| Scleritis, unlike episcleritis, is a severe destructive disease, sometimes
leading to the loss of an eye from deteriorating vision, severe pain, or
even (occasionally) perforation of the globe. Such changes, when
they occur, are rapid, so early diagnosis and effective treatment are
essential. Scleral disease can be diagnosed when the patient is first
seen if one remembers that whereas episcleritis rarely, if ever, involves
the scleral tissue, in scleritis the episclera is always involved. Therefore, attention
must be diverted to the sclera to detect the early
changes of scleral edema or necrosis. The onset of scleritis is usually gradual, building up over several days. By
the time patients seek advice, the clinical types can be distinguished
as anterior or posterior, or occasionally both. Anterior scleritis may be further subdivided into diffuse, nodular, or necrotizing. The last condition may present with signs of inflammation or with few
or no signs of inflammation (scleromalacia perforans). Long-term follow-up
of patients with scleral disease showed that only 8% of patients
changed from one type of disease to another during the course of this
disease, so although differentiation into these types does not usually
indicate an etiology, it does have a direct bearing on the prognosis
and the type of treatment to be used.10 Scleritis is most common in the fourth to sixth decades of life and occurs
more frequently in women than men (8:5). Necrotizing scleritis occurs
later than the other varieties, the mean age being 61 years. Scleritis
is bilateral in 52% of patients. In half of these, the condition
starts in both eyes simultaneously, with the rest becoming bilateral in 5 or
more years. ASSOCIATED SYSTEMIC DISORDERS In a review of 1200 patients with scleritis who have attended the Scleritis
Clinic at Moorfields Eye Hospital in London, an associated systemic
disorder was found in all patients with scleromalacia perforans, in
half of those with nodular and necrotizing disease, in a third of those
with diffuse anterior scleritis, and in only 10% of those with posterior
scleritis. Severe polyarticular rheumatoid arthritis and a case
of porphyria accounted for the patients with scleromalacia perforans. Forty percent of the patients with necrotizing scleritis had other connective
tissue disorders, but, surprisingly, only 21% of the patients with
diffuse anterior or nodular scleritis had rheumatoid arthritis or
other connective tissue disorders. This percentage is much lower than
that reported by other authors,7,11–13 but this may be because patients with the less severe scleral disease
are referred to us only if the etiology is in doubt, thus biasing the
results. Twelve percent of the patients with diffuse anterior and nodular
scleritis had ankylosing spondylitis, and in a further 15% the scleritis
followed an attack of herpes zoster ophthalmicus. A variety of
other conditions, including syphilis, tuberculosis, gout, Reiter's
disease, IgA nephropathy, and erythema nodosum, were thought to be definite
etiologic factors because with appropriate treatment the eye changes
disappeared. Fowler investigated a random selection of the patients
with scleritis at Moorfields Eye Hospital and found only 7%, all
of whom were young males, who did not have any other detectable physical
abnormality.14 Forty percent of these patients had hypertension, which in some cases
required treatment. It was thought that the hypertension could have been
a manifestation of a generalized arteritis, but this was convincingly
demonstrated in only 19% of these patients. PATHOLOGY The pathology of scleritis has received much attention in the past.6,8,15–19 Although certain inferences can be drawn from pathologic specimens of
eyes removed because of pain, perforation, or mistaken diagnosis, these
eyes have been severely damaged from advanced disease. Unfortunately, biopsies
of scleral lesions have proved to be unsatisfactory, at best
yielding material of limited diagnostic value and at worst leaving an
area of exposed choroid that will not heal. Consequently, they should
not be performed. Scleritis usually affects the anterior segment of the eye, possibly because
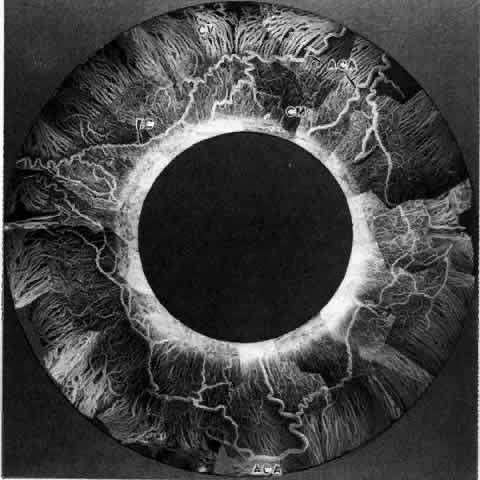
this is the area with the best blood supply, but with sluggish flow
through the vessels (Fig. 19). The sclera is thickened and roughened in the affected area, which appears
to be sharply demarcated from the rest of the sclera. However, tissue
obtained at surgery during the course of grafting of areas adjacent
to necrotic tissue shows marked pathologic changes.20,21 The area of affected sclera may be swollen, excavated, or frankly ulcerated
with undermined edges covered with a thin layer of fibrous tissue. However, spontaneous
perforation is extremely unusual and, where seen
in pathologic specimens, has usually occurred at the time of removal
of the eye. A posterior scleritis often occurs as an extension of anterior
disease; but, as in Figure 20, most of the inflammation (in some cases all of the inflammation) is in
the posterior segment and the exudative detachments and subretinal granulomas
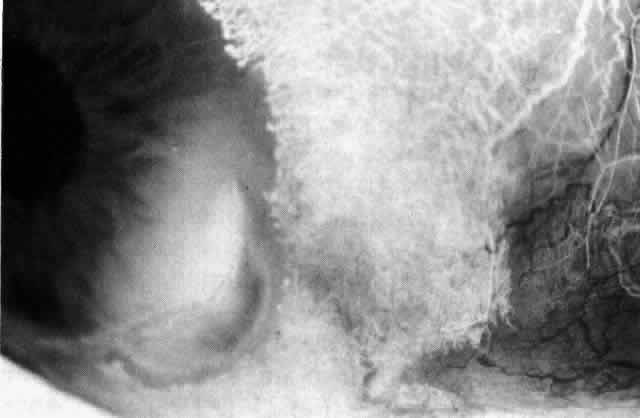
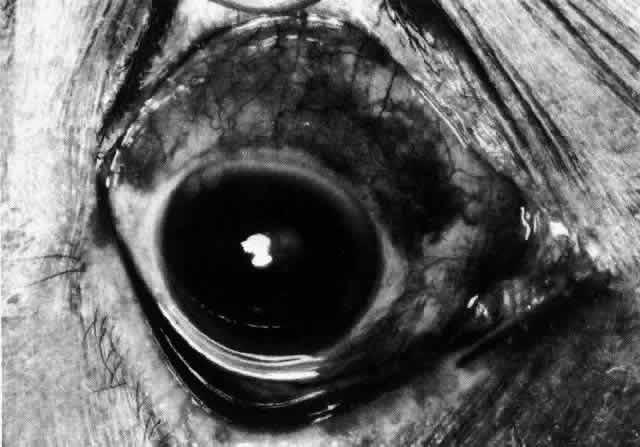
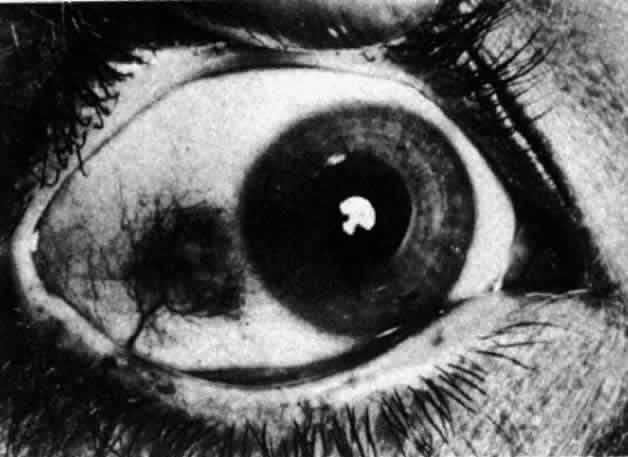
can be mistaken for malignant melanoma. 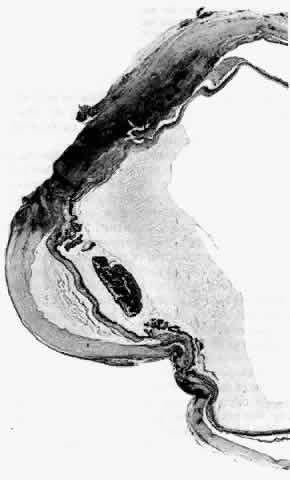 Fig. 19. Anterior necrotizing scleritis. The eye was removed because of loss of
vision and intractable pain. No form of steroid was given to this patient
because of a severe Pseudomonas infection of the chest. (Courtesy of Professor N. Ashton) Fig. 19. Anterior necrotizing scleritis. The eye was removed because of loss of
vision and intractable pain. No form of steroid was given to this patient
because of a severe Pseudomonas infection of the chest. (Courtesy of Professor N. Ashton)
|
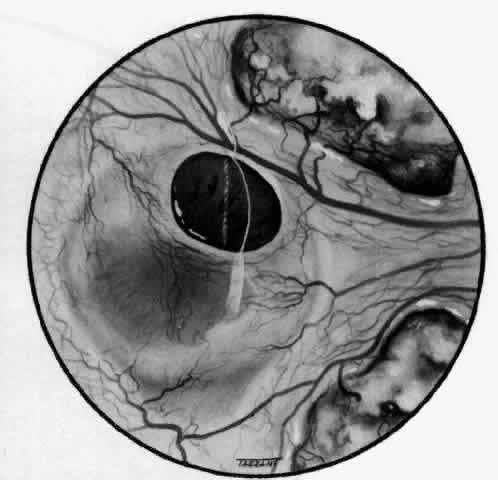
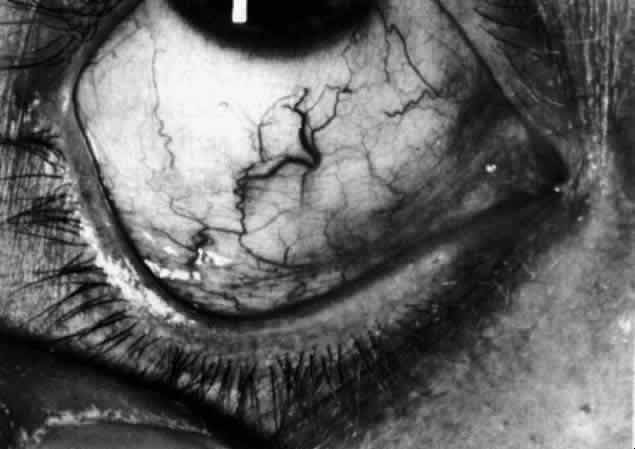
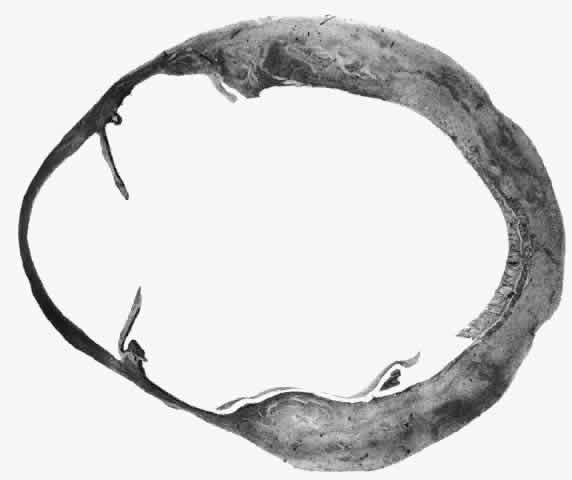 Fig. 20. Posterior scleritis. This eye was removed because of loss of vision and
pain, mistakenly diagnosed as malignant melanoma. (Courtesy of Professor N. Ashton) Fig. 20. Posterior scleritis. This eye was removed because of loss of vision and
pain, mistakenly diagnosed as malignant melanoma. (Courtesy of Professor N. Ashton)
|
What is clinically represented solely by inflammation and edema is histopathologically
a granulomatous lesion of the sclera, the center of which
consists largely of plasma cells, lymphocytes, and mast cells (Figs. 21 through 23). Foster and colleagues have identified the cellular subsets and glycoproteins
in both necrotizing and non-necrotizing scleritis.22 This shows an active T-cell inflammatory response with a high CD4/CD8 ratio
and increased HLA/DR and CD14, indicating a macrophage-induced response
that would lead to granuloma formation. Remote from the granuloma, the
fibrocytes of the sclera become activated, the proteoglycan adjacent
