaborators,65 and on work published by Krill and Deutman,66 the classification of cone dystrophies listed below can be used as an aid in the understanding of these conditions.
- Stationary forms:
- Rod monochromatism
- Autosomal recessive Complete
- Autosomal recessive Incomplete
- Autosomal recessive Complete
- X-linked Blue Cone monochromatism
- Miscellaneous forms
- Achromatopsia with normal visual acuity
- Oligocone trichomacy
- Achromatopsia with normal visual acuity
- Rod monochromatism
- Progressive forms: cone-rod dystrophies
The two forms, complete and incomplete rod monochromatism,66–68 probably reflect the total number of abnormal and missing cones. The presence of both forms of rod monochromatism in members of several affected families suggests that the complete and incomplete forms of rod monochromatism may represent different phenotypic expressions of the same genotype. Krill and Deutman66 have combined progressive cone dystrophy and progressive cone-rod dystrophy into one entity and refer to it as dominant macular degeneration. However, we shall refer to these dystrophies as progressive cone-rod dystrophies.
ROD MONOCHROMATISM: COMPLETE FORM
Synonyms: Typical monochromatism, complete achromatopsia
Introduction
Complete rod monochromatism is a rare form of congenital color defect. It is characterized by the presence of a marked generalized cone abnormality or complete absence of cones associated with a low level of visual acuity.64,65,69,70 The inheritance pattern is autosomal recessive. The mutations associated with rod monochromatism have not yet been completely identified. Arbour and colleagues72 have demonstrated linkiage of the disorder in an Iranian-Jewish pedigree to a centromeric region of chromosome 2. Rod monochromacy has also been reported with mutations in chromosome 14.73 Histopathologic study of donor eyes from patients with rod monochromatic has demonstrated the presence of cone-like structures within the retina.73 There is some controversy as to the nature and distribution of these cones. Two separate reports, one by Larsen74 and a second by Falls and co-workers75 found a normal total number of cones but with abnormal shapes. Harrison and colleagues76 reported abnormally shaped cones, reduced in total number throughout the retina.
Clinical Findings
The dystrophy is first detected in early youth as an absence of color vision with associated nystagmus, photophobia, and hemeralopia.74 Both the photophobia and the nystagmus tend to disappear as the patient becomes older, especially after 15 years of age. The patients often present with a high degree of hypermetropic refractive error. The clinical findings include a pronounced abnormality of color perception found on all color tests and a visual acuity in the 20/200 range. The vision is sometimes better for near distances and in dim illumination and worse in bright illumination. Visual fields may be normal or slightly constricted, particularly to colored targets, and a central scotoma can sometimes be demonstrated as the patient grows older. Nystagmus may make testing difficult. The nystagmus is pendular in youth, may decrease at near distances, and tends to decrease or disappear with time. The pupils respond slowly to light and paradoxic responses have been recorded during dark adaptation.76 Fundus changes are absent or minimal and, when present, are limited to the fovea. When present, these changes may include an abnormality or absence of the foveal reflex and, occasionally, a discrete, fine, granular disturbance of the retinal pigment epithelium within the central macula. The fovea has also been described as being hypoplastic.
No medical treatment is available, although tinted glasses help to decrease photophobia in bright illumination and may aid in slightly improving the visual acuity. Red-tinted contact lenses have also been tried with some success. Though central acuity remains diminished, the dystrophy is not progressive.
Ancillary Tests
Fluorescein angiography may show mild retinal pigment epithelium (RPE) transmission defects when a disturbance of the retinal pigment epithelium is present. Rod monochromats fail to recognize any plates on the common plate tests such as the Ishihara and Hardy-Rand-Ritller (HRR) tests. Rarely, residual cone function in rod monochromats can be demonstrated. A Stiles Crawford effect may be demonstrated and the dark adaptation curve maybe biphasic. Although Sharpe and Nordby reported residual cone function with ERG testing, these patients may have occult cases of incomplete achromatopsia or even progressive cone rode dystrophy.
