REGIONAL CHOROIDAL ATROPHIES
Regional Choriocapillaris Atrophies
CENTRAL AREOLAR CHOROIDAL DYSTROPHY (CENTRAL AREOLAR CHOROIDAL SCLEROSIS).
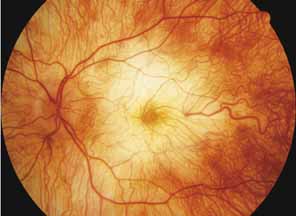
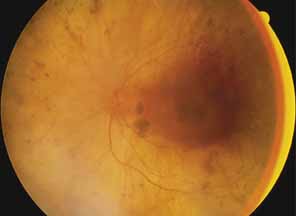
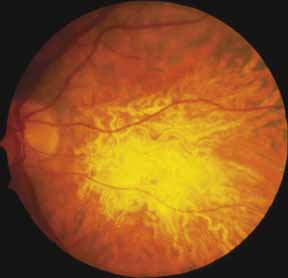
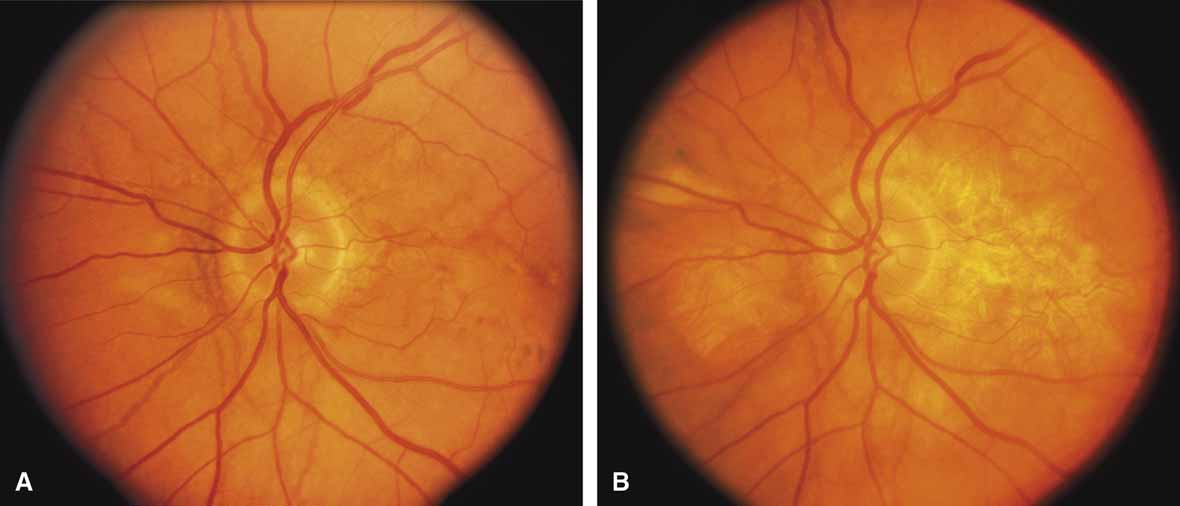
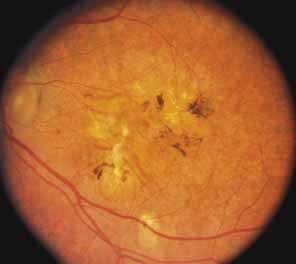
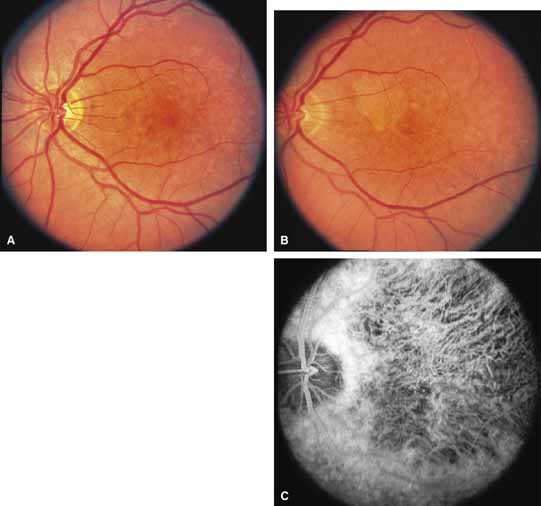
First described by Nettleship12 in 1884, central areolar choroidal dystrophy (CACD) (MIM No. 215500, Phenotype catalog number (MIM) from McKusick VA: Mendelian Inheritance in Man. Catalogs of Human Genes and Genetic Disorders. 12th ed. Baltimore: Johns Hopkins University Press, 1998 [http://www.ncbi.nlm.nih.gov/Omim/]), as it is most appropriately called,13 has been described under many names, including central areolar choroidal sclerosis, central areolar choroidal atrophy, central angiosclerosis, and central senile choroiditis.14,15 The disorder can be autosomal dominant or autosomal recessive, and phenocopies can occur from many other diseases, including mutations of peripherin/RDS16–19 and advanced stages of macular dystrophies (see later text). A locus for CACD has been identified on chromosome 17p.20 The earliest symptoms result from pericentral scotomas and include difficulty reading, poor dark adaptation, reduced visual acuity, and glare sensitivity. The earliest fundus findings are subtle and include pigment epithelial and choriocapillaris lesions in the macula (Fig. 1A) that enlarge and eventually form the punched-out central atrophic lesions typical of this disease (Fig. 1B). Histopathology shows fibrotic scarring with absence of choriocapillaris, retinal pigment epithelium, and overlying photoreceptors in the affected areas.21 The Ganzfeld electroretinogram is usually normal early in the course but may become mildly to moderately abnormal for cone and rod responses late in the course of disease when extensive atrophy of the choroid and secondarily the pigment epithelium and neurosensory retina occurs. Recent studies using the multifocal ERG have indicated that the abnormality of retinal function extends beyond the borders of the visible atrophy and is consistent with presynaptic photoreceptor dysfunction.22 The EOG can be normal or mildly abnormal depending on the extent of associated retinal pigment epithelial dysfunction.
