TABLE 1. Neurofibromatosis Classification/Subtypes
| Major type | Subtype |
| Neurofibromatosis type 1 (NF1) | von Recklinghausen's disease |
| Peripheral neurofibromatosis | |
| Multiple neurofibromatosis | |
| Neurofibromatosis type 2 (NF2) | |
| Bilateral acoustic neurofibromatosis | |
| Central neurofibromatosis22 | |
| Rare forms of Neurofibromatosis | Mosaic/segmental20,22 |
| Familial spinal neurofibromatosis | |
| Familial intestinal neurofibromatosis | |
| Schwannomatosis | |
| Autosomal dominant café au lait spots | |
| Autosomal dominant neurofibromas | |
| Watson syndrome (pulmonary stenosis, café au lait patches) | |
| Noonan/neurofibromatosis syndrome22 |
NEUROFIBROMATOSIS TYPE 1
Epidemiology
Neurofibromatosis type 1 (NF1) accounts for 90% of the neurofibromatoses and shows autosomal dominant inheritance. Its prevalence is 1/3500 to 1/5000.20,21 (Prevalence is an estimate of the frequency of a condition in the general population; incidence is an estimate of the number of new cases over a given period of time, typically 1 year.) Although the penetrance of NF1 is close to 100% by age 5 years, its expressivity even within the same family may be highly variable. (Penetrance is the proportion of individuals with the defective gene who show clinical evidence of the disorder; expressivity is the degree of variability in clinical findings among individuals with similar mutations.) The NF1 gene has a high mutation rate, with up to half of newly diagnosed cases representing de novo mutations.22
Systemic Features
NF1 can involve any organ system.
CUTANEOUS FINDINGS.
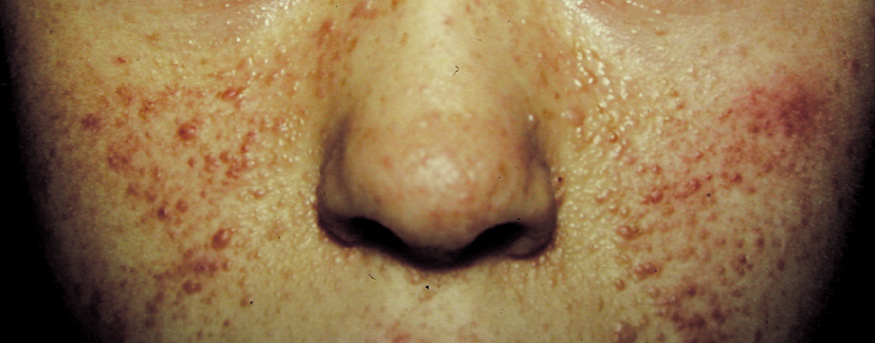
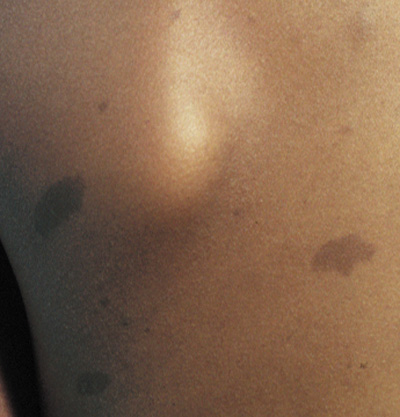
- Café au lait spots: Evenly pigmented flat brown macules that generally appear in the first
year of life, are present in 95% of adults with NF1, and tend
to fade in the elderly.23 One or two café au lait spots are not uncommon in normal patients.24 Overall, 75% of children with six or more café au lait spots
develop other clinical findings of NF1 by 5 years of age (Fig. 1).24 A regular border differentiates café au lait spots in NF1 from
the café au lait spots of McCune Albright syndrome (fibrous
dysplasia and precocious puberty). Café au lait spots may
be an isolated dominant condition in rare instances.25
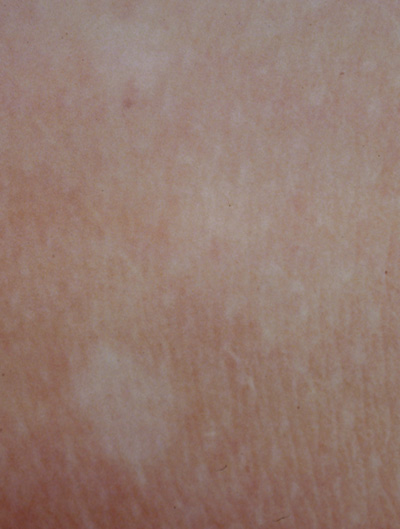
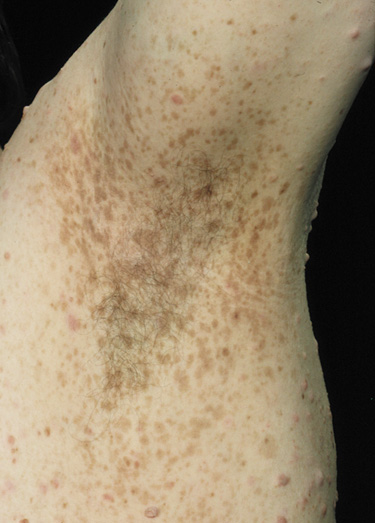
- Freckling in non–sun-exposed areas: the axilla, inguinal region, and the submammary
folds (typically after age 20 years). Freckling may
not have developed by the time café au lait spots appear (first
year of life). Eighty percent to 90% of NFI patients
show axillary or inguinal freckling by 5 years (Fig. 2).25,26
- The skin overlying dermal neurofibromas and plexiform neurofibromas
is often abnormal and will be discussed with nerve-associated tumors below.