Electrophysiologic Tests
The diagnosis is based on the findings of a congenital complete color defect with a nonprogressive subnormal visual acuity in the 20/200 range, normal scotopic ERG, absent or markedly subnormal photopic ERG, and a normal EOG.66–68
ROD MONOCHROMATISM: INCOMPLETE FORM
Synonym: Incomplete achromatopsia
Introduction
The clinical findings in incomplete achromatopsia are the same as those in the complete form, but they are usually less severe.64,66,69,70 Patients often have some residual color discrimination. The inheritance pattern is autosomal recessive. Photophobia and nystagmus may be absent, and visual acuity is less severely impaired than in individuals with the complete form. Unlike complete rod monochromats, individuals with the incomplete form have residual cone function mediated by photosensitive pigments other than rhodopsin, which provide the basis for some color perception.67,68
Clinical Findings
The diagnosis is based on the clinical findings of a congenital incomplete color defect with a nonprogressive subnormal visual acuity in the 20/60 to 20/200 range.66–68 Fundus changes are absent or minimal and, when present, are similar to those found in the complete form of rod monochromatism.
Ancillary Tests
Patients with incomplete rod chromatism demonstrate normal scotopic ERG response, nonrecordable or subnormal photopic ERG response, and a normal EOG.
BLUE CONE MONOCHROMATISM
Synonym: Atypical monochromatism
Introduction
Blue cone monochromatism, which demonstrates an X-linked recessive pattern of inheritance, is similar to the autosomal recessive form of rod monochromatism in its functional defects and clinical characteristics, although the two forms may be differentiated on the basis of spectral sensitivity and color matching. Female carriers of blue cone monochromatism may have an abnormal macular appearance and demonstrate abnormal photopic and flicker ERGs.
Clinical Findings
These male patients present with reduced acuity, nystagmus, and photophobia. Affected individuals are myopic, with fundus findings reflecting a tilted optic disc with a normal macular appearance.
Ancillary Tests
Under low photopic illumination levels, the blue cone monochromat will show dichromatic color vision. Hansen short wavelength perimetry can also be used to distinguish between blue cone and rod monochromats.
MISCELLANEOUS FORMS OF CONE MONOCHROMATISM
Synonyms: Atypical achromatopsia, achromatopsia with normal visual acuity, oligocone trichromacy.
Achromatopsia with normal visual acuity is a rare form of total color defect present in youth, reported in approximately 1 in every 100 million people64,69,77,78 It is characterized by minimal or no evidence of cone abnormality and relatively normal or only slightly decreased vision. The inheritance pattern is uncertain. Clinically, there is no photophobia or nystagmus, and the visual fields are usually normal. The fundus examination is also normal. Patients frequently have some degree of color vision, depending on the saturation of colors used, the luminance level, and the size of the test field. On routine color testing, however, these patients are found to be totally color defective. The ERG and the EOG are both normal.
Diagnosis is based on the clinical findings of congenital complete color blindness without amblyopia and a normal ERG and EOG. The differential diagnosis includes other forms of complete color defect without amblyopia, that is, the protan and deutan defects. No medical treatment is available, although the prognosis is good because the dystrophy is nonprogressive.
Oligocone trichromacy, first recognized by van Lith80 in a patient with reduced acuity, reduced photopic electroretinogram, and a normal appearing fundus, show good color discriminination. There appears to be a decreased photopigment concentration in these patients with normal regeneraration rates. Thus, it has been proposed that this disorder arises from a reduced cone population of all cone types. Its classification is ambiguous because many feel these patients should be classified as incomplete achromats versus oligocone trichromats.