|
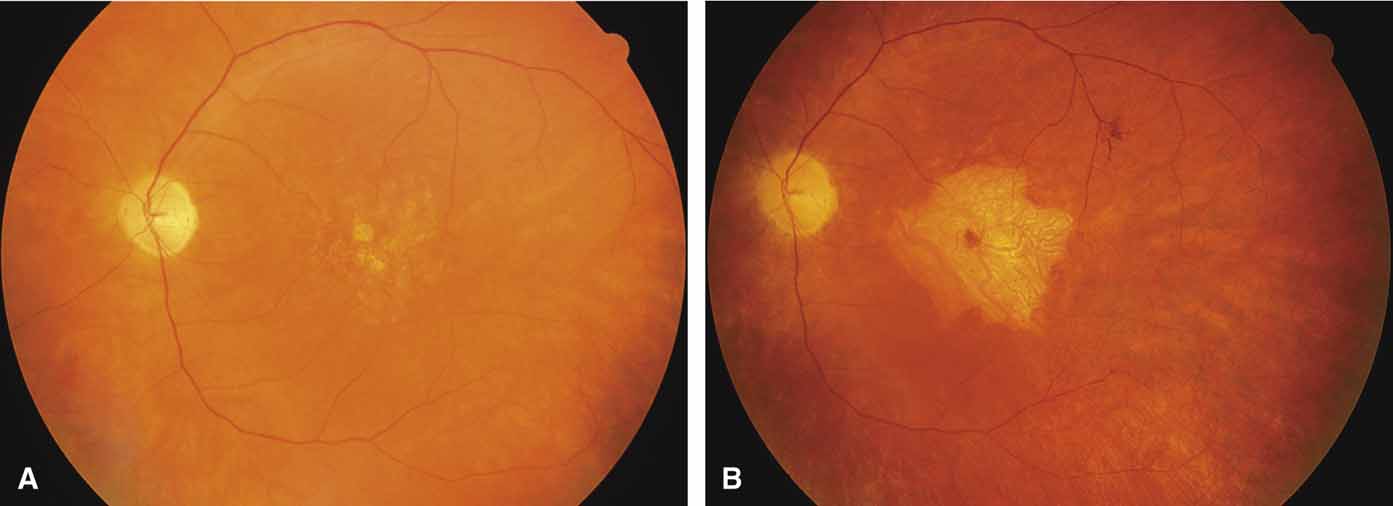
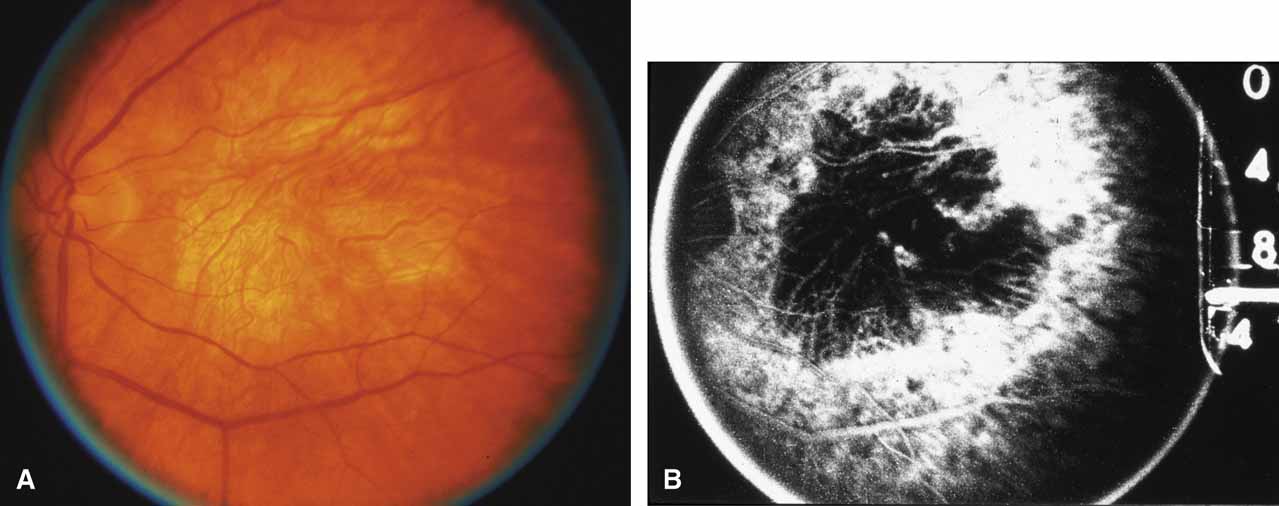
Other forms of central choroidal dystrophy exist that do not show the discrete oval lesions of typical central areolar choroidal sclerosis. These forms of central choroidal choriocapillaris atrophy often present with progressive pigment epithelial mottling and patchy choriocapillaris atrophy initially limited to the macula (Figs. 2A and 2B). With time, the atrophy enlarges and eventually encompasses the entire posterior pole (Fig. 2C). For this form of central choroidal atrophy, a gradual transition usually occurs from atrophic central pigment epithelium and choriocapillaris to essentially normal retina and choroid in the peripheral fundus.
|
BIETTI'S CRYSTALLINE DYSTROPHY.