to them becomes altered, and the collagen fibrils of the sclera
become unraveled (Figs. 23 and 24). These changes appear to take place prior to the invasion of the stroma
by cells of the granuloma.20 The vessels in and around the necrotic area show medial necrosis and perivascular
cuffing with lymphocytes, and endothelial swelling with microvascular
occlusion. Ninety-six percent of the specimens examined by
Foster and associates show a microangiopathy characterized by a neutrophil
infiltrate in and around the vessel wall.22–23 This is most obvious at the center of the lesion where there may be occlusion
of the vessel, thrombosis, or even aneurysm formation (Fig. 25). From these pathologic investigations, clinical observations, animal
experiments, and the results of fluorescein angiography, it would appear
that the scleral inflammation is initiated either by trauma (be it
accidental or surgical)23–25 or by bacterial or viral infection. If circulating immune complexes are
present because of the poor blood flow, they become precipitated in
and around the vessel walls in the area of inflammation. In other patients, a
persistence of tissue damage will lead to autoimmunization. Damage
to the endothelial cells of the microvasculature leads to changes
within the vessels detectable on angiography and to catabolic changes
in the surrounding tissues. These changes, in turn, allow the granulomatous
response that is seen in histopathologic sections, the first detectable
change being in the scleral fibrocytes and the proteoglycan and
collagen remote from the site of cellular infiltration. 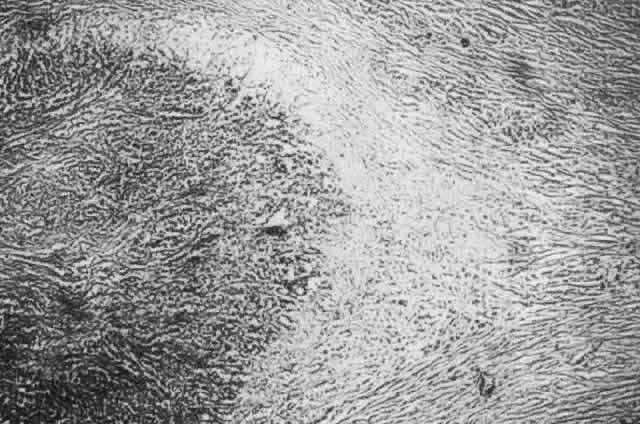 Fig. 21. Advancing edge of a granulomatous reaction. Scleral fibers are split and
separated by edema and then disrupted when invaded by the granuloma Fig. 21. Advancing edge of a granulomatous reaction. Scleral fibers are split and
separated by edema and then disrupted when invaded by the granuloma
|
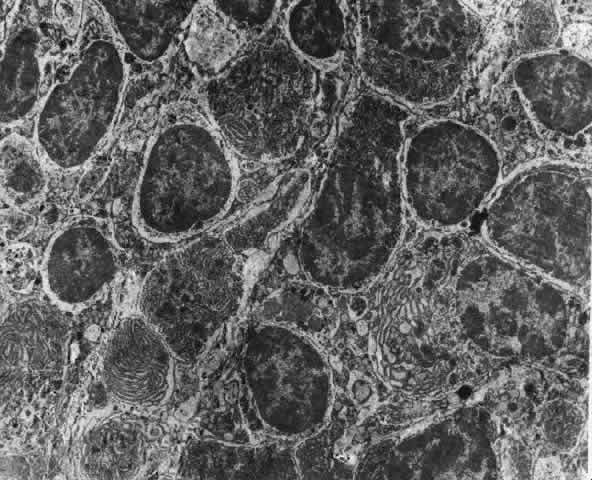 Fig. 22. Electron micrograph of an area of active scleritis showing the plasma cell
infiltrate suggestive of an immune response. Note aggregated plasma
cells, with the characteristic whorled rough endoplasmic reticulum, in
the process of degeneration, releasing organelles and nuclear debris
into the extracellular matrix. (Uranyl acetate and lead citrate. X3000) (Courtesy of Dr. R. Tripathi) Fig. 22. Electron micrograph of an area of active scleritis showing the plasma cell
infiltrate suggestive of an immune response. Note aggregated plasma
cells, with the characteristic whorled rough endoplasmic reticulum, in
the process of degeneration, releasing organelles and nuclear debris
into the extracellular matrix. (Uranyl acetate and lead citrate. X3000) (Courtesy of Dr. R. Tripathi)
|
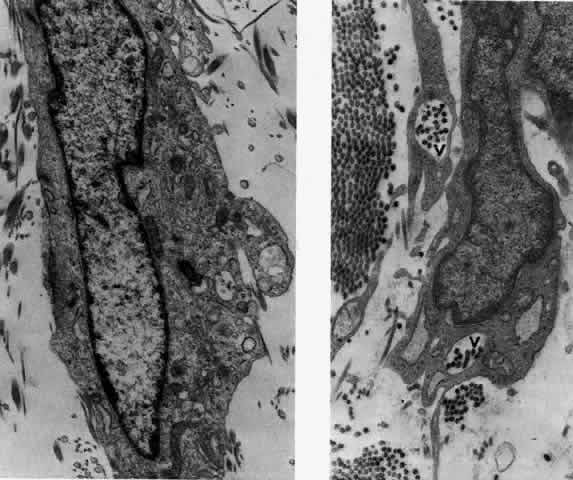 Fig. 23. Electron micrographs of scleral stroma at the periphery of an area of ulceration
in a patient with necrotizing scleritis. The left shows an active
fibroblastic cell, and the right shows collagen fibrils within intracellular
vacuoles (V) in the fibroblastic cell. (Left X15,375; right
X15,375) (Watson PG, Young RD: Changes at the periphery of a lesion necrotizing
scleritis: Anterior segment fluorescein angiography correlated with electron
microscopy. Br J Ophthalmol 68:781–789, 1984) Fig. 23. Electron micrographs of scleral stroma at the periphery of an area of ulceration
in a patient with necrotizing scleritis. The left shows an active
fibroblastic cell, and the right shows collagen fibrils within intracellular
vacuoles (V) in the fibroblastic cell. (Left X15,375; right
X15,375) (Watson PG, Young RD: Changes at the periphery of a lesion necrotizing
scleritis: Anterior segment fluorescein angiography correlated with electron
microscopy. Br J Ophthalmol 68:781–789, 1984)
|
 Fig. 24. Electron micrograph of scleral stroma at the periphery of an ulcer in necrotizing
scleritis (same patient as in Figure 23) showing swelling and unraveling of collagen fibrils (arrows) in longitudinal
section (X29,270) and in transverse section (inset, X44,000). Fibrils
of all diameters are affected. (Watson PG, Young RD: Changes at the periphery of a lesion necrotizing
scleritis: Anterior segment fluorescein angiography correlated with electron
microscopy. Br J Ophthalmol 69:656–663, 1985) Fig. 24. Electron micrograph of scleral stroma at the periphery of an ulcer in necrotizing
scleritis (same patient as in Figure 23) showing swelling and unraveling of collagen fibrils (arrows) in longitudinal
section (X29,270) and in transverse section (inset, X44,000). Fibrils
of all diameters are affected. (Watson PG, Young RD: Changes at the periphery of a lesion necrotizing
scleritis: Anterior segment fluorescein angiography correlated with electron
microscopy. Br J Ophthalmol 69:656–663, 1985)
|
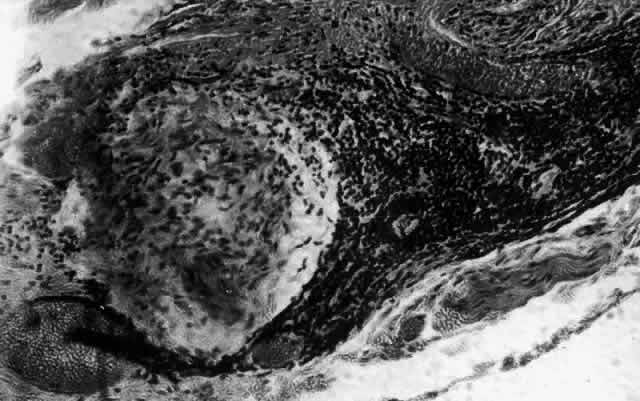 Fig. 25. Intense lymphocytic reaction and infiltration around a medium-sized arteriole
and nerve. Fig. 25. Intense lymphocytic reaction and infiltration around a medium-sized arteriole
and nerve.
|
CLINICAL MANIFESTATIONS Lacrimation and photophobia are more common in scleritis than in episcleritis. However, they
are not always clearly related to the severity of
the scleritis or to the keratitis and uveitis that may accompany it. The pain of scleritis is its most dominant feature and is the symptom that
causes the patient to seek medical advice. The exception to this is
scleromalacia perforans occurring in long-standing rheumatoid arthritis, which
may be entirely pain free. Pain, when it occurs, may be localized
to the eye, but in 66% of patients it is much more diffuse, radiating
to the temple, the jaw, and the sinuses. It is boring in nature, severe
enough to prevent sleep, accompanied by malaise, and only temporarily
relieved by analgesics (Fig. 26). The pain is particularly severe in those patients suffering from progressive
necrotizing scleritis with overlying inflammation; eyes have
been removed for this reason alone. The pain can be a diagnostic problem, particularly
in the early stages of posterior scleritis before the
vision becomes affected. Patients with posterior scleritis are often
referred to neurologists and others because of the severity of the headache
or ophthalmoplegia. The pain is probably caused by distention of
sensory nerve endings as a result of edema. In the necrotizing disease, the
severity of the pain is increased by the destruction of the nerve
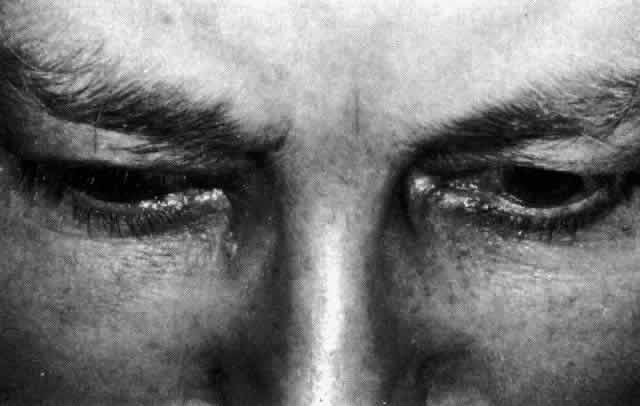
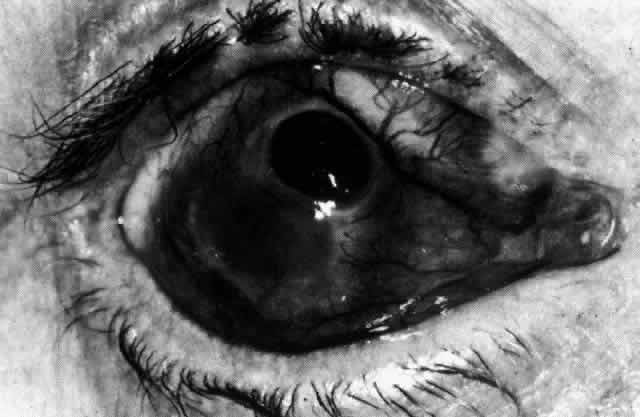
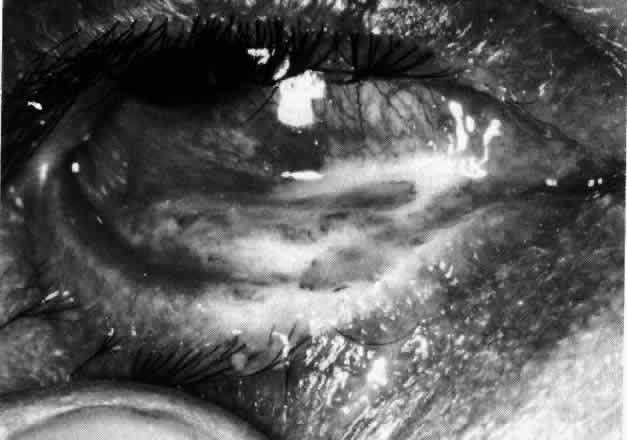
endings that takes place. 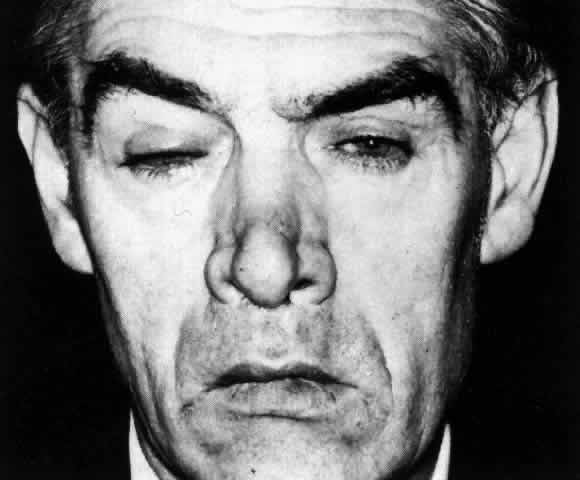 Fig. 26. Severe ptosis produced in a severe diffuse anterior scleritis. Pain radiated
to temple and face and was severe enough to prevent sleep. Fig. 26. Severe ptosis produced in a severe diffuse anterior scleritis. Pain radiated
to temple and face and was severe enough to prevent sleep.