|
|
NEUROLOGIC FINDINGS.
- Cutaneous neurofibromas are benign tumors associated with peripheral nerves. There is evidence
for a single-cell origin despite multiple cell types within the tumors, although
the cell of origin is unclear.27,28 Neurofibromas are typically soft and mobile on palpation. The overlying
skin may be dimpled and have a violacious color due to dilated capillaries. Neurofibromas
often enlarge and increase in number during puberty
or pregnancy. In all, 95% of adults over age 30 with NF1 have
neurofibromas, compared with less than half of adolescents and rarely
in infants. Neurofibromas occur predominantly on the trunk—only 20% of
patients have lesions on the head or neck. Less common
subcutaneous peripheral neurofibromas are palpable as firm tumors along
the trunks of peripheral nerves.22,25,29
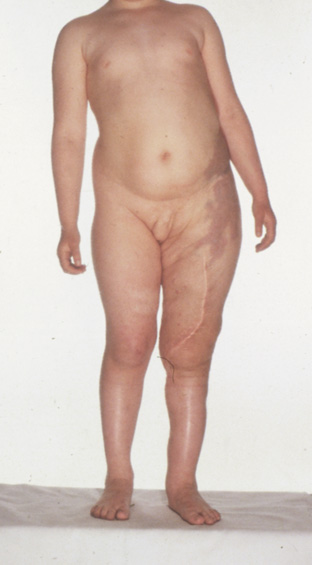
- Plexiform neurofibromas are subcutaneous tumors with ill-defined borders. The overlying the skin
is often hypertrophic, hyperpigmented, and may have excessive hair
growth. They affect 25% to 30% of individuals with NF1, most
commonly on the trunk, less commonly on the limbs, head, and neck. They
may carry about a 5% lifetime risk for malignant transformation (neurofibrosarcoma).25,29,30 Plexiform neurofibromas are a surgical challenge. They do not respect
tissue planes, making complete removal difficult. They regrow when not
fully excised and are not radiosensitive.25
- Brain tumors. Gliomas will be discussed below. Other brain tumors occur infrequently (<1%) in NF1. With the exception of brainstem astrocytomas
and optic gliomas, the outcome of brain tumors in children
affected with NF1 is similar to children without NF1.25 Brainstem and optic gliomas show slower progression in NF1.31 Vestibular schwannomas and spinal meningiomas, known complications of
NF2, are probably no more common in NF1 patients than in the general population.22,25
- Cognitive impairment. Thirty percent to 60% of children with NF1 have learning difficulties, which
are typically mild and nonprogressive. These include visual-spatial
problem-solving difficulty, language disorders, and attention
deficit disorder. The reason for this is not clear but may be related
to unidentified bright objects (UBOs) seen on T2-weighted
brain magnetic resonance imaging (MRI) scans. These hyperintensities
are common in young patients with NF1 but decrease with advancing
age. The histopathologic correlate of UBOs is unclear. It has
been postulated that the prevalence of learning difficulties in children
with NF1 may be related to heterozygosity of the NF1 gene (i.e., the gene may have additional functions that affect cognition when the
full complement of its gene product is not expressed in the central nervous
system).25,32–34
SKELETAL/VISCERAL FINDINGS.