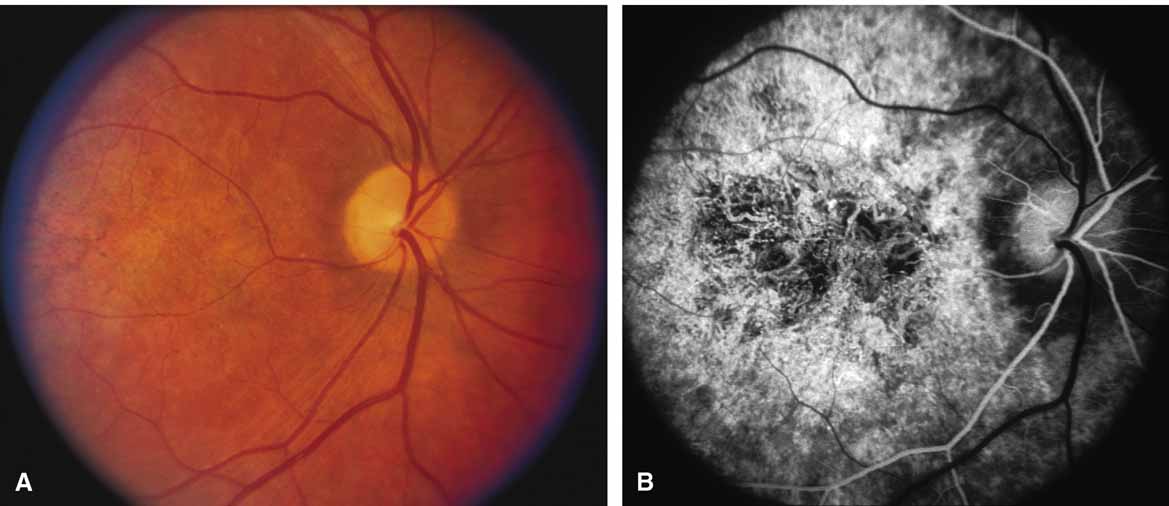
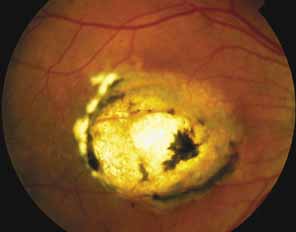
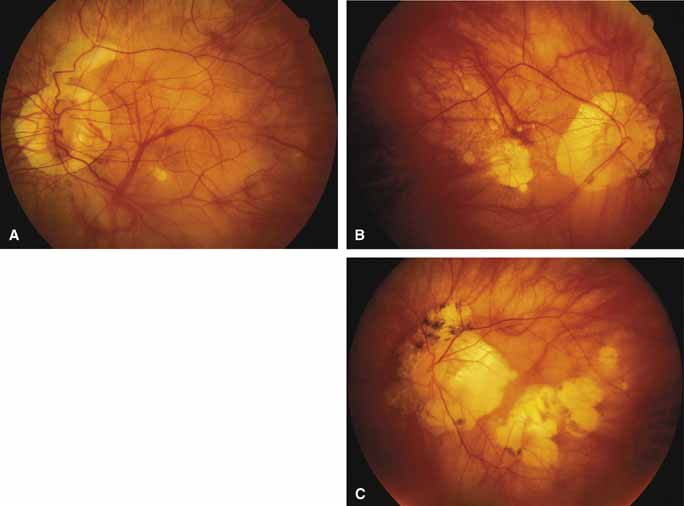
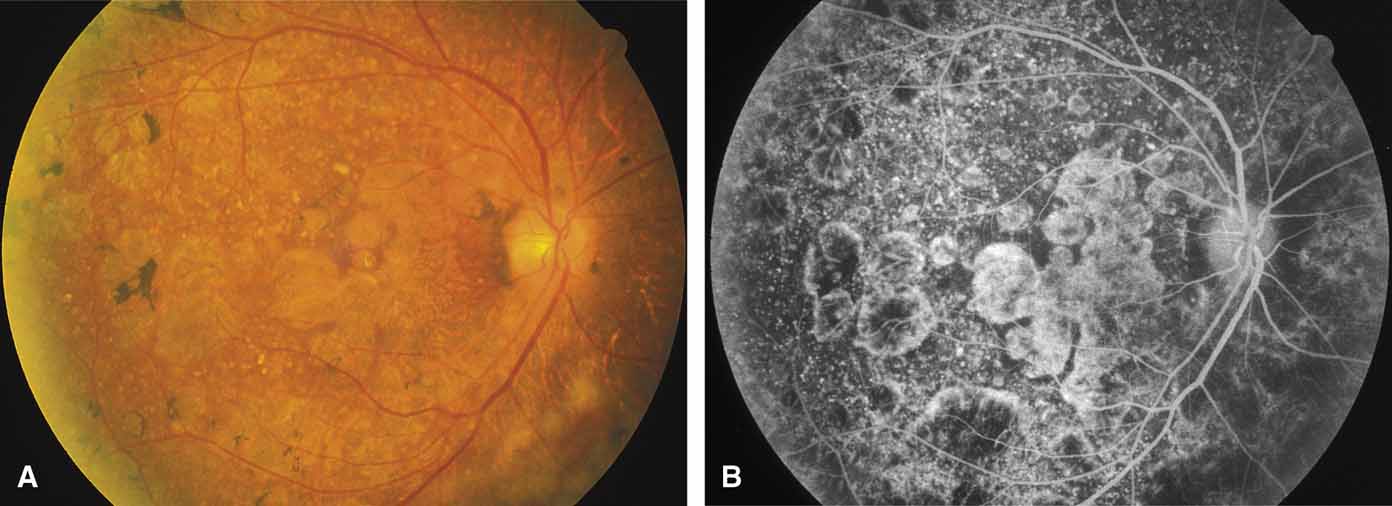
Bietti's crystalline dystrophy (MIM No. 210370) of the cornea and retina is an autosomal recessive disorder that is characterized by the presence of crystals of unknown composition in the stroma of the peripheral cornea and at several layers of the retina (Figs. 3A and 3B).23 The disease can be subdivided into regional and diffuse forms, and the lack of any reports of the two patterns in the same family suggests genetic heterogeneity and not just variable expressivity.24,25 The regional form begins in midlife as pericentral scotomas that cause difficulty reading and reduced central visual acuity. Peripheral retinal function is retained and the electroretinogram and electrooculogram are normal or near normal even in moderately advanced disease. The fundus appearance and fluorescein angiogram reveal regional loss of pigment epithelium and choriocapillaris limited to the posterior pole (Figs. 3C and 3D). The finding of abnormal crystals in leukocytes indicates that this is a systemic metabolic disorder.24
|
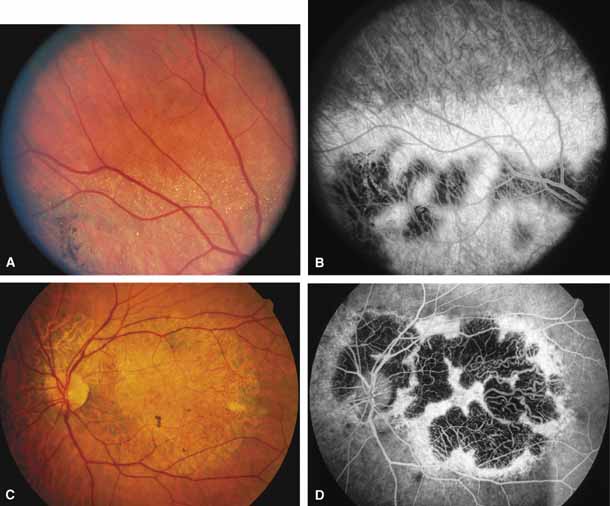
The diffuse form presents with loss of peripheral visual field, symptoms of poor night vision as well as reduced visual acuity, and diffuse loss of pigment epithelium and choriocapillaris on funduscopy (Fig. 4) and on fluorescein angiogram. The ERG is profoundly abnormal early in the course of the disease, and visual impairment eventually becomes much more severe in the diffuse than in the regional form of the disease.
|
|
REGIONAL TOTAL VASCULAR CHOROIDAL ATROPHIES.