|
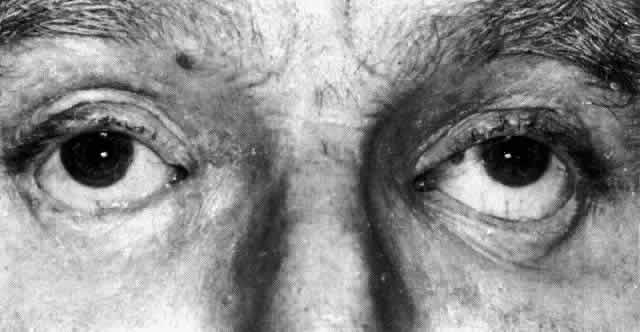
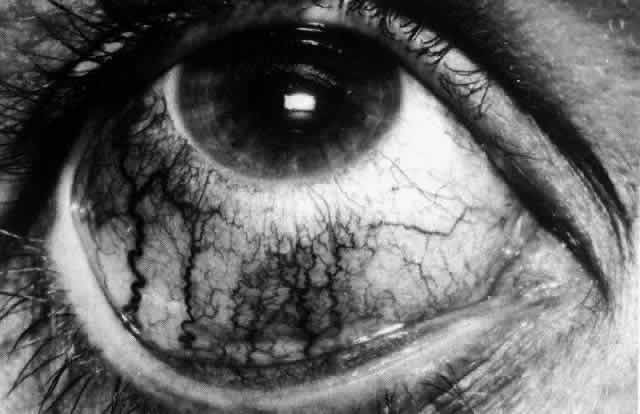
The inflammation of the eye is a prominent feature. The inflammation has
a bluish-red hue in contrast to the brighter red of episcleritis and
may be sectorial or diffuse. The severity of inflammation seems to depend
on the amount of episcleral tissue present. Therefore, it is more
prominent in younger people and is least prominent in those with rheumatoid
arthritis in whom the episcleral tissue almost disappears. Each of the various types of scleritis can be distinguished by its typical
clinical appearance. Because the pathologic change is in the sclera, there
is always edema and/or necrosis of that tissue. This gives rise
to an overlying episcleral edema and to congestion that may be very
severe and may need blanching with epinephrine 1:1000 or phenylephrine 10% to
detect the underlying edema. The sclera that is edematous is pushed forward, and the deep episcleral
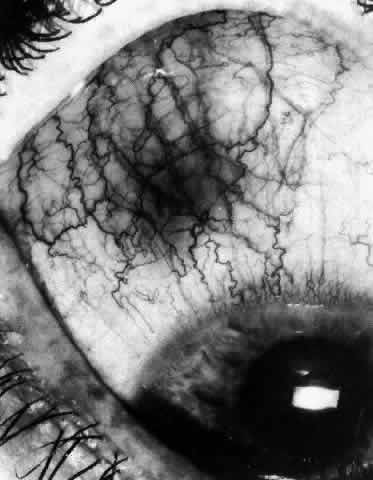
network is more congested than the superficial networks (Figs. 27 and 28). It is usually easy to ascertain by simple observation that the patient
has scleritis and not episcleritis. However, it is not as easy to ascertain
whether the patient has early necrotizing scleritis. It is in
these patients that fluorescein angiography has considerable value, because
the first changes are detectable in the ocular vasculature. Prompt
and adequate treatment can prevent these changes from becoming irreversible. 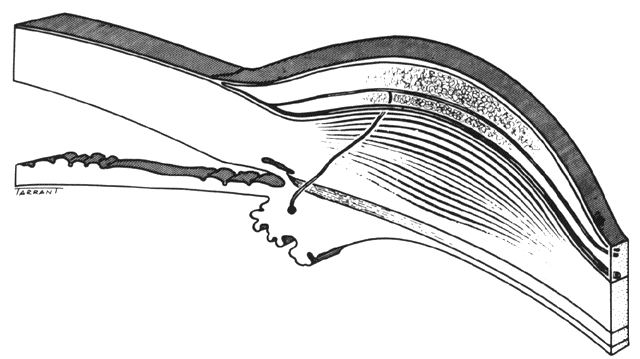 Fig. 27. In scleritis, maximum congestion occurs in deep episcleral plexus, which
is bowed forward by underlying scleral edema. Episcleral tissue is slightly
infiltrated and superficial plexus is slightly congested (see Fig. 14). (Watson PG, Hayreh S, Awdry P: Episcleritis and scleritis. Br J Ophthalmol 52:278–279, 1968) Fig. 27. In scleritis, maximum congestion occurs in deep episcleral plexus, which
is bowed forward by underlying scleral edema. Episcleral tissue is slightly
infiltrated and superficial plexus is slightly congested (see Fig. 14). (Watson PG, Hayreh S, Awdry P: Episcleritis and scleritis. Br J Ophthalmol 52:278–279, 1968)
|
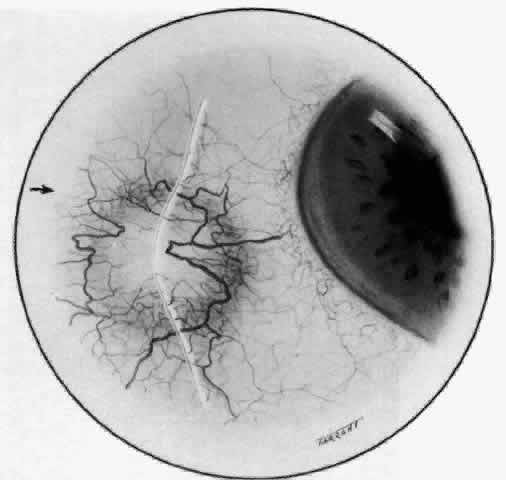 Fig. 28. Nodular scleritis. Both the anterior conjunctival slit and the deep scleral
slit are displaced forward by the scleral edema. There is little
separation between these two beams, indicating that all the edema is in
the sclera and not in the overlying episclera. (Watson PG, Hayreh S, Awdry P: Episcleritis and scleritis. Br J Ophthalmol 52:278–279, 1968) Fig. 28. Nodular scleritis. Both the anterior conjunctival slit and the deep scleral
slit are displaced forward by the scleral edema. There is little
separation between these two beams, indicating that all the edema is in
the sclera and not in the overlying episclera. (Watson PG, Hayreh S, Awdry P: Episcleritis and scleritis. Br J Ophthalmol 52:278–279, 1968)