- Bone dysplasia25
- Scoliosis affects a significant number of NF1 patients (10% to 15%) and may be severe enough to require surgical
correction
- Thinning of long bones, especially medial bowing of the tibia, may
predispose to fractures and pseudoarthrosis
- Congenital absence of the greater wing of the Sphenoid bone
- Scoliosis affects a significant number of NF1 patients (10% to 15%) and may be severe enough to require surgical
correction
- Hypertension.22 There is increased incidence of aortic coarctation and pheochromocytomas. Retroperitoneal
neurofibromas may compress the renal artery. These
complications may manifest as hypertension in affected children
Ophthalmic Features
Virtually any part of the visual system may be affected by NF1: the bony orbit, extraocular muscles, orbital nerves, eyelids, conjunctiva, cornea, uvea, retina, optic nerves, chiasm, and optic radiations.
OCULAR FEATURES.
Conjunctiva.
Neurofibromas of the conjunctiva are infrequent, affecting about 2% of patients with NF1, often on the perilimbal conjunctiva.35
Cornea.
Enlarged corneal nerves have been reported in association with NF1 but are more commonly seen with multiple endocrine neoplasia syndrome (MEN-IIb).36,37
Uveal Tract.
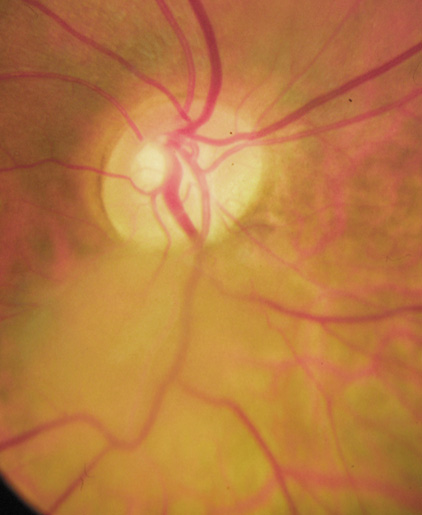
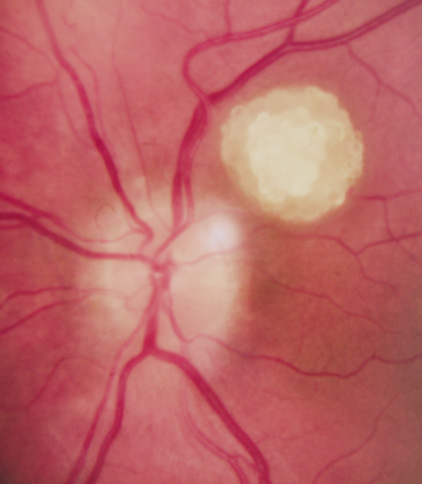
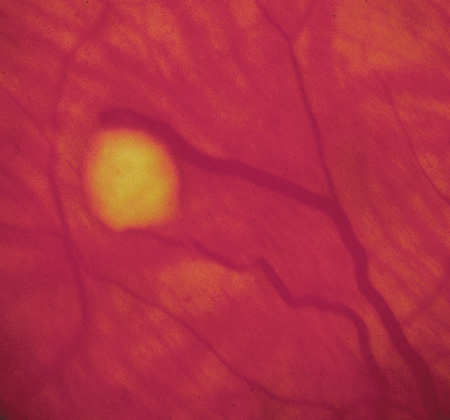
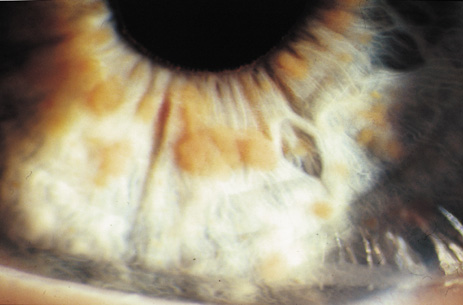
Lisch nodules are hamartomas (a tumor involving only those tissue elements normally found at the involved site) of the iris pigment epithelium. They are dome-shaped discrete lesions, are typically light brown in color, and may also be found in the angle.38 Lisch nodules appear earlier (33% at 2.5 years, 50% at 5 years of age)39 than neurofibromas. They are benign and can help to confirm diagnosis in children who may have café au lait spots as the only other clinical finding. Lisch nodules are present in nearly all adults with NF139,40 but are rare in NF2 (Fig. 3).41,42 Diffuse nodular iris nevi (also known as iris mamillations) should not be confused with Lisch nodules. Its clinical significance is not well established.43,44
|
Like Lisch nodules of the iris, pigment epithelial hamartomas may affect the choroid. In one series, flat pigmented lesions of the choroid were found in 35% of patients with NF1. Fluorescein angiography suggests these are choroidal nevi.45The entire uveal tract may be thickened by a diffuse neurofibroma, thought to be hamartomatous hyperplasia of Schwann cells. This may result in the development of glaucoma.46,47
Retina.