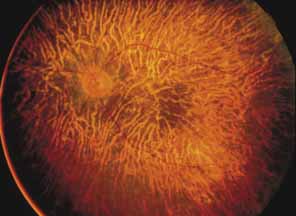
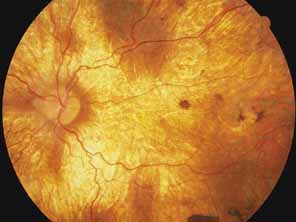
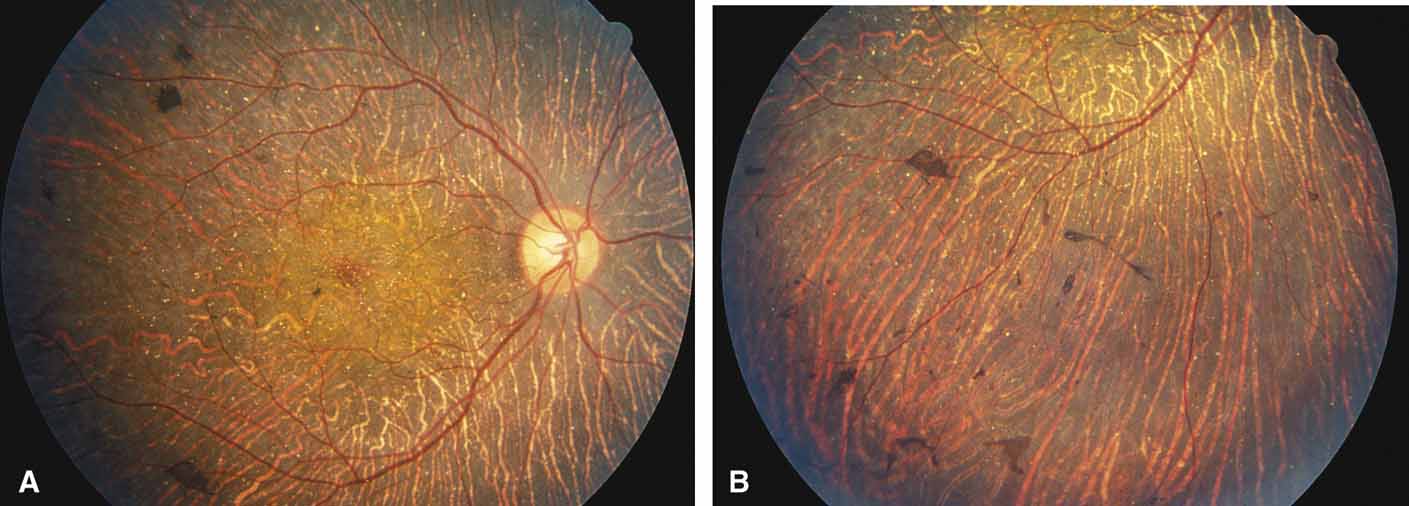
Helicoidal peripapillary chorioretinal degeneration (also called atrophia areata and peripapillary chorioretinal degeneration, Iceland type). First described in Iceland by Sveinsson26 in 1939 as choroiditis areata and renamed as helicoidal peripapillary chorioretinal degeneration (MIM No. 108985),27,28 this autosomal dominant disorder is characterized by peripapillary tongue-shaped patches of total vascular choroidal atrophy that extend radially away from the disc. Signs of edema or inflammation have not been described, distinguishing this disorder from serpiginous choroidopathy. Brazitikos and Safran29 believe that the peculiar fundus lesions are the result of tearing and retraction of the retinal pigment epithelium, and possibly Bruch's membrane, followed by atrophy of the choroid and retina. The areas of atrophy bear no relation to the pattern of the retinal vessels. The ERG suggests focal rather than diffuse retinal dysfunction initially at the level of the RPE and later affecting the sensory retina.30 Magnússon,31 who used the term atrophia areata, studied a large pedigree of 26 affected individuals in whom the disorder was inherited as an autosomal dominant trait (Fig. 5). Myopia and astigmatism were usually present, and the visual acuity was extremely poor in later years. Occasionally, the disorder has been called central gyrate atrophy, but this name should be discouraged because of the confusion with gyrate atrophy of the choroid and retina with hyperornithinemia. Serum amino acid levels are normal in helicoidal peripapillary chorioretinal degeneration.31
|
This clearly dominantly inherited disorder should not be confused with the nonfamilial condition called serpiginous choroidopathy, which has unfortunately been reported in the past (and in some reviews is still classified) under the name geographic helicoid peripapillary choroidopathy.32
PROGRESSIVE BIFOCAL CHORIORETINAL ATROPHY.
Progressive bifocal chorioretinal atrophy is a rare autosomal dominant disorder mapped to chromosome 6q33,34 that was first described by Douglas, Waheed, and Wyse35 in a large Scottish family in 1968. Thirty-three of 91 family members were affected. The authors classified the disease into three stages (Fig. 6). Stage 1, which lasts from birth to age 14 years, involves a temporal focus of atrophy of retinal and choroidal tissue in the macula that continues to slowly enlarge with time. The upper, nasal, and lower edges of the temporal lesion are well defined, whereas the temporal edge is serrated and indistinct. Stage 2, which lasts from age 15 years to age 45 years, is associated with a second focus of atrophy nasal to the disc, hence the term bifocal in the name. At the end of this stage, the nasal focus is about three disc diameters in size. Stage 3, which occurs after 45 years of age, is associated with further expansion of the temporal and, especially, the nasal focus, leaving only a vertically oriented one- to two-disc-diameter swath or band of intact retina and choroid extending from the disc to each equator superiorly and inferiorly. Visual acuity was markedly subnormal compared with early years, but no patients lost all vision. All patients had coarse nystagmoid eye movements that precluded measurement of visual fields. Most patients were myopic. The ERG b-wave amplitude was subnormal but not unrecordable by a non-Ganzfeld single-flash technique.
|
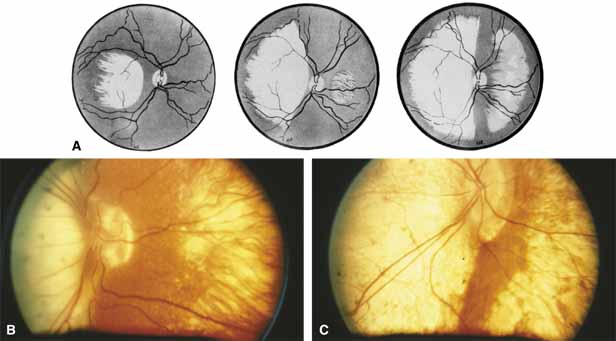
SORSBY'S FUNDUS DYSTROPHY.