PROGRESSIVE CONE-ROD DYSTROPHIES
Synonym: Progressive cone dystrophy
Introduction
The progressive cone-rod dystrophies constitute a relatively rare group of cone disorders that, for now, may be regarded as a single entity demonstrating variable expressivity.65,66,80–82 An attempt, however, has been made by one investigator to separate the group into two types based on electrophysiologic and psychophysical testing.71
This dystrophy is characterized by the presence of progressive color defects and a progressive decrease in visual acuity. There is usually a gradual decrease in visual acuity during the first or second decades of life, followed by a gradual loss of color vision. Frequently, these patients may go undiagnosed for years and they may be mistakenly believed to have a functional disorder.
The inheritance pattern is usually autosomal dominant, although autosomal recessive and X-linked recessive pedigrees have been described.83,84 Even within these inheritance modes, multiple different locations of mutations have been documented. Autosomal dominant progressive cone dystrophy has been mapped to chromosome 19q13.3, 17q12-p13, and to specific mutations of the peripherin/RDS gene on chromosome 6p.85–87 In patients with X-linked recessive progressive cone dystrophy, several loci have been mapped, including Xp21-p11.1, Xq27, and Xq28.88
No consistent metabolic or systemic abnormality has been found in patients with the progressive cone or cone-rod dystrophies, although there have been isolated reports of an association with low α-L-fucosidase activity in serum and leukocytes and chromosomal translocation.89 Histopathologic study reveals a complete disappearance of the photoreceptors in the macular and paramacular regions, with associated degeneration and disappearance of the retinal pigment epithelium.90 Likewise, there may be atrophy of the outer retinal layers in the periphery. These findings, together with the results of electrophysiologic and psychophysical testing, indicate that this dystrophy may be a form of outer segment photoreceptor degeneration and may be regarded, in some ways, to be the cone-rod counterpart to the rod-cone dystrophies (retinitis pigmentosa).
Clinical Findings
The progressive cone-rod dystrophies are characterized by selective involvement of the cones with the gradual development of color defects and a decrease in visual acuity. Photophobia is extremely common, along with hemeralopia. Night blindness (nyctalopia) is relatively rare but, when present, is usually associated with the more advanced stages of the dystrophy. Patients may develop total achromatopsia with visual acuities in the 20/200 range.
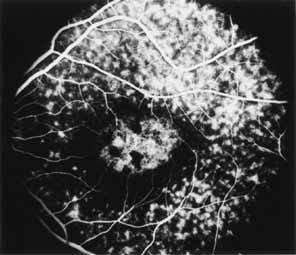
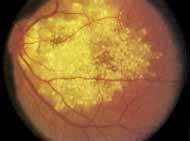
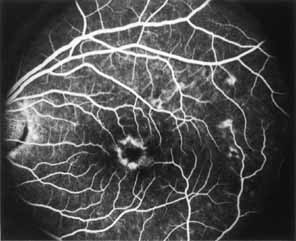
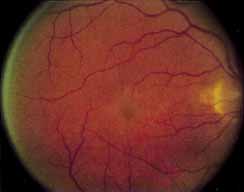
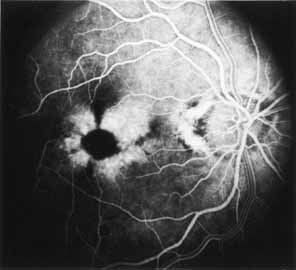
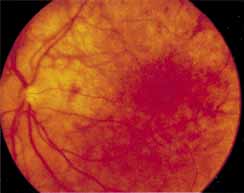
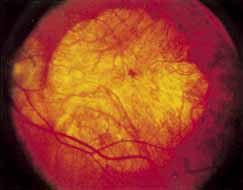
During the early stage of the dystrophy, when patients demonstrate a slight-to-moderate decrease in visual acuity and minimal color defects, there are minor or no visible fundus abnormalities. At most, the foveal reflex may be absent and there may be some increased granularity of the retinal pigment epithelium in the macula. Later, there is a decrease of visual acuity to the 20/400 range, oval atrophy of the macular retinal pigment epithelium (“beaten bronze” atrophy), and associated choroidal atrophy (Fig. 3). A characteristic bull's-eye maculopathy, similar to that seen in patients with chloroquine retinopathy, may also be seen.90 Photophobia, occasional nyctalopia, incomplete-to-complete color defects, and a central scotoma are often present. The symmetry of the process in both eyes is remarkable.