Retinal involvement in NF1 is unusual. Astrocytic hamartomas (similar to those in tuberous sclerosis), retinal capillary hemangiomas, and combined hamartoma of retina and retinal pigment epithelium (RPE) occur.48,49
EYELID AND ORBIT.
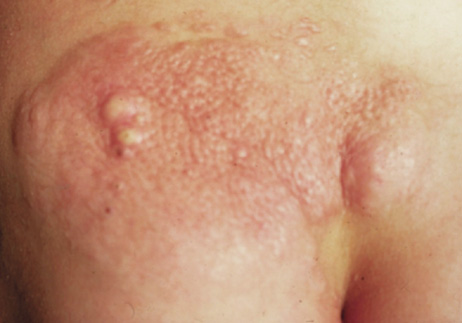
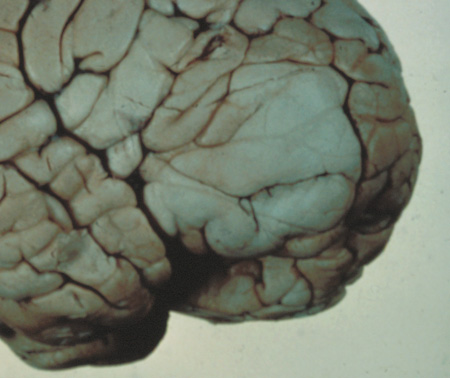
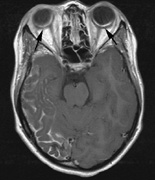
Orbital neurofibromas may arise from any nerve in the orbit, especially the trigeminal nerve.50 Classically the triad of visual loss, optic atrophy and optociliary shunt vessels have been associated with optic nerve sheath meningiomas,51 but these findings may be nonspecific for optic nerve compression,52,53 particularly in children. If a child with a presumed diagnosis of NF1 is found to have an optic nerve meningioma, NF2 should be considered.
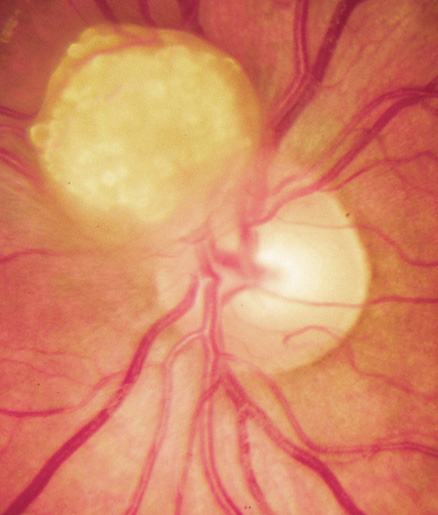
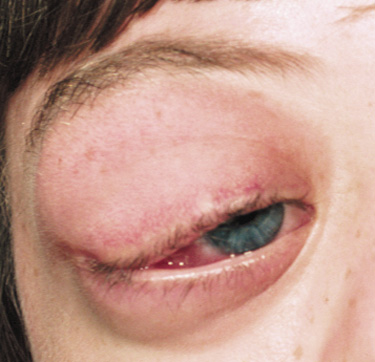
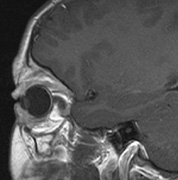
The eyelids are frequently involved in NF1. The lateral portion of the upper lid is prone to develop a plexiform neurofibroma, producing a sinusoidal or S-shaped deformity of the lid margin.54–56 Amblyopia develops from ptosis and astigmatism (Fig. 4).