|
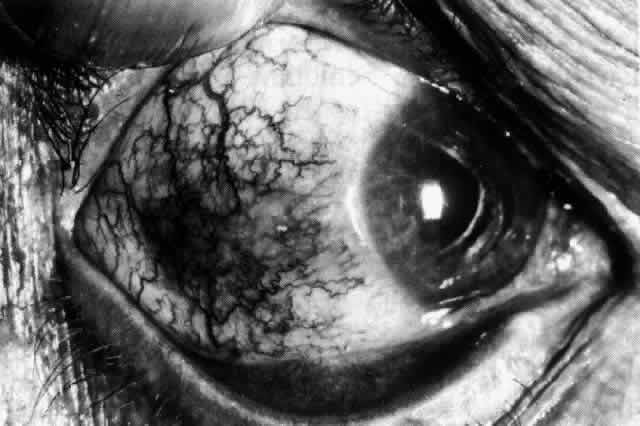
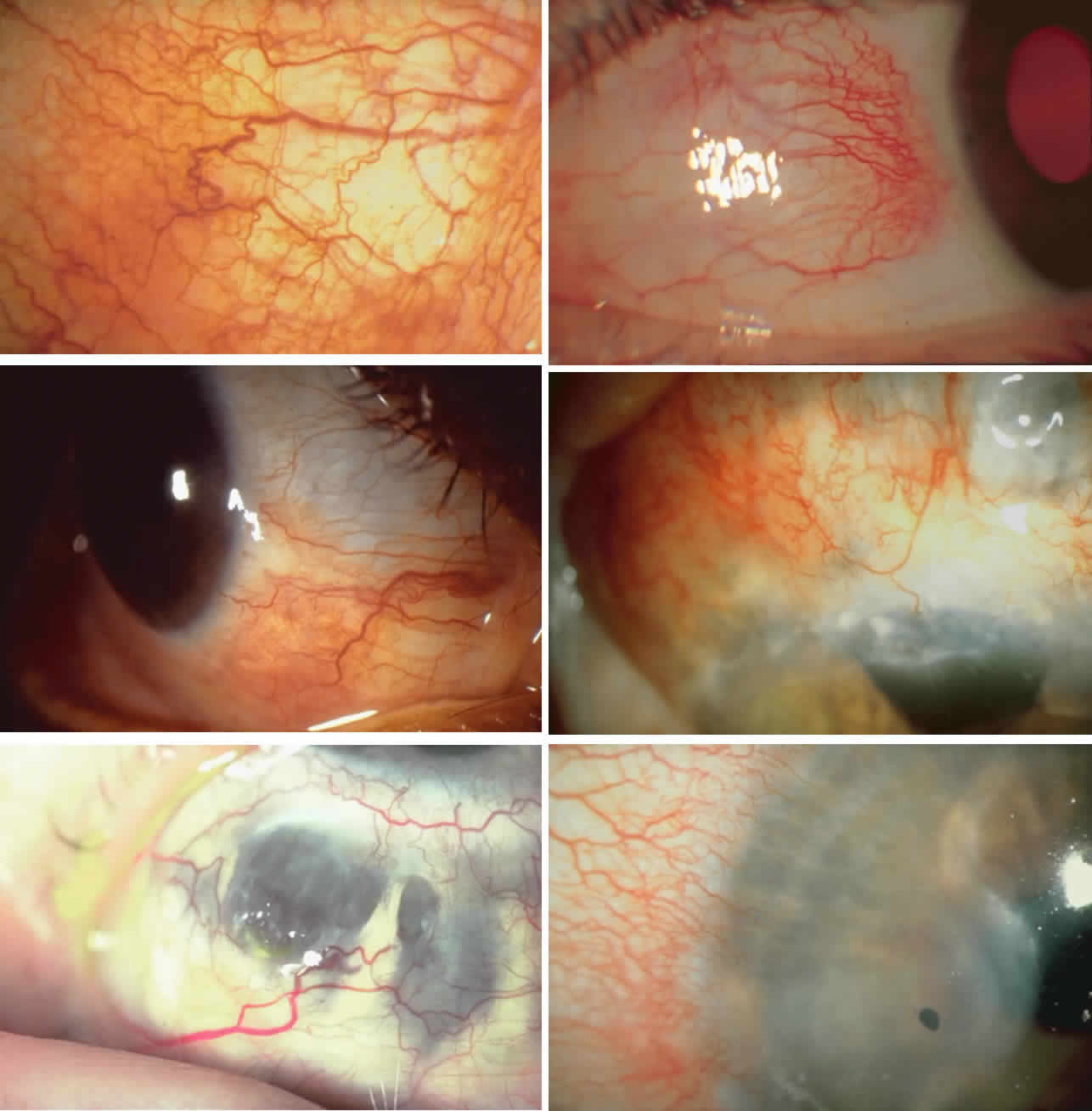
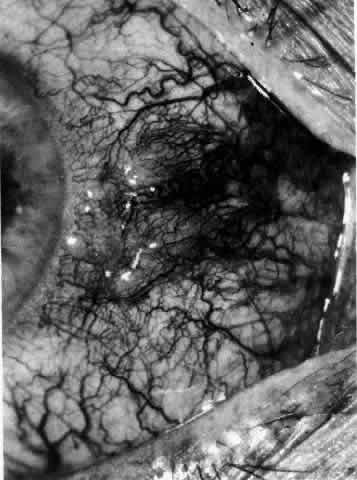
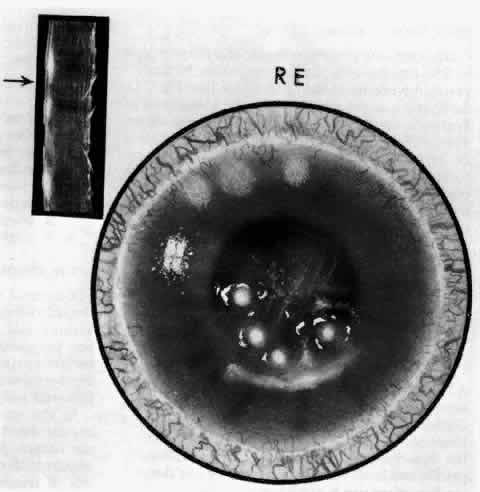
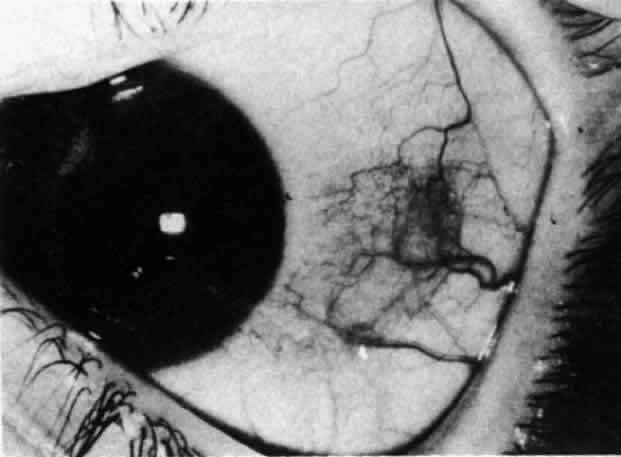
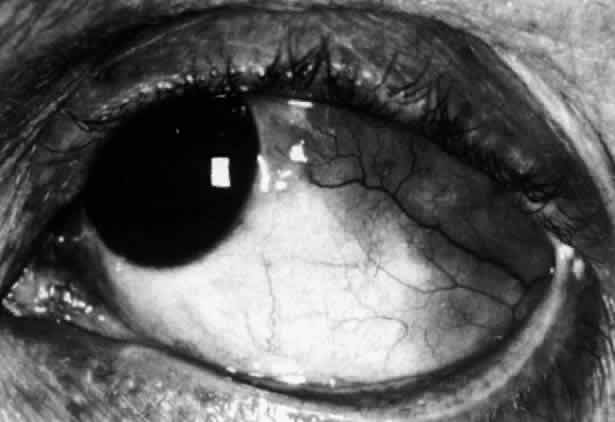
Diffuse Anterior Scleritis Diffuse anterior scleritis is the most common and least severe type of
scleritis. The inflammation is widespread, and it may involve either a
small segment or the whole of the anterior segment, sometimes with such
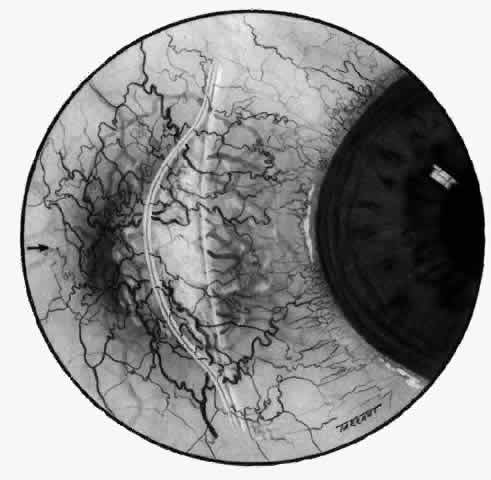
severe overlying inflammation as to justify the name “brawny” scleritis (Fig. 29). On slit lamp examination, the vascular pattern of both deep and superficial
layers may be distorted, so that the normal radial pattern of
the vessels is lost; large anastomotic channels develop, leading to beading
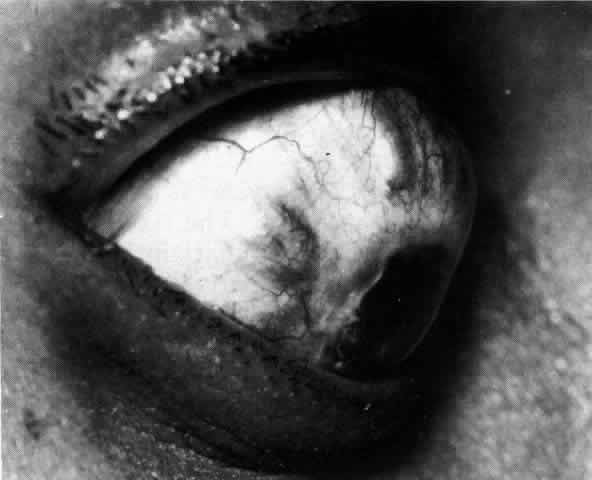
and tortuosity of the remaining vessels (Figs. 30 and 31; Color Plate 1C). 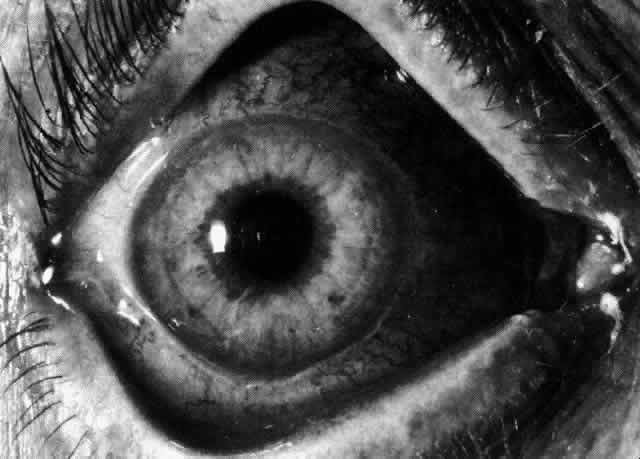 Fig. 29. Intense inflammation, edema, and conjunctival chemosis that accompany acute
diffuse anterior scleritis. Fig. 29. Intense inflammation, edema, and conjunctival chemosis that accompany acute
diffuse anterior scleritis.
|
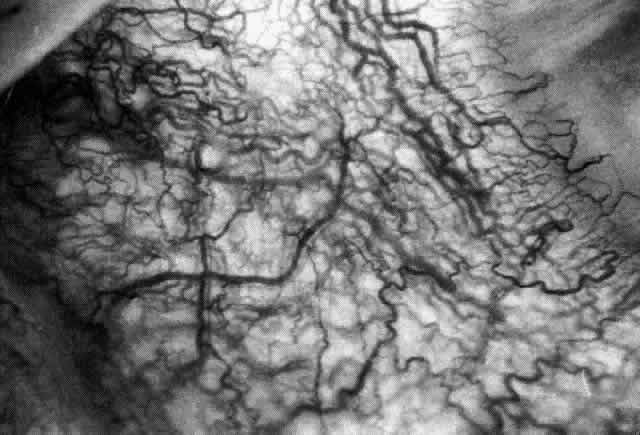 Fig. 30. Diffuse anterior scleritis. During the acute attack, the vessels are dilated
and distorted. New vessels or large vessels not normally seen have
appeared adjacent to the limbus. Fig. 30. Diffuse anterior scleritis. During the acute attack, the vessels are dilated
and distorted. New vessels or large vessels not normally seen have
appeared adjacent to the limbus.
|
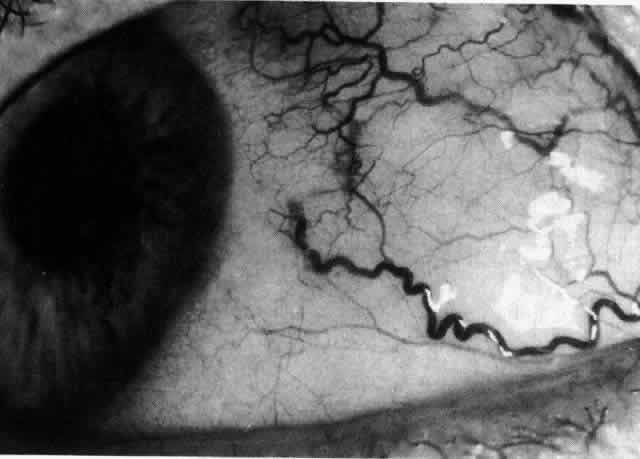 Fig. 31. Diffuse anterior scleritis after treatment. The dilated abnormal blood
vessels remain even though no inflammation remains. Fig. 31. Diffuse anterior scleritis after treatment. The dilated abnormal blood
vessels remain even though no inflammation remains.
|
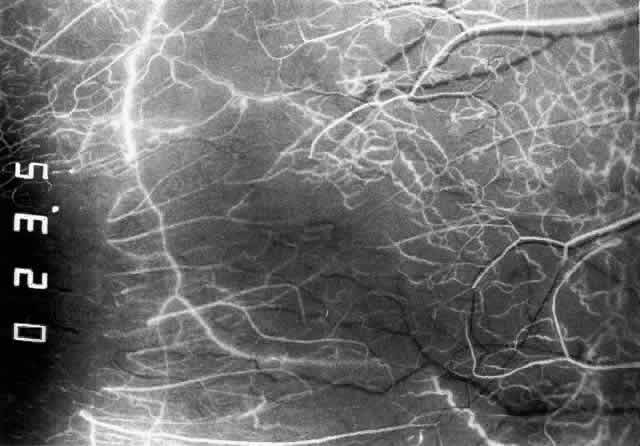
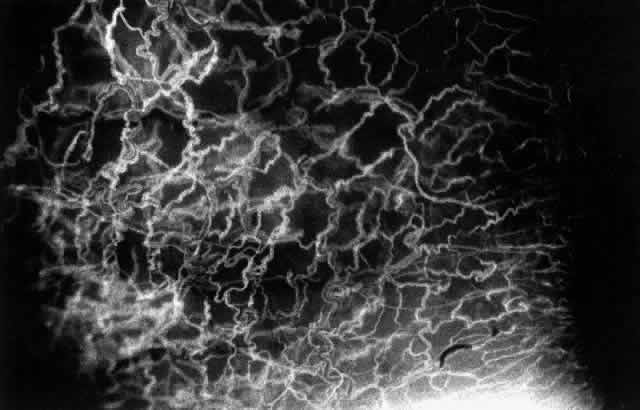
In this relatively benign form of scleral inflammation, the fluorescein
angiogram reveals a rapid flow pattern in which the transit time of the
dye is very rapid (as in episcleritis) (Figs. 32 and 33). Subtle changes occur in the capillary network, and abnormal leaking
vessels appear after prolonged inflammation. These changes do not disappear
after the inflammation subsides or is treated (see Fig. 31). 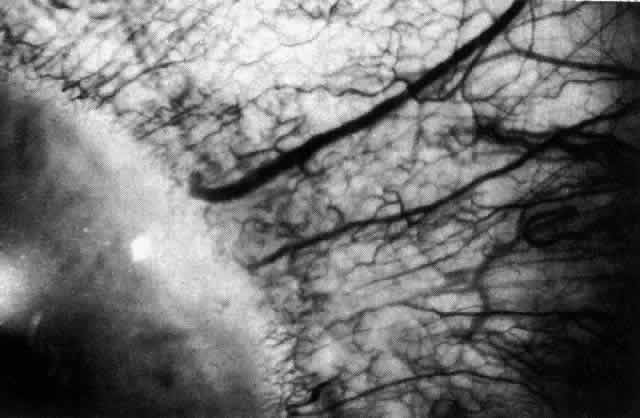 Fig. 32. Diffuse anterior scleritis associated with corneal edema. The limbal vessels
are grossly dilated in association with generalized scleral edema. The
cornea adjacent to these vessels is edematous. Fig. 32. Diffuse anterior scleritis associated with corneal edema. The limbal vessels
are grossly dilated in association with generalized scleral edema. The
cornea adjacent to these vessels is edematous.