|
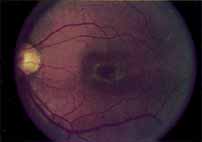
In the early stages of the dystrophy, the optic disc, retinal vessels, and peripheral retina are usually normal. The patient may have a visual acuity of 20/200 and normal color vision by standard testing and, yet, generalized cone dysfunction can be demonstrated by color field testing. In the late stages, there may be temporal optic disc pallor, attenuation of the retinal arterioles, and granular or bone spicule peripheral retinal pigmentary changes. Not infrequently, the peripheral pigmentary abnormalities may be regional. This probably reflects regional rod involvement, as evidenced by ERG, which may demonstrate a reduction in amplitude without a change in latency of the rod responses.72,80 At this stage, contracted peripheral fields may be found on both white and color target testing.
No medical treatment is available. The prognosis for vision is guarded and poorer than for the other cone dystrophies because both cone and rod function is impaired in the macula and retinal periphery. Accordingly, central vision is decreased and color vision impaired in association with contraction of the peripheral visual fields. As the dystrophy progresses, vision may decrease to the 20/400 level or less and may also be accompanied by contracted peripheral fields and a total color defect. Patients with poor central vision should be referred to low visual aids. Severe photophobia in adults and children has been reduced with the use of appropriately tinted spectacles or contacts. The tint will often depend on the dystrophy as some patients with relatively well-preserved color vision require neutral tints and other more severely affected patients require red tints similar to those in rod monochromats. Miotic drops have been used in some patients with severe photophobia but are not generally well tolerated or advised.
Ancillary Tests
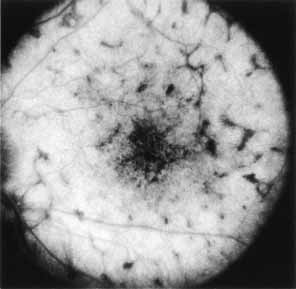
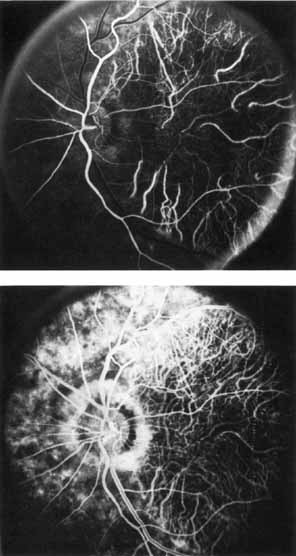
Fluorescein angiography demonstrates increased transmission of choroidal fluorescence in the macula during early phases of the study, without late leakage of dye or fluorescein staining. In addition, an annular pattern of hyperfluorescence is often seen in the macula, highlighting the bull's-eye pattern seen on fundus examination (Fig. 4). Visual field defects include central scotoma, peripheral field loss, and ring scotoma.
|

Electrophysiologic Testing
In the early stages of the dystrophy, electrophysiologic testing reveals normal photopic and scotopic ERG responses. With progression of the disease, there is a moderate-to-marked loss of cone function, associated with a slight-to-moderate loss of rod function, markedly decreased or absent photopic responses and mildly decreased scotopic responses. Unusual variants with supernormal scotopic responses to suprathreshold stimuli have been described in some pedigrees.91,92 The EOGin the early stage of the dystrophy is normal. With more widespread involvement of the retina, a reduction of the light rise develops and the EOG becomes a sensitive indicator of the progress of the dystrophy. The EOG first becomes subnormal, and is then followed by the presence of a subnormal ERG. Dark adaptation curves late in the course of the dystrophy are monophasic with slightly elevated rod thresholds.