|
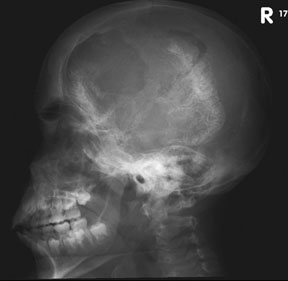
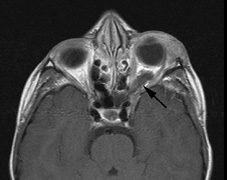
Growth of a plexiform neuroma may be directed posteriorly, infiltrating orbital tissues and involving the bony walls. Congenital plexiform neuromas of the orbit are often associated with absence of the sphenoid wing (Fig. 5). Intracranial pulsation can be transmitted to the orbit causing pulsatile proptosis or enophthalmos.57–59 Plexiform neuromas of the lid and orbit are difficult to treat; complete excision is difficult and potentially disfiguring. The tumors are vascular and may bleed copiously at surgery. They are not radiosensitive and there is also risk of inducing malignant transformation. Trials with antiangiogenic chemotherapeutic agents (thalidomide, interferon alpha) and mitotic-signaling pathway blockers (inhibition of Ras by Farnesyl protein tranferase inhibitor) are in progress.60,61
|
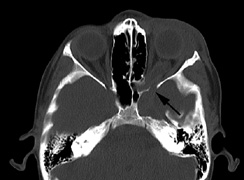
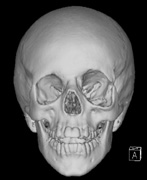
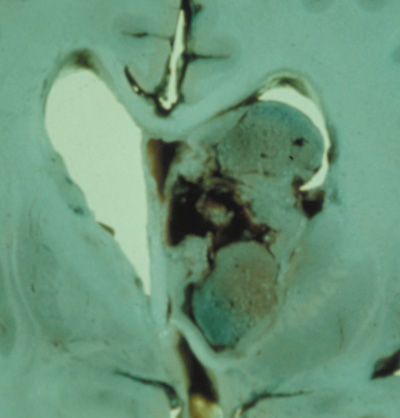
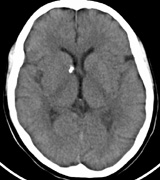
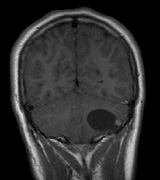
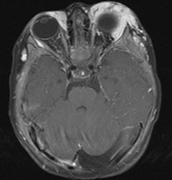
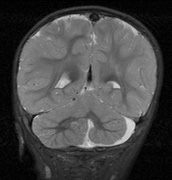
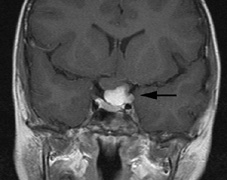
OPTIC NERVE GLIOMAS
Optic pathway gliomas arise from the astrocytes of the optic nerve. Most are pilocytic astrocytomas and typically remain intradural, extending in the subdural space. Malignant transformation is rare. There appear to be two growth patterns62:
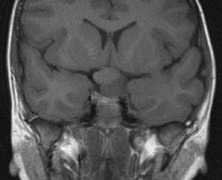
- Perineural growth pattern, correlating with a diagnosis of NF1. The tumor expands in the subarachnoid
space and the optic nerve is compressed as a central ribbon. On
T2-weighted MRI, this may be seen as a low-intensity core with surrounding
high-intensity rim. Increased tortuosity of the optic nerve is also
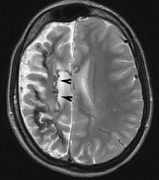
associated with this growth pattern (Fig. 5).63
- Intraneural growth pattern, correlating with the absence of NF1.