|
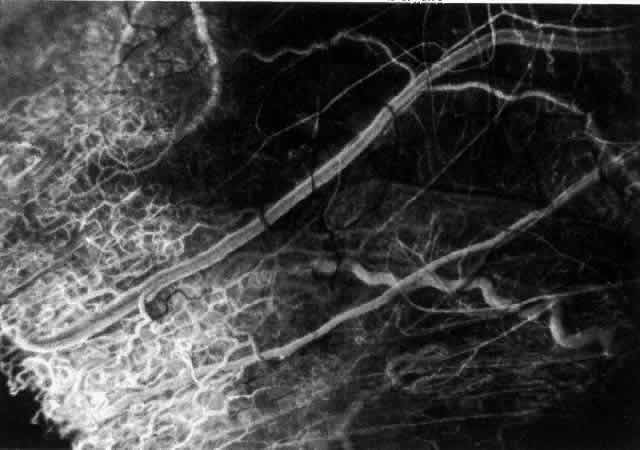 Fig. 33. Fluorescein angiogram of patient in Figure 32 four seconds after the appearance of the dye. This is a very rapid transit
time. All the limbal capillaries are completely full, and all the
major episcleral vessels contain fluorescein. Note that the very large
vessel is a vein, and the narrow vessel below it is an artery. The deep
vessels are distorted, and some are abnormal in configuration. Fig. 33. Fluorescein angiogram of patient in Figure 32 four seconds after the appearance of the dye. This is a very rapid transit
time. All the limbal capillaries are completely full, and all the
major episcleral vessels contain fluorescein. Note that the very large
vessel is a vein, and the narrow vessel below it is an artery. The deep
vessels are distorted, and some are abnormal in configuration.
|
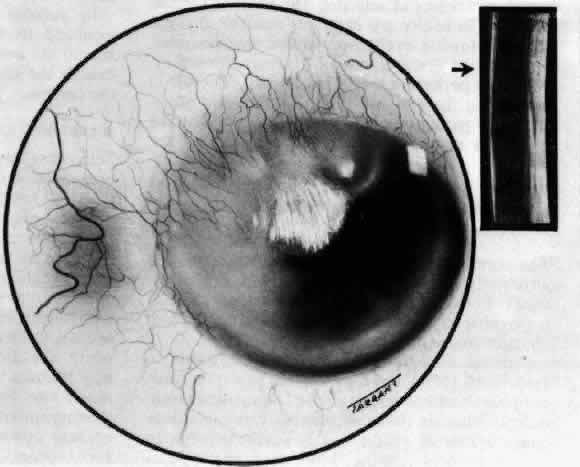
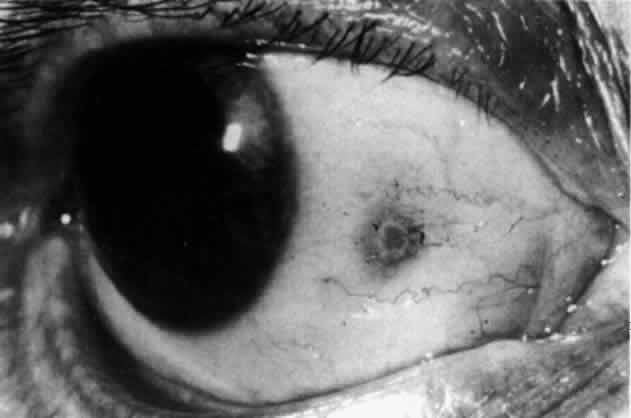
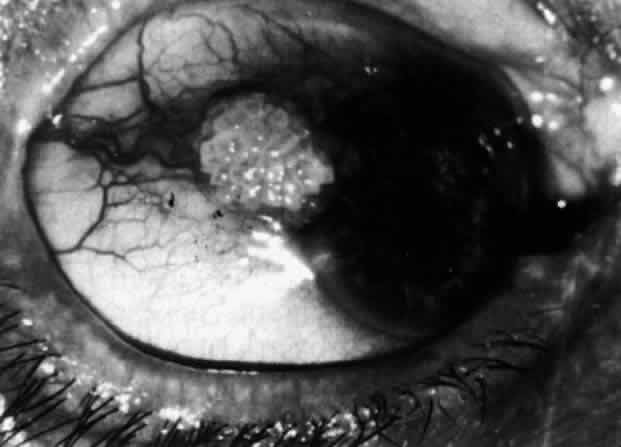
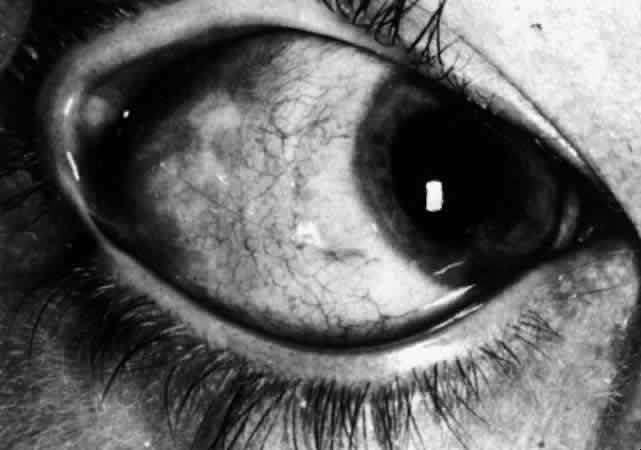
Nodular Anterior Scleritis Although patients with nodular anterior scleritis resemble those with nodular
episcleritis on cursory examination, detailed examination reveals
marked differences. The nodule or nodules (they may be multiple) consist
of scleral tissue that is immovable episclera is tightly adherent
to the nodule, which is tender to the touch. Although the sclera sometimes
becomes transparent below the nodule, it does not become necrotic, nor
does the condition extend beyond the site of the nodule, as occurs
in necrotizing scleral disease (Fig. 36). (see Fig. 28; Figs. 34 and 35). The edematous 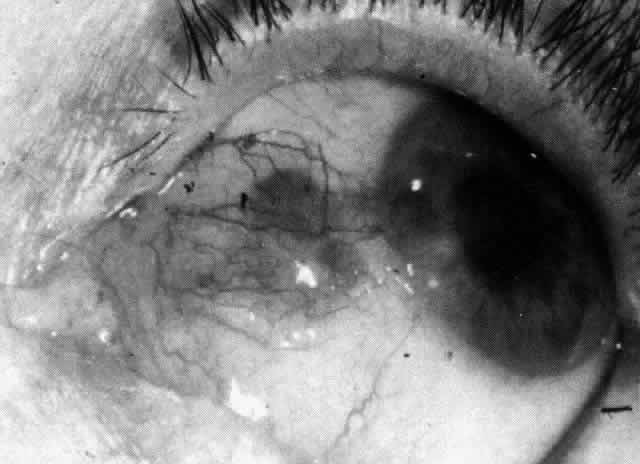 Fig. 36. Increased scleral transparency that occurred at the same site resulting
from recurrent attacks of nodular scleritis after herpes zoster ophthalmicus. Fig. 36. Increased scleral transparency that occurred at the same site resulting
from recurrent attacks of nodular scleritis after herpes zoster ophthalmicus.
|
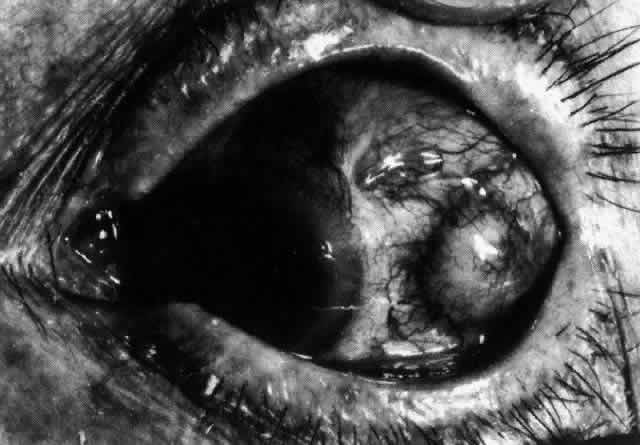 Fig. 34. Scleral edema has displaced all the vessel layers forward. Area surrounding
the nodule is acutely inflamed. Fig. 34. Scleral edema has displaced all the vessel layers forward. Area surrounding
the nodule is acutely inflamed.