Differential Diagnosis
The diagnosis is based on the findings of progressive loss of central and color vision beginning in the first or second decades of life, with associated photophobia, a characteristic bull's-eye macular lesion, markedly decreased or absent photopic ERG responses, mildly decreased scotopic ERG responses, a mildly abnormal dark adaptation curve and, in the late stages, peripheral retinal pigmentary changes with retinal arteriolar attenuation.
The differential diagnosis includes rod monochromatism, pericentral and sine pigmento forms of retinitis pigmentosa (rod-cone dystrophy), Stargardt's disease, central areolar choroidal dystrophy, chloroquine retinopathy, and congenital optic atrophy. Finally, certain syndromes are associated with cone dystrophies including Bardet-Biedl syndrome, Alstroms's syndrome, and Pierre-Marie ataxia with cone rod dystrophy.
PERICENTRAL (INVERSE) RETINITIS PIGMENTOSA (ROD-CONE DYSTROPHY)
Synonyms: Pericentral Pigmentary retinopathy, pericentral Pigmentary dystrophy, Peripapillary Pigmentaryretinal degeneration, pericentral Pigmentary degeneration
Introduction
Pericentral retinitis pigmentosa (rod-cone dystrophy) is characterized by pigmentary disturbances similar to those occurring in classic retinitis pigmentosa (rod-cone dystrophy), but which occur solely in the pericentral retina with no involvement of the peripheral retina.20 Grondahl94 described twenty-eight patients from four families, some demonstrating an autosomal dominant pattern of inheritance. Other previous studies from Traboulsi, O'Neill and Maumenee have reported an autosomal recessive mode of inheritance.95
Clinical Findings
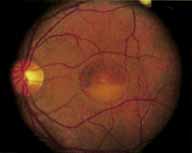
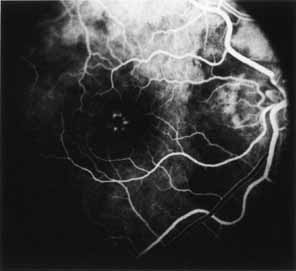
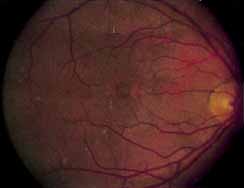
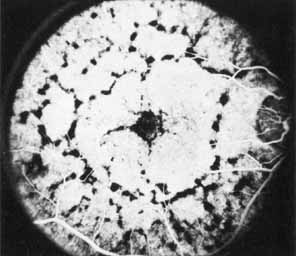
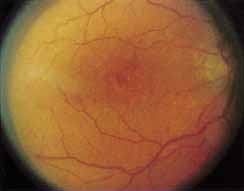
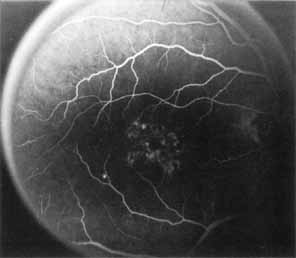
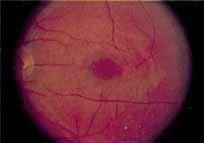
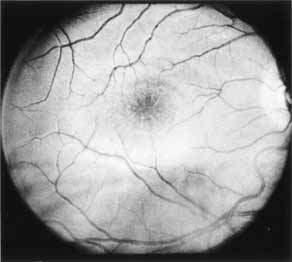
Vision is usually normal in the early stage and patients are often diagnosed on routine fundus examinations in the second and third decades of life. In later stages, vision may be markedly reduced to the 20/200 level or worse. Color vision is normal early on, but frequently becomes defective and may be accompanied by a progressive decrease in central vision. Nyctalopia is often present. The pigmentary changes may take the form of bone spicules or scattered black dots and are the earliest signs of the dystrophy (Fig. 5 and 6). Because the fovea is lacking in vessels, no bone spicules are present in this region. Atrophy of the retinal pigment epithelium may also be present. In contradistinction to retinitis pigmentosa (rod-cone dystrophy), the disc and retinal vessels are usually normal until late in the course of the disease.