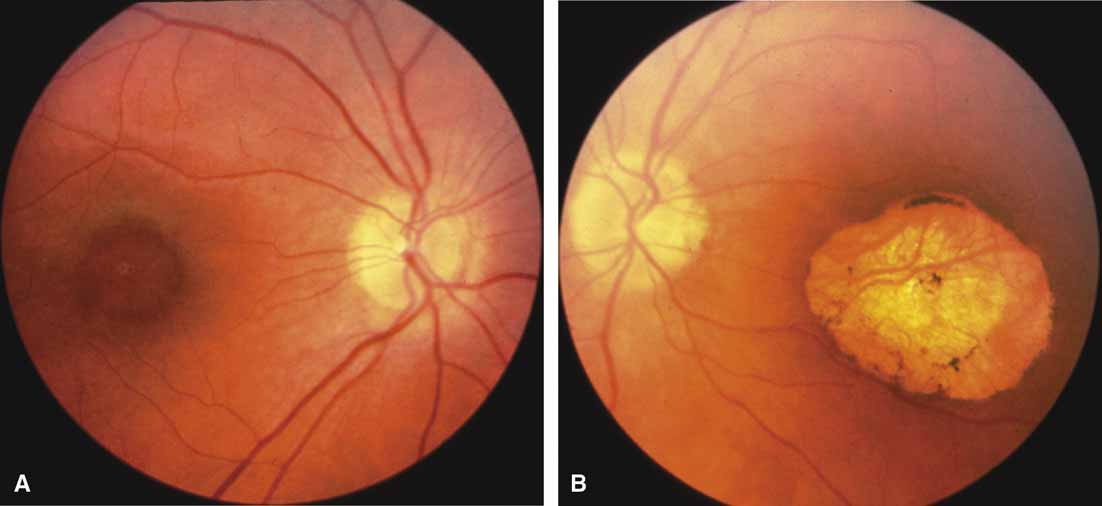
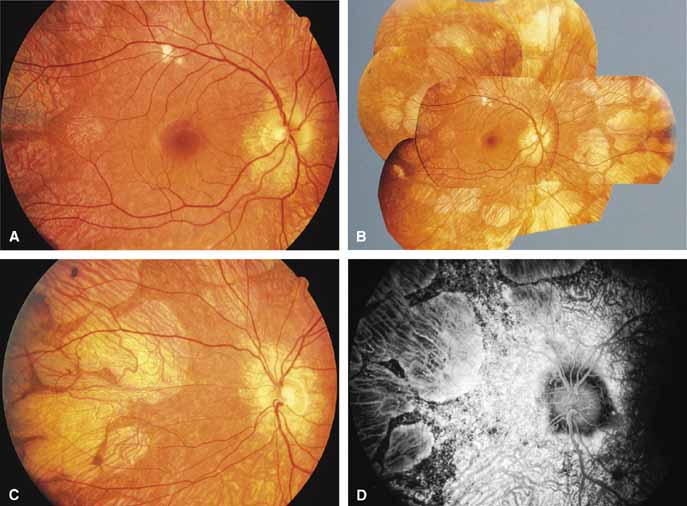
Sorsby's fundus dystrophy (MIM No. 136900), previously called Sorsby's pseudoinflammatory macular dystrophy, is a highly penetrant, autosomal dominantly inherited disorder characterized by a tritan color vision defect, drusen-like subretinal deposits, and pigment epithelial atrophy in younger individuals followed by choroidal neovascularization, hemorrhage, subretinal fibrosis, and choroidal atrophy in later years (Fig. 7).14,36–38 Loss of vision usually commences after the age of 50 years. The disease in late stages progresses to involve the peripheral retina. More recent studies of members of Sorsby's original family demonstrated angioid streaks and yellow plaque-like subretinal deposits, features distinguishing this disorder from dominant drusen.39,40 Visual prognosis is poor because of the tendency of the disease process to involve the macula. The ERG is generally normal except in the most advanced stages when extensive areas of the retina are involved.41 Linkage studies assigned the gene for Sorsby's dystrophy to 22q13-qter.42 Weber et al.43 found mutations in the gene for the tissue inhibitor of metalloproteinase-3 (TIMP3), which is located on the long arm of chromosome 22, in patients with Sorsby's fundus dystrophy. This finding suggests that a defect in maintenance and renewal of Bruch's membrane by the altered gene product is an important underlying pathologic event in Sorsby's fundus dystrophy.
|
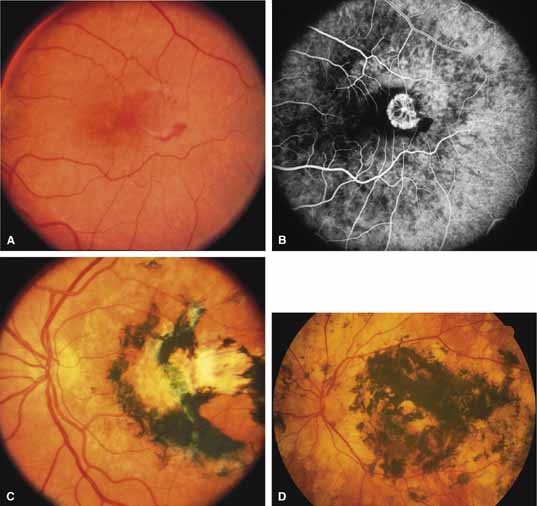
SERPIGINOUS CHOROIDOPATHY.