|
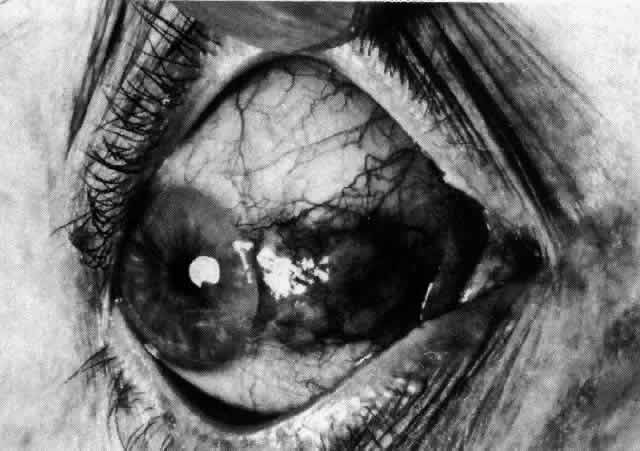 Fig. 35. Multiple scleral nodules. Surrounding inflammation is deep and intense. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5. London, Churchill-Livingstone, 1975) Fig. 35. Multiple scleral nodules. Surrounding inflammation is deep and intense. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5. London, Churchill-Livingstone, 1975)
|
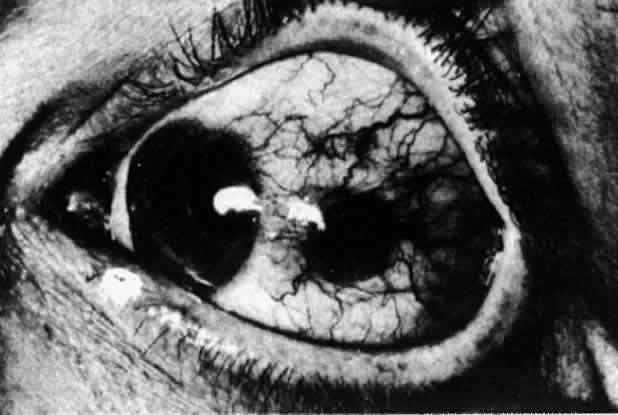
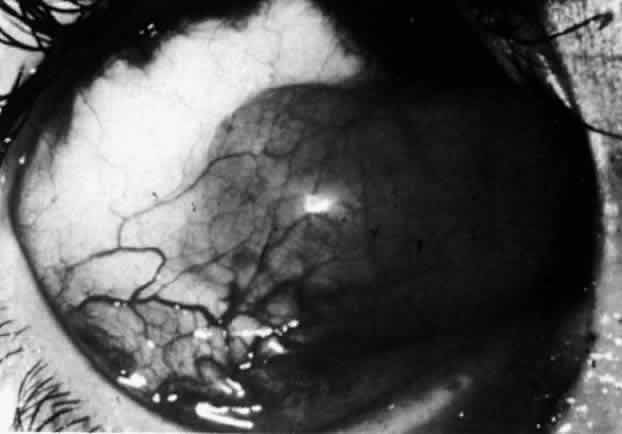
The angiogram is similar to that of diffuse anterior scleritis (i.e., there is a rapid filling pattern and deep scleral leakage of dye).26 Necrotizing Anterior Scleritis with Inflammation Patients with necrotizing anterior scleritis with inflammation not only
suffer extremes of discomfort but are in serious danger of losing an
eye. Therefore, it is of great importance that the condition be detected
early and treated adequately. (It is of equal importance that those
varieties of scleral inflammation that are not destructive to the eye
should not be treated with drugs that are themselves dangerous.) Accurate
diagnosis is the key. Necrotizing scleritis accompanied by inflammation is always painful, waking
the patient at night, increasing in intensity day by day, and leading
to severe distress. The sclera is swollen, and the overlying inflammation
is localized to the center of a lesion or to either end of an
extending lesion (Fig. 37; Color Plate 1D). After inflammation, the sclera becomes transparent so that the underlying
choroidal pigment becomes visible when viewed in daylight (Fig. 38). These areas may be invisible with the slit lamp. The area of inflammation
extends outward around the globe from the original site of inflammation, often
joining with other areas of scleritis that have subsequently
appeared. If the inflammation is not suppressed, the process will
progress around the globe until the whole anterior segment is involved (Fig. 39). 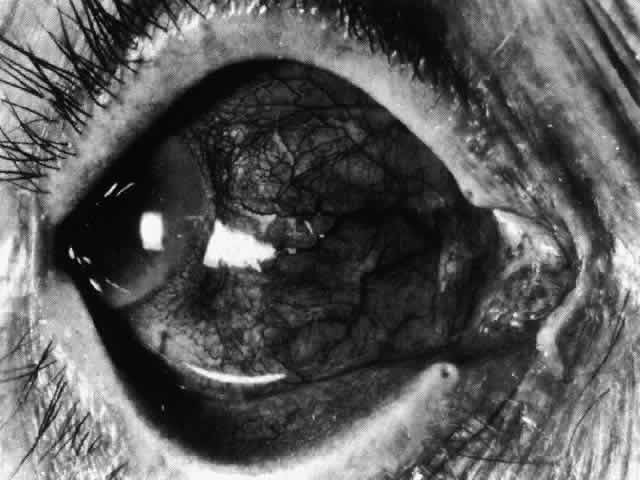 Fig. 37. Necrotizing anterior scleritis. Early stage in which there is diffuse, intense, scleral
congestion in one segment of the globe, and anomalies
of the vascular pattern. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5, pp 77–87. London, Churchill-Livingstone, 1975) Fig. 37. Necrotizing anterior scleritis. Early stage in which there is diffuse, intense, scleral
congestion in one segment of the globe, and anomalies
of the vascular pattern. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5, pp 77–87. London, Churchill-Livingstone, 1975)
|
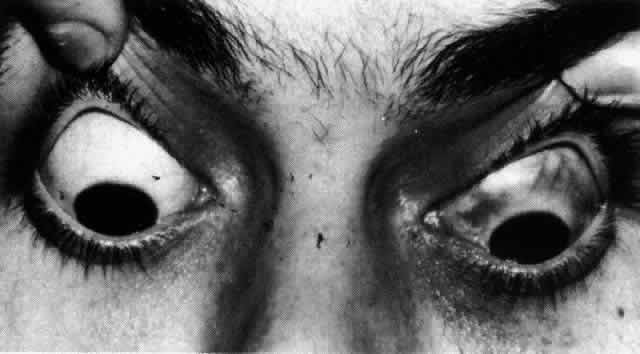 Fig. 38. Diffuse circumferential increased scleral pigmentation in left eye of a
young patient after treatment of a necrotizing anterior scleritis. Subtle
color changes of this type can be differentiated from similar changes
of increased transparency only if the patient is examined in daylight. Fig. 38. Diffuse circumferential increased scleral pigmentation in left eye of a
young patient after treatment of a necrotizing anterior scleritis. Subtle
color changes of this type can be differentiated from similar changes
of increased transparency only if the patient is examined in daylight.
|
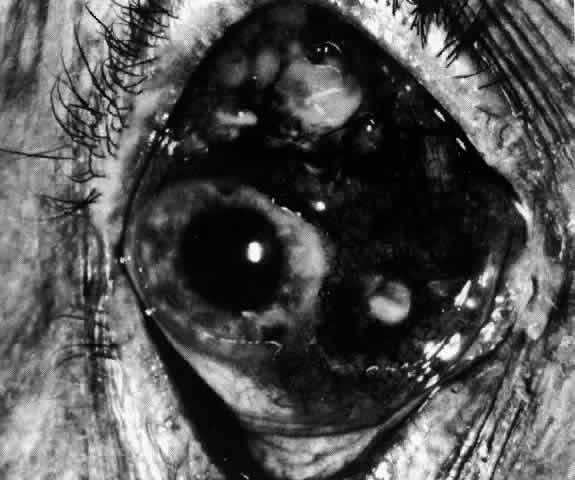 Fig. 39. Necrosis occurs in areas behind the advancing edge, which is at 4 o'clock. The
sclera at 6 o'clock is so far not affected. Fig. 39. Necrosis occurs in areas behind the advancing edge, which is at 4 o'clock. The
sclera at 6 o'clock is so far not affected.
|
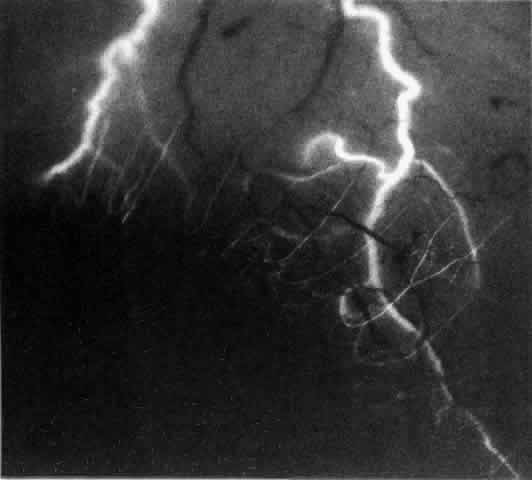
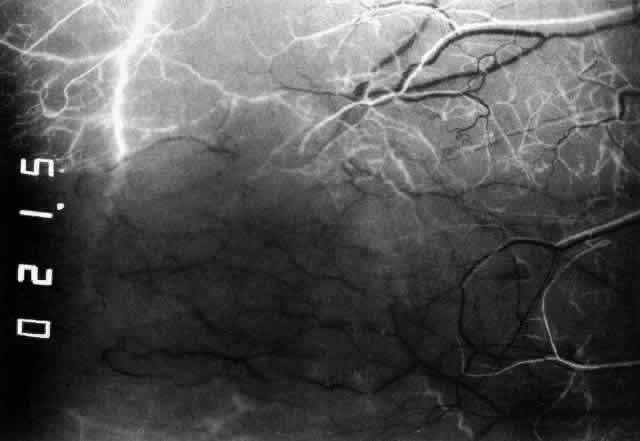
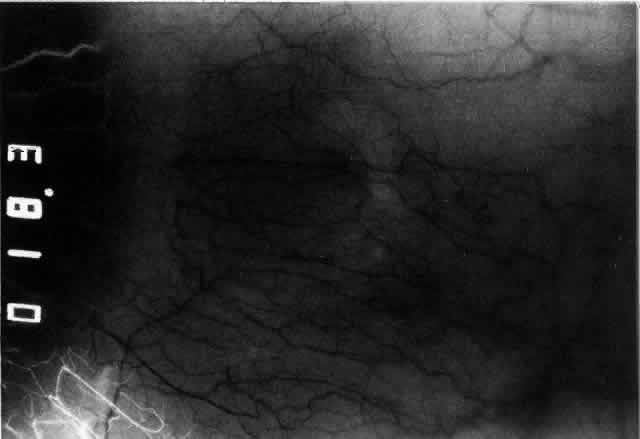
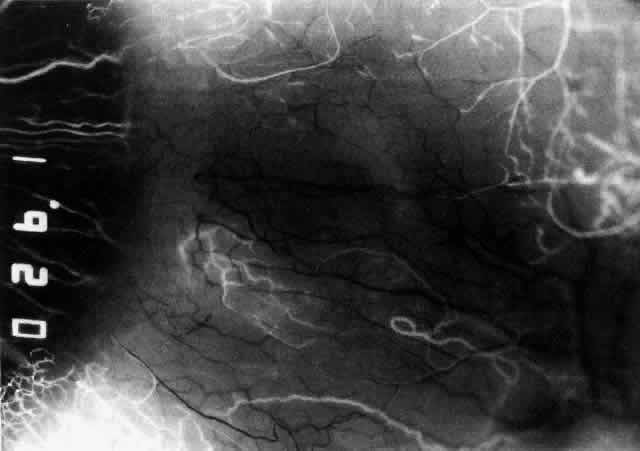
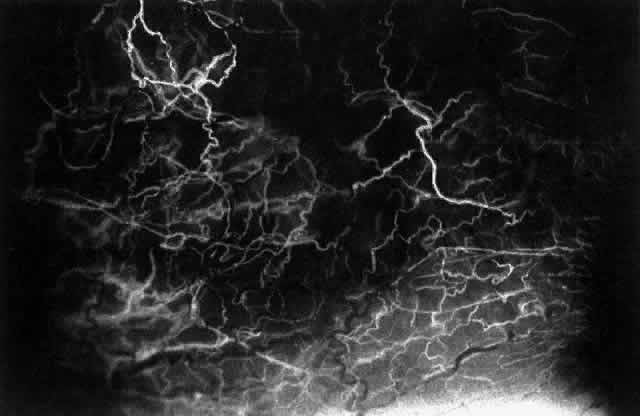
The characteristic features of necrotizing scleritis on fluorescein angiography
are hypoperfusion and, eventually, nonperfusion of the vascular
networks (Figs. 40 through 43).26 The initial changes are on the venous side of the capillary network; the
transit time of the dye increases even if the eye is red and congested. If
the disease process persists or has been present for a long time, thrombosis
and permanent vaso-occlusive changes occur. These vessels (or
the occluded capillary network) are bypassed by the opening of
anastomotic channels. New vessels in a granuloma give rise to deep intrascleral
leakage of dye (see Fig. 43). Conjunctival and episcleral involvement by the destructive change is
late but is always preceded by vaso-occlusive changes that can sometimes
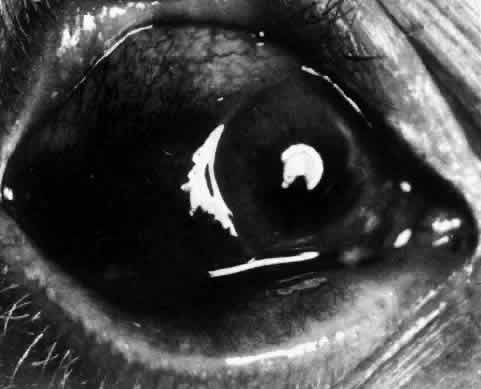
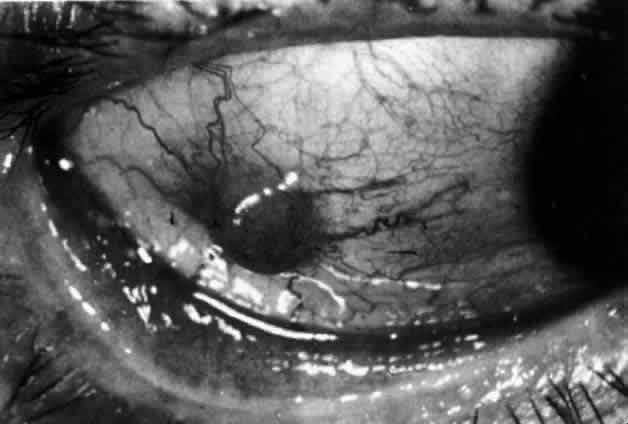
be detected with use of the red-free light on the slit lamp (Figs. 44 and 45). 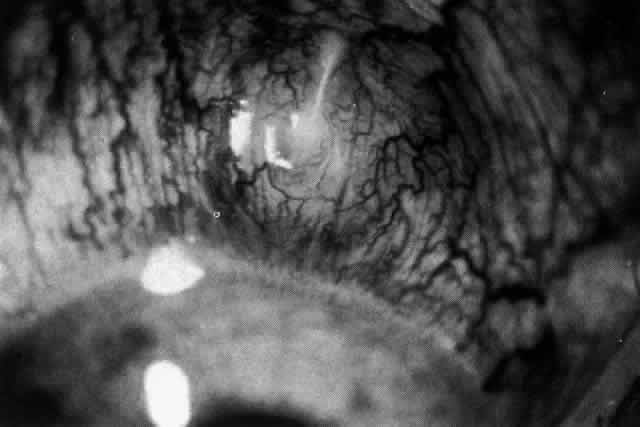 Fig. 40. Early necrotizing scleritis. There is characteristic yellow discoloration
of the sclera underlying the conjunctiva at a point of necrosis. In
this instance a small filament of tissue has penetrated the conjunctiva. Fig. 40. Early necrotizing scleritis. There is characteristic yellow discoloration
of the sclera underlying the conjunctiva at a point of necrosis. In
this instance a small filament of tissue has penetrated the conjunctiva.