|
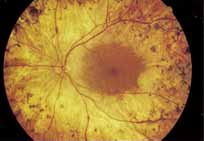
|
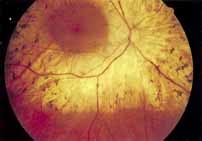
Durlu and colleagues96 described macular complications occurring in two patients including a full thickness macular hole and central retinal artery occlusion. As is the case with retinitis pigmentosa, many forms of therapy have been attempted, none of which have been successful.
Ancillary Tests
Tritan color defects have been reported on Ishihara and Farnsworth-Munsell testing. Despite the common presenting symptom of nyctalopia, the dark adaptometry is normal or slightly delayed.96 The angiogram in the early course of the disease is often unremarkable without evidence of retinal pigmentary derangement.
Electrophysiologic Tests
The primary lesion is thought to reside in the photoreceptors and the pigment epithelium, but due to the localized nature of the process, the EOG and ERG are often normal or only slightly subnormal.11,21,96
Differential Diagnosis
The differential diagnosis includes classic retinitis pigmentosa (rod-cone dystrophy), progressive cone-rod dystrophy, the late stage of fundus flavimaculatus, and inflammatory and infectious causes of chorioretinitis. The ERG should enable one to differentiate this dystrophy from the progressive cone-rod dystrophies and classic retinitis pigmentosa (rod-cone dystrophy). The retinopathy of neuronal ceroid lipofuscinosis and olivo- pontoceerebeallar ataxia potentially involves the central macula in a similar presentation.96,97
DOMINANT CYSTOID MACULAR DYSTROPHY
Introduction
Deutman and Pinckers reported a 4-year-old female with severe hyperopia and diminished acuity.98 In addition to a pigmentary retinopathy, cystoid edema was a prominent feature.98–102 Deutman and colleagues98 further characterized three of five families with the disorder. An additional report describes an affected Greek family.101 Since that time, the gene for dominant cystoid macular dystrophy has been localized to chromosome 7p.102
Clinical Findings
Dominantly inherited cystoid macular edema is characterized in the first and second decades with a prolonged course of macular cystoid change and progressive loss of central vision. The pathogenesis and prevalence of this dystrophy are as yet undetermined. Hyperopia from +2.00 to +10.00 D is seen along with hypopigmentation within the central macula. Macular atrophy is a fairly prominent finding late in the disease. Occasionally, a bull's-eye pattern of atrophic change may be seen. Vitreous cells, strands, and veils, and peripheral retinal pigmentary disturbances ranging from a slight hyperpigmentation and depigmentation to the formation of bone spicule changes have also been reported. Late in the disease, vision may be reduced to the 20/200 to finger counting range and a relative-to-absolute central scotoma may be present. No known treatment is available.
Ancillary Tests
Fluorescein angiography reveals leakage from perifoveal capillaries with dye pooling in a cystoid pattern. Later in the disease, transmission defects are seen in areas of RPE atrophy. In some cases, fluorescein leakage from optic disc capillaries has also been noted. Color testing in some patients often demonstrated blue yellow defects likely secondary to macular edema and in chronic cases red green deficiencies secondary to photoreceptor damage.
Electrophysiologic Studies
The ERG has been normal in all patients studied to date. The EOG and results of dark adaptometry, however, are subnormal in most, but not all, patients. The subnormal EOG would indicate widespread dysfunction of the retinal pigment epithelium.