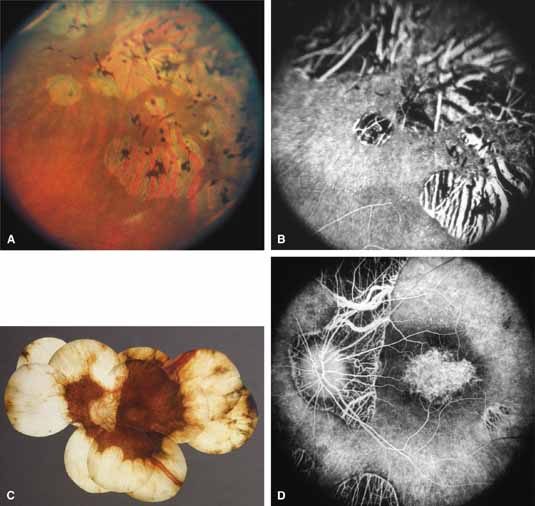
Serpiginous choroidopathy, also called serpiginous choroiditis, is a recurrent, progressive, destructive chronic degeneration of the choroid and retinal pigment epithelium that begins around the optic nerve and extends through the posterior pole.44 Unfortunately, some cases of this disorder have been reported under the name geographic helicoid peripapillary choroidopathy, and this has led to confusion with the clearly dominantly inherited genetic disorder first described in Iceland by Sveinsson26 in 1939 as choroiditis areata and subsequently renamed helicoidal peripapillary chorioretinal degeneration.27,28 The etiology of serpiginous choroidopathy is unknown, but the disease is not thought to be genetic. The disease starts as a gray, cream, or greenish discoloration and edema of the retinal pigment epithelium, followed by extension of the lesion, usually away from the disc, in a stepwise fashion (Figs. 8A and 8B). Vitritis is present in one-third of cases. Periods of quiescence or activity can be separated by months to years. Subretinal fibrous scarring, atrophy of the choroid, and hyperpigmentation in adjacent tissues can be prominent features (Figs. 8C and 8D). The prognosis for retention of central vision is poor because the disease process will often involve the macula. Systemic immunosuppression may be effective in prolonging remission and improving the visual outcome.45
|
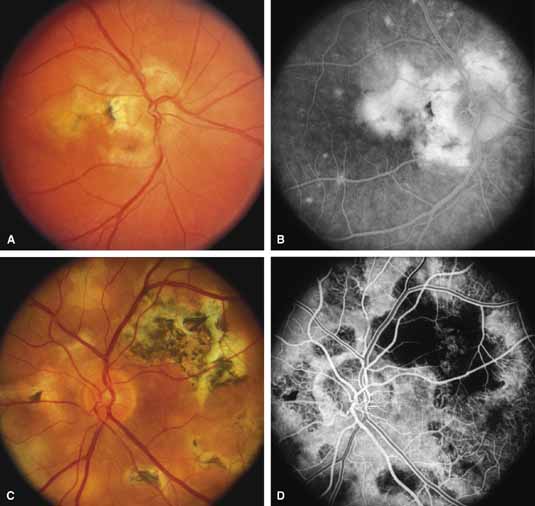
Although originally described as separate disorders, serpiginous choroidopathy and geographic choroidopathy are believed by most authors to be variations of a single disease process.32,46–48 Thus, there appear to be two distinct conditions only—serpiginous choroidopathy, which is an acquired nongenetic disorder, and helicoidal peripapillary chorioretinal degeneration (MIM No. 108985), which is a dominantly inherited disorder.
CENTRAL GYRATE ATROPHY.
Some clinicians have used the term central gyrate atrophy as a label for the fundus appearance of marked total vascular choroidal atrophy in the posterior pole. Central gyrate atrophy appears to not be a distinct entity but probably represents the atrophic changes that can occur in the posterior pole in advanced malignant myopic degeneration, the end stage of serpiginous choroidopathy, or the end stage of helicoidal peripapillary chorioretinal degeneration. Use of the term central gyrate atrophy for such fundus appearances should be avoided because it leads to confusion with the metabolic disorder gyrate atrophy with hyperornithinemia.
DIFFUSE CHOROIDAL ATROPHIES
Diffuse choriocapillaris atrophies
DIFFUSE CHORIOCAPILLARIS ATROPHY.
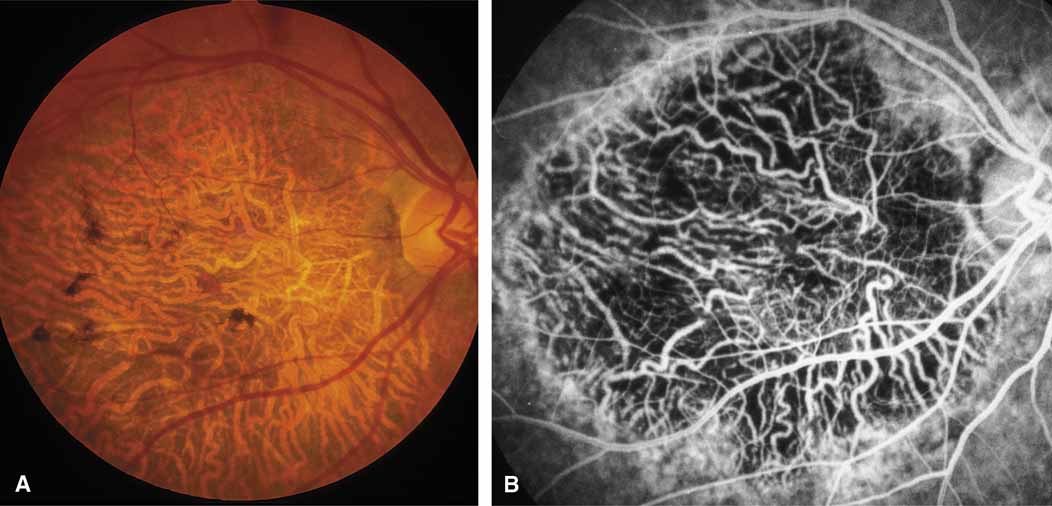
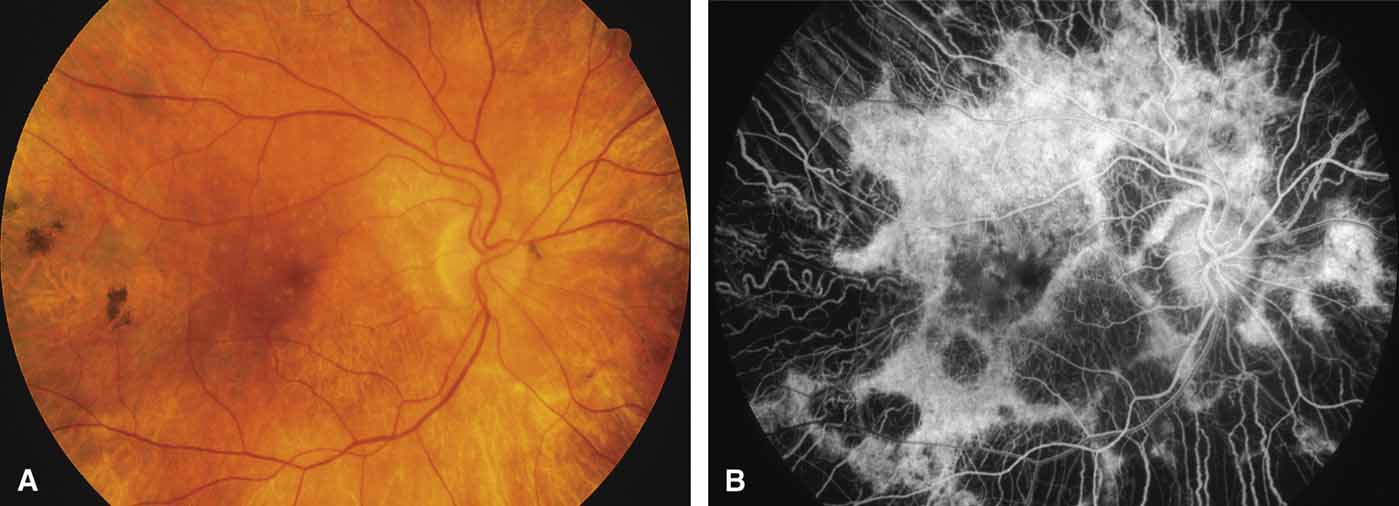
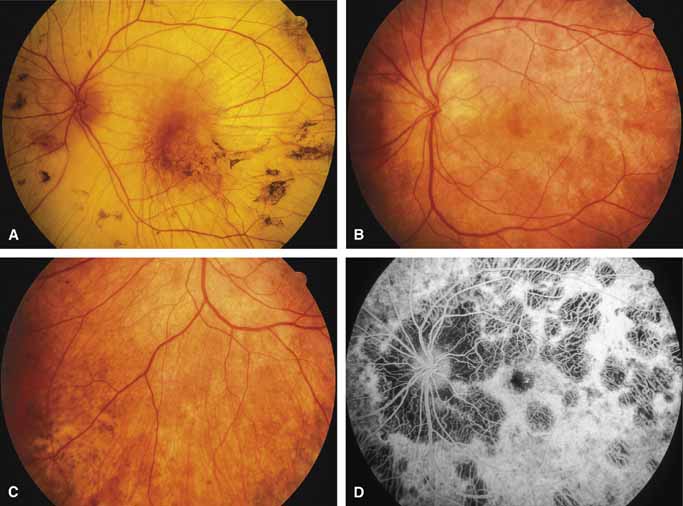
Diffuse choriocapillaris atrophy, also called diffuse choroidal sclerosis and diffuse choroidal angiosclerosis, can be inherited either as an autosomal dominant or, less commonly, as a recessive trait. Certain mutations of the peripherin/RDS gene have also been associated with diffuse choriocapillaris atrophy.18 The disorder is characterized by progressive thinning and loss of choriocapillaris beginning in midlife and resulting in severe loss of vision by later years (Fig. 9). Although the disease in most families is quite consistent among affected individuals, both diffuse and regional central choriocapillaris atrophy have been reported in the same family, suggesting that these two disorders are interrelated.11 Symptoms may include night blindness, loss of central vision, and loss of peripheral vision—the latter feature distinguishing this disorder from the regional form of central choriocapillaris atrophy. Fluorescein angiograms show thinned or absent choriocapillaris with prominent medium and large choroidal vessels throughout the retina. Eventually, mild pigment dispersion and clumping occur in the peripheral retina as the retinal pigment epithelium and overlying retina become affected, but the picture is still dominated by the atrophy of the choroid. The electroretinogram, except in the earliest stages, is usually severely abnormal and eventually becomes unrecordable. The EOG is abnormal early in the disease.