|
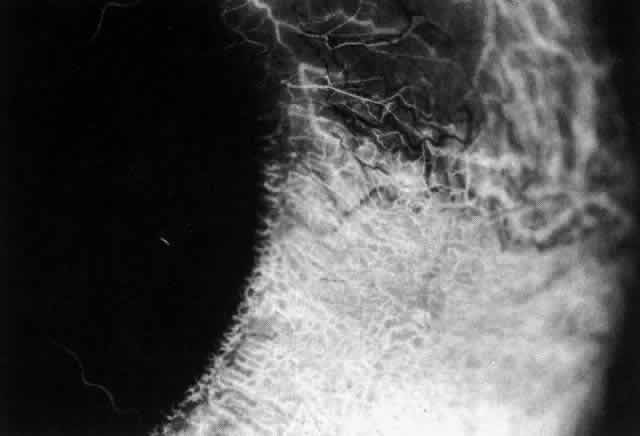 Fig. 41. Late stage of fluorescein angiogram adjacent to the site of necrosis in
the same patient as in Figure 40. Although the eye is uniformly congested, the area near the necrosis shows
vascular shutdown, whereas the rest of the conjunctiva and episclera
is normally perfused. Fig. 41. Late stage of fluorescein angiogram adjacent to the site of necrosis in
the same patient as in Figure 40. Although the eye is uniformly congested, the area near the necrosis shows
vascular shutdown, whereas the rest of the conjunctiva and episclera
is normally perfused.
|
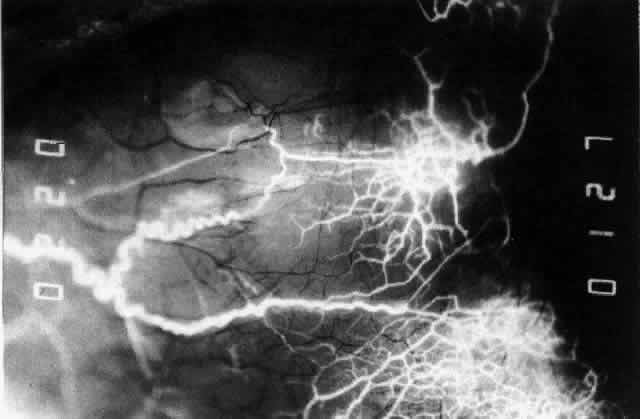 Fig. 42. Late arterial phase of fluorescein angiogram in a patient with necrotizing
scleritis. All the vessels except the main trunk and the vessels around
the limbal perforating vessels are occluded and remain unperfused
throughout the angiogram. Fig. 42. Late arterial phase of fluorescein angiogram in a patient with necrotizing
scleritis. All the vessels except the main trunk and the vessels around
the limbal perforating vessels are occluded and remain unperfused
throughout the angiogram.
|
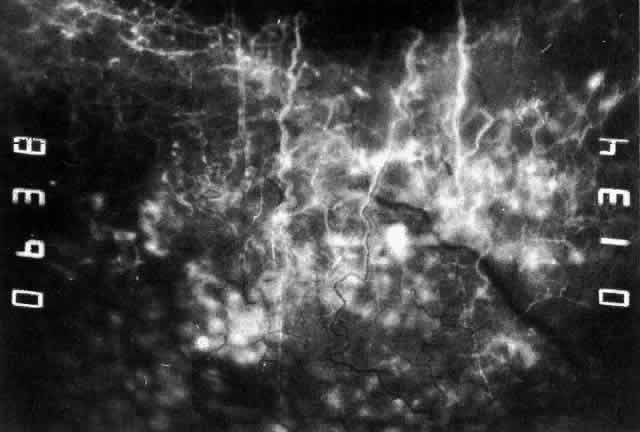 Fig. 43. Late venous phase of angiogram of a patient with necrotizing scleritis
showing late deep leakage from vessels on the surface of the sclera and
leakage of the capillary network at the limbus and the vessels draining
it, together with poor or absent perfusion of the remaining vessels. Fig. 43. Late venous phase of angiogram of a patient with necrotizing scleritis
showing late deep leakage from vessels on the surface of the sclera and
leakage of the capillary network at the limbus and the vessels draining
it, together with poor or absent perfusion of the remaining vessels.
|
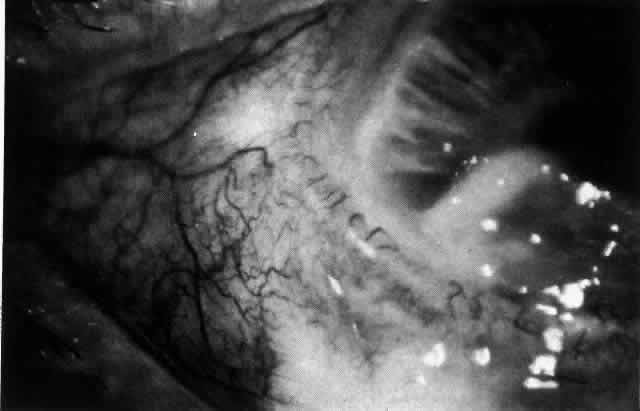
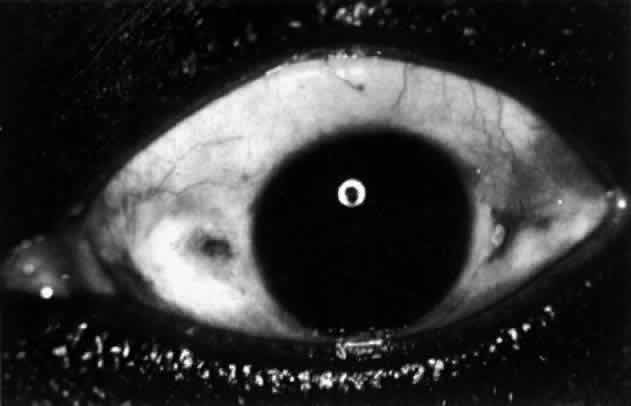
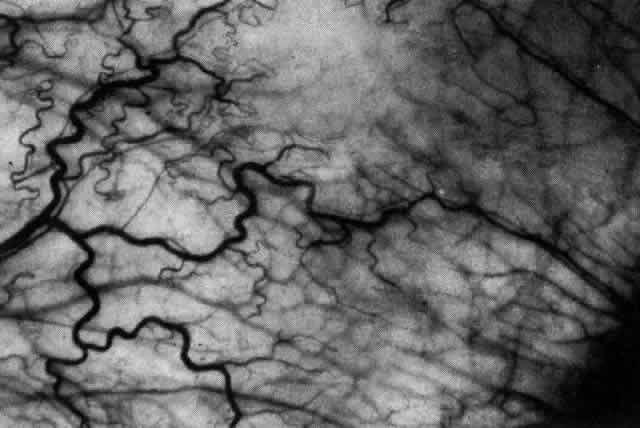 Fig. 44. Necrotizing scleritis. An avascular patch is seen in red-free light. If
left untreated, this will progress to the situation found in Figure 45. Fig. 44. Necrotizing scleritis. An avascular patch is seen in red-free light. If
left untreated, this will progress to the situation found in Figure 45.
|
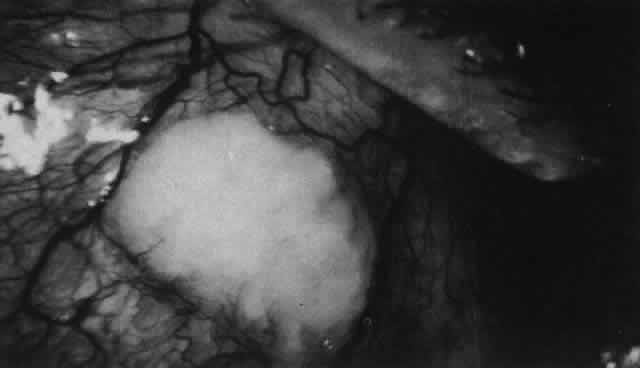 Fig. 45. Necrotizing scleritis. An area of necrosis is evident in the eye of this
patient with localized Wegener's granulomatosis. The conjunctiva
adjacent to the white necrotic tissue becomes adherent to the underlying
episclera. Fig. 45. Necrotizing scleritis. An area of necrosis is evident in the eye of this
patient with localized Wegener's granulomatosis. The conjunctiva
adjacent to the white necrotic tissue becomes adherent to the underlying
episclera.
|
Uveitis, lens changes, glaucoma and other serious complications such as
central vein occlusion do not seem to occur until the disease process
affects the whole circumference of the eye.27 Sometimes scleral thinning, as well as increased transparency, occurs, but
unless the intraocular pressure rises above 40 mm Hg, staphylomas
are extremely rare. As the disease is brought under control, the necrotic
areas are absorbed or sequestered, leaving an ectasia with the underlying
uvea exposed or covered with a thin film of conjunctiva or episclera (Fig. 46). If the defect is small, new collagen will cover it (Figs. 47 and 48). If the defect is large or if it is thought to have been the source of
a persisting antigenic stimulus, the necrotic tissue may need to be
excised and then covered by scleral grafts. However, this procedure is
usually performed for aesthetic reasons rather than because the vision
is endangered. The underlying disease process is not affected by the
presence of a scleral graft, which has to be covered by conjunctiva and, preferably, episclera
if it is to survive. Surgery must never be undertaken
until the disease process has been suppressed. Temporary gluing
may be used in perforated eyes until this has been achieved. 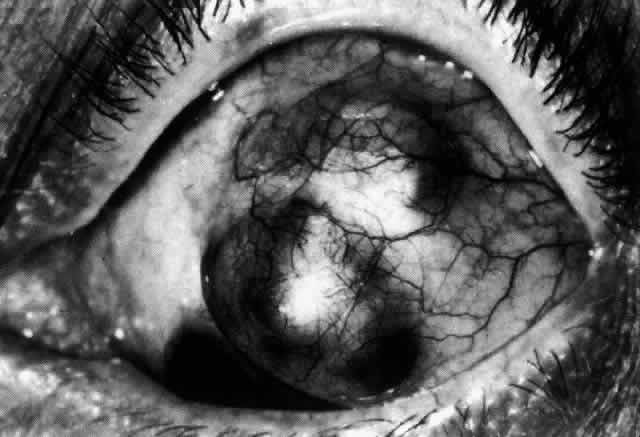 Fig. 46. Localized anterior staphyloma after necrotizing anterior scleritis and
seconday glaucoma, during the acute phase of which the intraocular pressure
rose to 50 mm Hg. Fig. 46. Localized anterior staphyloma after necrotizing anterior scleritis and
seconday glaucoma, during the acute phase of which the intraocular pressure
rose to 50 mm Hg.