The incidence of optic pathway gliomas in NF1 is difficult to estimate, as the majority are asymptomatic and do not affect vision. In NF1, radiographic evidence of an optic nerve glioma occurs in 15% of patients.64,65 Most tumors are neither suspected historically nor detected by ophthalmic examination.65 Even when optic nerve gliomas become clinically detectable, visual function often remains stable in the absence of any intervention.66,67 Thus routine computed tomography (CT) imaging, even in patients with a known optic nerve glioma, is controversial. The potential risks of repeated radiation exposure in a child with a tumor-suppressor gene defect is a consideration. MRI studies are more revealing than CT but may require sedation or anesthetic and are costly. An MRI is often recommended at the time of diagnosis, but in most cases routine “follow-up” MRIs are unnecessary. Annual ophthalmic clinical examination for relative afferent pupillary defect (RAPD), visual acuity, visual fields, color vision, and funduscopy is usually all that is required, as no therapeutic action is likely to be taken unless there is significant and progressive visual involvement.
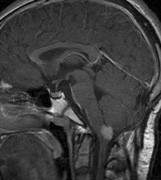
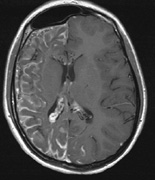
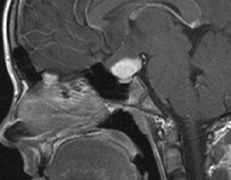
Optic nerve gliomas may involve the optic chiasm and be associated with endocrine disorders or nystagmus.68–70 Surgical excision of chiasmal gliomas (Fig. 6) carries a high risk of visual loss. Invasion of the hypothalamus or the third ventricle carries a poor prognosis, with greater than 50% 15-year mortality rate.71 One review of radiation treatment for chiasmal gliomas collated data from small case series and found no significant long-term improvement in visual function, progression, or mortality with radiation treatment.71 Other reports suggest that radiation doses over 4500cGy improve symptoms and slow progression of chiasmal gliomas over several years.72 Adequate tumor coverage by radiotherapy results in irradiation of normal brain and nearly all children need hormone replacement.73 Chemotherapy is an alternative.74,75
|
Diagnostic Criteria
The NIH has defined 7 criteria,22 at least two of which must be present:
- A first-degree relative with NF1
- At least two Lisch nodules (Fig. 3)
- At least five café au lait spots ≥ 5 mm before puberty or
at least 6 café au lait spots ≥ 15 mm after puberty (Fig. 1)
- Axillary or inguinal freckling (Fig. 2)
- Optic glioma (Fig. 5)
- A “distinctive” bone lesion: sphenoid bone dysplasia or thinning of long bone cortex (with or without pseudoarthrosis)
- At least two neurofibromas of any type or one plexiform neurofibroma
In young children, nearly half of sporadic cases fail to meet NIH criteria by 1 year of age, but meet them by age 8 to 12 years. The clinical features appear in the following order: café au lait spots, axillary freckling, Lisch nodules, neurofibromas.26 The inclusion of UBOs on MRI also aids the diagnosis of very young children25 but may require a general anesthetic.
Genetics and Screening
All family members of patients with NF1 should be screened. Lisch nodules are present in 90% or more of patients with the disease, so ophthalmic examination is essential. Annual ophthalmic examination with visual acuity, visual field, color vision, and fundoscopy is recommended for affected individuals.
The NF1 gene (17q11.2) was cloned in 1990.76,77 Its protein, neurofibromin, is expressed throughout the body but particularly in the central and peripheral nervous system.12 It is a tumor-suppressor gene with autosomal dominant inheritance, high penetrance, and variable expressivity. NF1 has been called a neurocristopathy, but the disease is not limited to neural crest derived tissues.12,78
Neurofibromin has structural similarity to GTPase, activating proteins that inhibit the function of G proteins such as Ras. Inhibition of Ras by neurofibromin has been shown in vitro and in vivo. NF1-deficient tumors show increased levels of Ras-GTP. Increased Ras function has been shown in a number of human tumors.12,79–81
NEUROFIBROMATOSIS TYPE 2
Neurofibromatosis type 2 (NF2) was described in 1822 by the Scottish surgeon Wishart.82 Initially, NF1 and NF2 were lumped together despite their differences.83 The identification of different genes re-established them as separate entities.84
Epidemiology
The NIH consensus conference22 estimated an incidence of 1:50,000 but there are varying estimates internationally.84 NF2 is autosomal dominant with full penetrance by age 60.85 In all, 10% of affected patients are symptomatic by age 10, 50% by 20 years, and 80% by 30 years.86 Some sporadic cases reflect parental mosaicism.87,88
Systemic Features
Several studies have established the clinical picture of NF2.85,89–91 Patients may become severely disabled because of the combination of deafness and imbalance from vestibular schwannomas, weakness from multiple spinal tumors, and poor sight from cataracts, optic nerve meningiomas, and retinal hamartomas.