|
|
DIFFUSE TOTAL VASCULAR CHOROIDAL ATROPHIES.
Gyrate atrophy of the choroid and retina with hyperornithinemia from ornithine aminotransferase deficiency. Gyrate atrophy of the choroid and retina (MIM No. 258870) is an autosomal recessive disease that is associated with a 10- to 20-fold elevation of plasma and tissue levels of ornithine.49–52 Vitamin B6 responsive and nonresponsive forms exist. Although the disease is found worldwide, the greatest number of patients, nearly 50% of those reported (all B6 nonresponsive), are of Finnish descent.52 In the first or second decade of life, patients experience night blindness and begin to develop irregular round areas of total vascular choroidal atrophy (Fig. 10A).50 These lesions enlarge and coalesce with time, forming extensive atrophy in the periphery that is associated with constriction of the peripheral visual field (Figs. 10B and 10C). The classic appearance of the fundus is that of a sharp transition from more normal retina to nearly complete atrophy of the choroid and retina (Figs. 11A and 11B).50,53 Central vision may be lost from cystoid macular edema, epiretinal proliferation, or macular involvement in the atrophic process (Figs. 11C and 11D).50 Many patients will develop clinically significant posterior subcapsular cataracts. The ERG and EOG are markedly abnormal from early in the course of the disease.54
|
|
The underlying defect of gyrate atrophy involves ornithine aminotransferase (OAT), which is a mitochondrial matrix enzyme that is pyridoxal phosphate dependent. The OAT gene is located at chromosome 10q26. Although the majority of patients have the form of gyrate atrophy that does not respond to vitamin B6 or pyridoxine, some individuals (less than 5%) show partial vitamin B6 responsiveness. This is shown in vitro by an increase in enzyme activity when assayed with additional pyridoxine and in vivo with a 50% reduction of plasma ornithine levels with oral vitamin B6 supplementation.55,56 Histologic abnormalities occur also in muscle (subsarcolemmal deposits in type II muscle fibers) and other tissues (abnormal-appearing mitochondria in liver and iris).57–59 However, the predominant pathology and clinical symptoms occur in the choroid and retina (see elsewhere in these volumes).50,52 The exact mechanism of pathology for the ocular lesions in gyrate atrophy is unknown, but the three major theories are that, secondary to the OAT deficiency: (1) intracellular proline in the retina may be deficient; (2) creatine phosphate stores in the retina and choroid may be deficient, leading to cell dysfunction and cell death; and/or (3) elevated levels of ornithine may be toxic to the retinal pigment epithelium.60 The abnormal muscle inclusions appear to be the result of deficiency of creatine phosphate, but the role of creatine phosphate as an energy store in the eye is currently undefined.
The molecular basis of ornithine aminotransferase deficiency has been defined for many patients with gyrate atrophy.61–68 Two mutations of the OAT gene (Leu402-Pro and Arg180Thr) account for nearly all of the Finnish patients with nonpyridoxine-responsive gyrate atrophy.66 Loss of gene function occurs from deletions, insertions, splice site base-pair changes, and missense mutations. The rare pyridoxine-responsive patients with gyrate atrophy are homozygous for the Glu318SLys mutation or heterozygous for either the Val332Met mutation or the Glu318Lys mutation of the OAT gene, with the other allele inactivated by another mutation that may or may not be pyridoxine responsive.62,63,69
CHOROIDEREMIA (CHM)—TAPETOCHOROIDAL DYSTROPHY (TCD).