Differential Diagnosis
The condition should be differentiated from other hereditary retinal disorders that are associated with cystoid macular edema, especially retinitis pigmentosa (rod-cone dystrophy), and dystrophies characterized by the presence of foveal retinoschisis (which may be mistaken for cystoid macular edema) such as X-linked juvenile retinoschisis and familial foveal retinoschisis. Cystoid macular edema is seen after ophthalmic surgeries, pars planitis, and other ocular inflammatory disorders. Vascular disorders such as diabetic retinopathy, Coat's disease, and parafoveal telangectasis also cause cystoid macular edema. Finally, high dose nicotinic acid has been reported to cause cystoid edema without obvious leakage from perifoveal capillaries.103
FENESTRATED SHEEN MACULAR DYSTROPHY
Introduction
Fenestrated sheen macular dystrophy is an autosomal dominant condition with high penetrance. This is a rare disorder and only four affected pedigrees have been reported in the literature.104–106
Clinical Findings
Fenestrated sheen macular dystrophy is a slowly progressive disease that develops in childhood.103–107 The youngest reported case was in a 4-year-old girl. In the early stages, small, red demarcated lesions can be seen within the deep neurosensory retina106 It is characterized by the presence of a yellowish refractile sheen with red fenestrations within the macula (Fig. 7). The sheen, which is located between the retinal pigment epithelium and the retinal vessels, is also present within the foveal avascular zone and may be related to the macular luteal pigment. The fenestrations appear to be tiny windows within the sheen that allow increased visualization of the underlying pigment epithelium. The changes within the sensory retina are slowly progressive and are eventually accompanied by changes of the retinal pigment epithelium. By the third decade, an annular zone of hypopigmentation of the retinal pigment epithelium appears around the area of sheen and progressively enlarges to form a bull's-eye macular lesion (Fig. 8). An abnormal degree of RPE granularity in the posterior pole and the peripheral retina has also been reported in some affected individuals.106 During this period of change, visual acuity usually remains normal, although paracentral scotomas develop by the sixth decade of life. The prognosis for maintenance of good central vision is excellent and no treatment is available. Despite the ophthalmoscopically evident macular changes, central visual acuity remains normal.
|
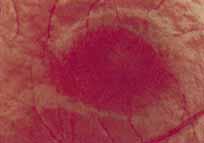
|
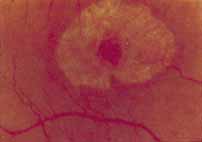
Ancillary Tests
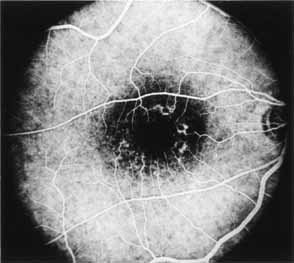
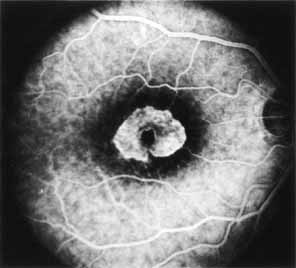
The fluorescein angiographic features of this dystrophy are dependent on chronicity of the disorder. Younger patients have essentially normal studies. Older patients demonstrate multiple punctate window defects in an annular zone ringed by an area of slightly subnormal choroidal fluorescence due to increased pigmentation of the retinal pigment epithelium (Fig. 9). The oldest reported patient, a 56-year-old male, had a large confluent annular transmission defect of the retinal pigment epithelium with normal perfusion of the underlying choriocapillaris (Fig. 10). This individual demonstrated progression of his lesion over an 18-month period.
|
|
Electrophysiologic Tests
Retinal function studies correlate roughly with age. Younger patients have normal ERGs and EOGs and a mild red-green color defect. Older patients have subnormal photopic ERGs and normal-to-subnormal EOGs.
Differential Diagnosis
The small red macular fenestrations in the early course of the disease appear similar to those described in acute macular neuroretinopathy. Patients with that disorder are often symptomatic, however. The differential diagnosis includes diseases with retinal pigment epithelial changes in an annular pattern including Stargardt's disease, progressive cone-rod dystrophy, benign concentric annular macular dystrophy, central areolar pigment epithelial dystrophy, and central areolar choroidal dystrophy.