CUTANEOUS FINDINGS.
Skin findings in NF2 may be subtle but not uncommon. Skin tumors were found in 60% and were the first presenting sign in more than 25% of patients.92 The skin tumors are predominantly schwannomas. A minority are neurofibromas and mixed types. Clinically differentiating the histological types may be difficult.92 They may present as dermal tumors or subcutaneous nodules of peripheral nerves.
NF2 plaques are well circumscribed, slightly raised, often pigmented, and hairy. They are a sensitive marker for NF222 and are more common on the limbs and trunk than the neck and face.92
Café au lait spots are not uncommon in NF2, but it is unusual to find more than five and most tend to be restricted to the trunk. Plexiform neurofibromas and axillary/inguinal freckling are uncommon.92,93
NEUROLOGIC FINDINGS.
- Schwannomas most commonly affect the fifth cranial nerve (CN), but
any cranial nerve may be affected. In contrast to vestibular
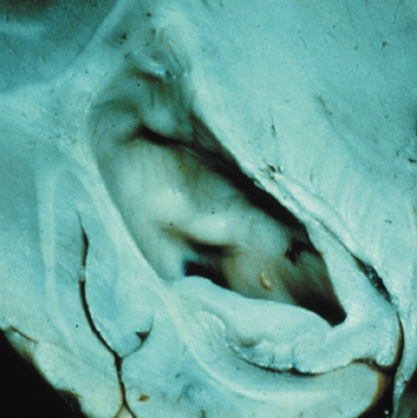
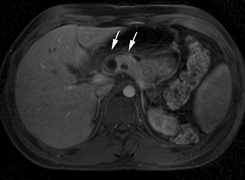
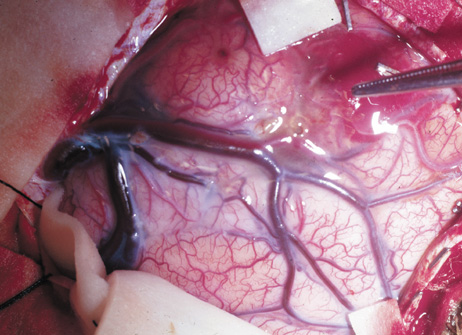
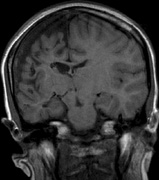
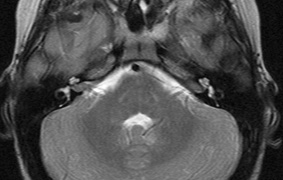
schwannomas (CN VIII), these typically do not grow large.84,90 Vestibular schwannomas (acoustic neuromas) are the classic finding
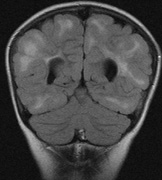
in NF2 (Fig. 7). The risk for malignant transformation is low, but may be higher
with radiation exposure.84,94
- Meningiomas are most likely to affect the spinal cord and supratentorial
part of the cranium.84
- Other tumors: low-grade ependymomas and gliomas may affect the cervical
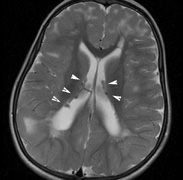
spine and Brainstem (Fig. 7).84
- Neuropathy: A generalized polyneuropathy affects 3% to 5% of
affected adults and is associated with an “onion-bulb” appearance
on nerve biopsy. In children, mononeuropathy, particularly
the facial nerve (CN VII), may be an early sign of NF2. Some
children may present with a poliomyelitis-like illness with lower-limb
muscle wasting.86,95,96
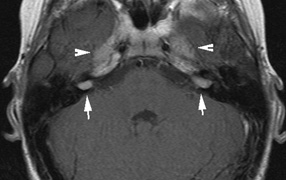
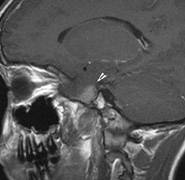
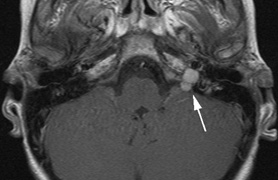
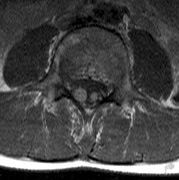
VISCERAL FINDINGS.