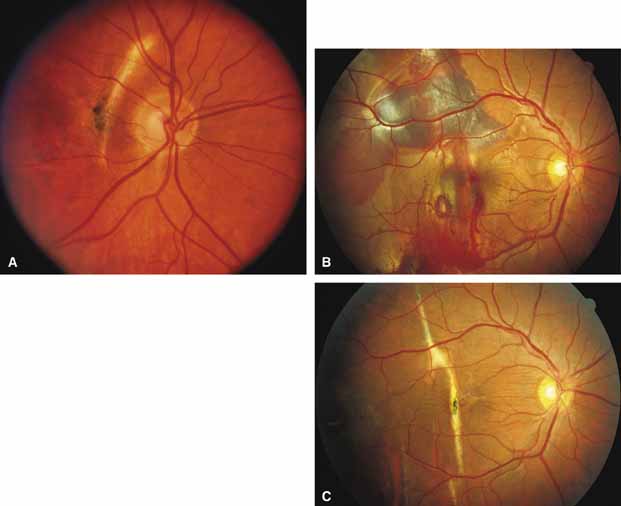
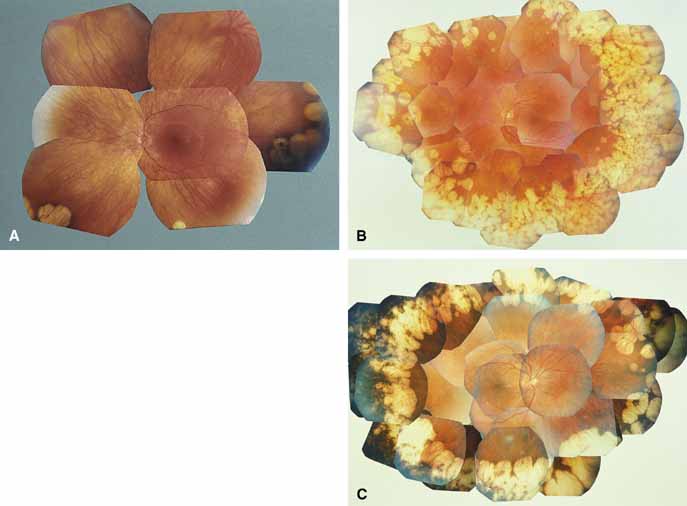
Choroideremia (gene symbol CHM; MIM No. 303100) is an X-linked disorder that is characterized by the onset of night blindness in the first or second decade of life followed by slowly progressive atrophy of the choroid and retina that usually results in legal blindness by midlife and near virtual blindness in later years. The disorder begins with a diffuse atrophic process involving retinal pigment epithelium and choriocapillaris (Figs. 12A, 12B, 12C, and 12D) but eventually results in near total vascular choroidal atrophy (Fig. 13A). The fundus does not show the sharp border or transition area that is characteristically seen with gyrate atrophy. The peripheral visual field is depressed and eventually becomes severely constricted. The ERG and EOG are abnormal early in the course of the disease.
|
|
Women who are carriers for choroideremia will nearly always show evidence of this on examination with patchy mottling and atrophy of the fundus with hyperpigmentation (Figs. 13B, 13C, and 13D). Random inactivation of one of the two X chromosomes, also called lyonization, results in the expression of the defect in a certain population of a carrier woman's retinal and choroidal cells, causing the carrier manifestations. These changes can occur in a patchy distribution throughout the fundus. Usually, carrier women are asymptomatic or, at most, experience only mild symptoms of poor night vision or pericentral scotomas, if the changes affect the posterior pole. Uncommonly, the carrier manifestations may cause considerable visual impairment, if the macular regions are involved. Histology has showed patchy loss of choroid, pigment epithelium, and photoreceptor outer segments, with abrupt transition from normal to abnormal areas.70 More diffuse abnormalities of the retinal pigment epithelium have been found throughout the fundus, suggesting that the defect may have a primary effect on this tissue.
The gene for choroideremia had been localized by linkage studies to Xq21.2 and has been found to encode a gene product, Rab escort protein-1 (REP-1),71 previously referred to as component A of Rab geranylgeranyl transferase (also called the CHM protein).72–74 REP-1 is involved in geranylgeranylation, which is the post-translational process of insertion of hydrophobic lipid side chains onto proteins to help them attach to membranes and to help them interact with other proteins.71,75 The gene for another similar protein, REP-2 (also called CHML protein), was subsequently found to be on the long arm of chromosome 1.76,77 The gene products of both are widespread throughout the body, but REP-1 appears necessary for the prenylation of a specific subset of Rab proteins essential for the retina and is the protein that is defective in choroideremia. Exactly how deficiency of REP-1 results in degeneration of the choroid is unknown, but loss of this protein would be expected to disrupt intracellular cycling of proteins between cytosol and membranes.71,75
A single point mutation (insertion of a T within a splice donor site of the intron downstream of exon C, changing the normal sequence of AGgtaag to AGgttaag) accounts for one-fifth of the world's population of patients with choroideremia.78 Five other point mutations have been reported, each of which results in the occurrence of a stop codon that leads to a truncated gene product.79 Deletions of the CHM gene account for approximately 12% of cases.78
LONG-CHAIN 3-HYDROXYACYL-COA DEHYDROGENASE (LCHAD) DEFICIENCY.
LCHAD deficiency is a recently described autosomal recessive disorder of mitochondrial fatty acid β-oxidation in which neonates or infants present with a Reye-like syndrome, cardiomyopathy, life-threatening episodes of hypoketotic hypoglycemia and cardiorespiratory arrest.80 Up to 90% of individuals share the same G1528C allele in the LCHAD gene.81 Patients can have recurrent muscle cramps with rhabdomyolysis during acute illnesses and can develop peripheral neuropathy. At least half, if not the majority, of the patients who survive infancy develop a severe, debilitating atrophy of the choroid and retina.82 The fundus and ERG may be normal at birth (Stage 1). Soon, however, pigment dispersion occurs in the RPE (Stage 2), followed by circumscribed chorioretinal atrophy, occlusion of choroidal vessels with accompanying reductions in acuity, visual field, and ERG amplitudes (Stage 3). Eventually, the posterior pole undergoes near total vascular choroidal atrophy (Fig. 14); however, the peripheral fundus is usually relatively spared. Finally, posterior staphylomas may develop (Stage 4). Cataract and progressive axial myopia are also features.82,83 Histopathologically, diffuse choroidal atrophy with loss of the choriocapillaris is seen, suggesting a primary fault at the level of the RPE and choriocapillaris with a secondary macrophage response.84 Dietary management of the disease involves avoidance of fasting, a high-carbohydrate diet, limitation of dietary long chain fatty acid intake, and supplementation of the diet with medium-chain triglycerides.85 The cause of the chorioretinal atrophy is unknown but may be the result of chronic damage to the retinal pigment epithelium and subsequently the choroid by the accumulation of toxic long-chain 3-hydroxyl fatty acids and/or acylcarnitines.85
|