NF2 does not appear to have significant visceral complications
Ophthalmic Features
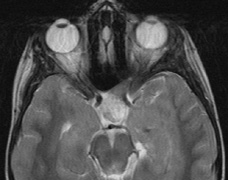
The most common ocular findings in NF2 include premature cataracts and retinal hamartomas. Ocular motor nerve deficits may also be associated with NF2.
Cataracts in NF2 were recognized in 1986.97 They are common, reported to be found in 69%,98 81%,91 85%,99 and 87%100of cases. They are typically mild98 and a lens opacity may be the initial manifestation of disease, as found in 10% (5 of 49 patients) in one study.98
Retinal hamartomas affect about 10% to 20% of patients (8% in one study91and 22% in another98) and may be associated with a more severe phenotype of NF2.91,98 Combined pigment epithelial and retinal hamartomas (CPERH) and epiretinal membranes have been reported.101–103 Lisch nodules are rare in NF2. The absence of Lisch nodules, in association with the presence of posterior subcapsular or cortical cataracts (Fig. 8) may aid the differentiation of NF1 and NF2.97,104,105 Optic nerve sheath meningiomas may cause significant visual impairment in the first years of life (Table 2).84,106
|
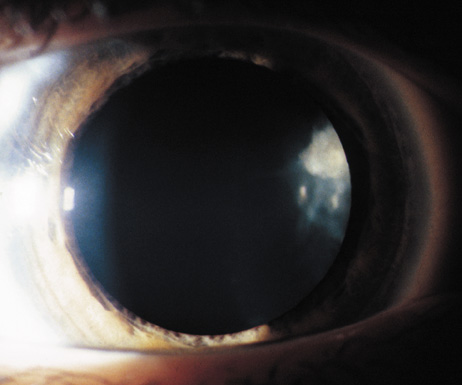
TABLE 2. Ocular Findings in Neurofibromatosis Type 298
Lens opacities (most common finding)
Posterior subcapsular cataract
Cortical wedge opacities
Lisch nodules (rarely)
Optic nerve sheath meningiomas
Retinal hamartomas
Combined pigment epithelial and retinal hamartomas
Epiretinal membranes
Optic disc gliomas
Motor nerve palsies
Diagnostic Criteria
The NIH scheme is most commonly used:95,107
- Bilateral eighth nerve schwannoma, or
- Positive family history of NF2, plus either unilateral eighth nerve schwannoma or two of the following:
- Neurofibroma
- Meningioma
- Glioma
- Schwannoma affecting other nerve(s)
- Juvenile lens opacity
- Neurofibroma
A significant proportion of cases (20% to 30%) may present with intracranial meningioma, spinal tumors, or cutaneous tumors. Vestibular schwannomas may account for as little as 15% to 30% of presenting signs in children. Thus, expanded criteria have been proposed to include individuals with multiple schwannomas/meningiomas who have not developed vestibular schwannomas as yet and do not have a clear family history.84
Genetics and Screening
Cataracts can affect vision in early life. For children at risk, annual ophthalmic examination is prudent. Central nervous system (CNS) tumors rarely become symptomatic before age 10 years. Vestibular schwannomas tend to grow faster in younger patients; therefore, asymptomatic at-risk individuals may benefit from MRI scanning every 2 years under age 20 and every 3 years over age 20. Epiretinal membranes and combined hamartomas should lead to investigation for NF2. An annual neurologic exam has also been recommended.84
The NF2 tumor-suppressor gene product is known as schwanomin108 or merlin109. The gene is located at 22q11.1–22q13.1 (Online Mendelian Inheritance in Man [OMIM] #607379).110 The NF2 gene shows homology to the highly conserved ezrin-radixin-moesin (ERM) family of membrane-cytoskeleton linking proteins. The NF2 protein is believed to interact with the actin cytoskeleton. Disruption of NF2 function has been linked to loss of cell-to-cell contact inhibition of growth and division.14