|
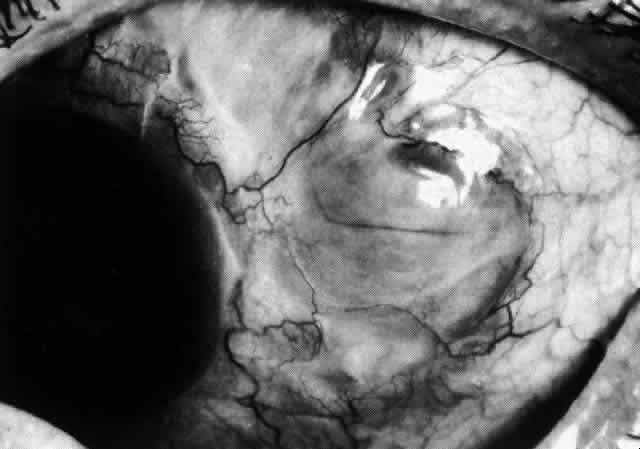 Fig. 47. Healing of a large scleral defect. Sclera is flat, without staphyloma formation. Newly
formed fibers are thin and radially arranged but adequate
to support a normal intraocular pressure. (Courtesy of Mr. HE Hobbs) Fig. 47. Healing of a large scleral defect. Sclera is flat, without staphyloma formation. Newly
formed fibers are thin and radially arranged but adequate
to support a normal intraocular pressure. (Courtesy of Mr. HE Hobbs)
|
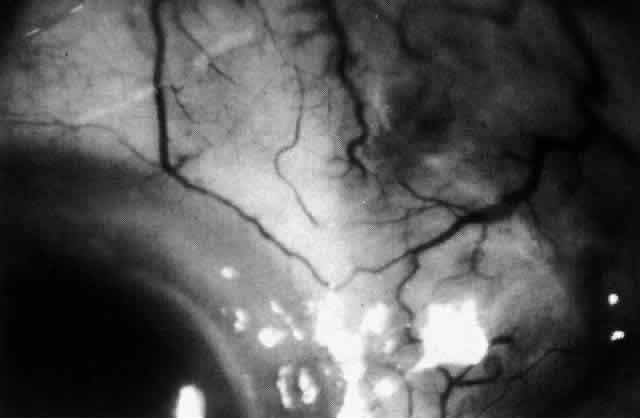 Fig. 48. Inactive necrotizing scleritis. The vessels have remained abnormal. The
sclera has been absorbed, leaving the deep, blue-gray choroid visible
beneath a very thin layer of conjunctiva. The adjacent cornea shows pitting
and lipid deposition because of the impaired venous drainage in
the adjacent episclera and conjunctiva. Fig. 48. Inactive necrotizing scleritis. The vessels have remained abnormal. The
sclera has been absorbed, leaving the deep, blue-gray choroid visible
beneath a very thin layer of conjunctiva. The adjacent cornea shows pitting
and lipid deposition because of the impaired venous drainage in
the adjacent episclera and conjunctiva.
|
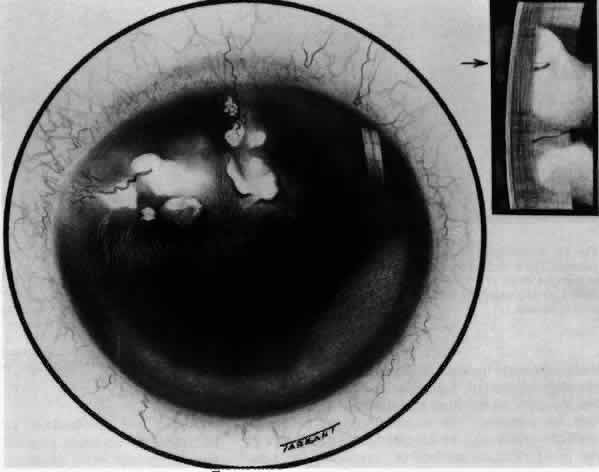
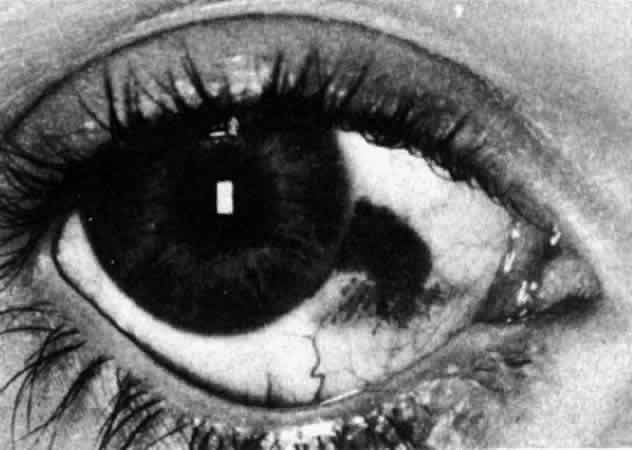
Necrotizing Anterior ScleritisdWithout Adjacent Inflammationd(Scleromalacia
Perforans) Necrotizing anterior scleritis without adjacent inflammation appears to
be a well-defined condition with little relation in clinical features
to necrotizing scleral disease, even though the pathology is similar
and the final result is the same. Scleromalacia perforans is characterized
by the almost total lack of any symptoms. It occurs almost exclusively
in patients with long-standing polyarticular rheumatoid arthritis, the
majority of whom are female (Figs. 49 and 50; Color Plate 1E). 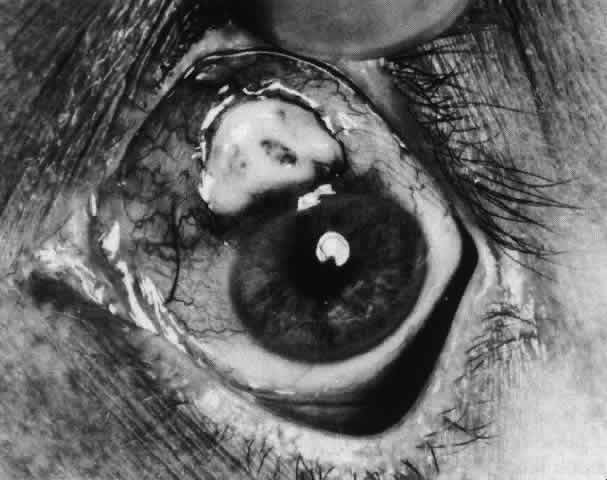 Fig. 49. A white necrotic plaque developing in an area of sclera with practically
no surrounding inflammation in a 60-year-old woman who had had Crohn's
disease for 17 years. Fig. 49. A white necrotic plaque developing in an area of sclera with practically
no surrounding inflammation in a 60-year-old woman who had had Crohn's
disease for 17 years.
|
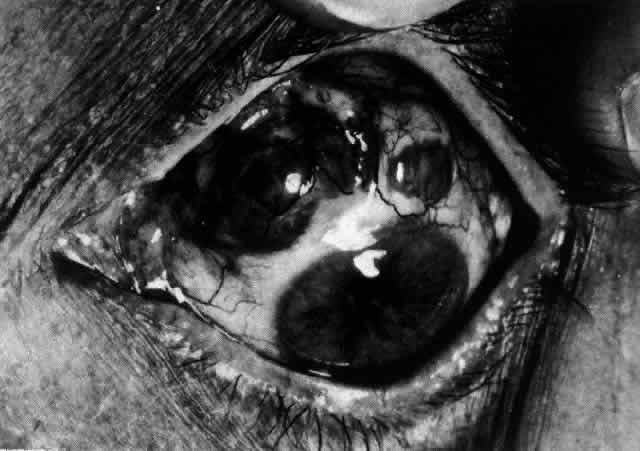 Fig. 50. Scleromalacia perforans after treatment. The very thin sclera is covered
by conjunctiva only and a few remaining large blood vessels. (Courtesy of Mr. HE Hobbs) Fig. 50. Scleromalacia perforans after treatment. The very thin sclera is covered
by conjunctiva only and a few remaining large blood vessels. (Courtesy of Mr. HE Hobbs)
|
The anterior sclera loses its covering of episclera and develops an area
of yellow-white necrotic slough over many months; this eventually separates
or is absorbed, leaving the underlying choroid covered by either
conjunctiva or nothing at all. As with necrotizing disease, the choroid
does not bulge into this ectatic area; but unlike necrotizing disease, spontaneous
healing of even small perforations is very limited once
the necrotic tissue has been removed (see Fig. 50). Fluorescein angiography is not helpful, except to indicate areas of vascular
closure in an otherwise extremely thin, atrophic episcleral tissue.4 The formation of a sequestrum appears to be caused by arteriolar closure
as opposed to the venular disease seen in the other forms of necrotizing
scleritis. Posterior Scleritis Because the posterior sclera is invisible, the diagnosis of posterior scleritis
is made only if the anterior sclera is also involved or some
other sign or symptom leads one to suspect it. Posterior scleritis is
much more common than previously suspected, as recent clinical and pathologic
studies have shown.19,28,29 There are two distinct forms of posterior scleritis. The first is usually
associated with an anterior scleritis. This granulomatous disorder, like
its anterior counterpart, can be diffuse, nodular, or necrotizing
in character and is associated with the connective tissue diseases. The
second form occurs in young patients of all races who are 9 to 40 years
of age. It is always diffuse in character but is not associated
with any systemic disorder. Both forms may cause uveitis if the inflammation
affects the ciliary body, and in both forms the patient may develop
exudative retinal detachments, choroidal folds, and swelling of
the disc (Figs. 51 and 52). The granulomatous type may also involve the structures outside the globe, causing
proptosis (Fig. 53), limitation of ocular muscle movement, and, uniquely, retraction of the
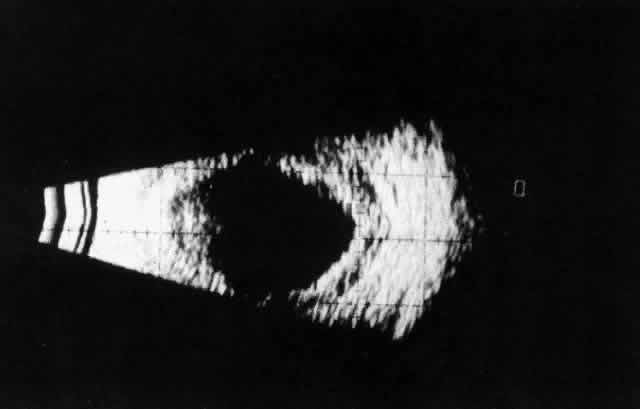
lower lid on attempted elevation of the eye (Fig. 54). Diagnosis is with B-scan ultrasonography. 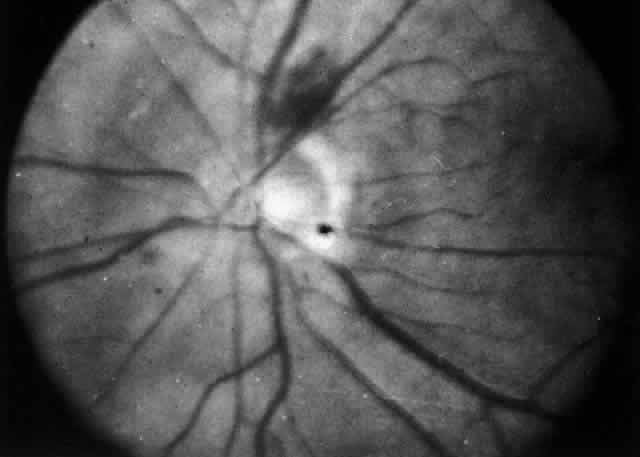 Fig. 51. Swelling of the optic nerve head and hemorrhage near the disc in a patient
with posterior scleritis. The poor quality of the photograph is partly
due to vitreous haze that accompanied the inflammation. Fig. 51. Swelling of the optic nerve head and hemorrhage near the disc in a patient
with posterior scleritis. The poor quality of the photograph is partly
due to vitreous haze that accompanied the inflammation.
|
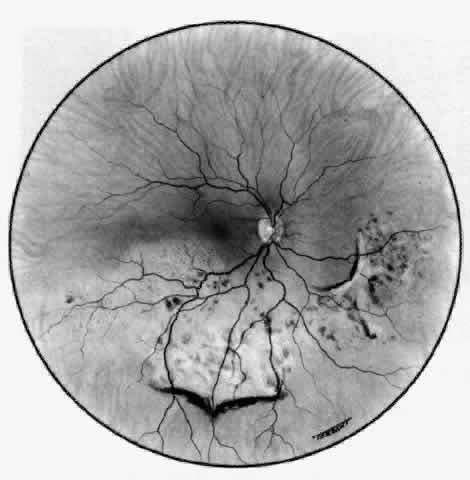 Fig. 52. Fundus appearance after resolution of exudative detachment in patient with
severe posterior scleritis. Macula was affected and vision much impaired. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5. London, Churchill-Livingstone, 1975) Fig. 52. Fundus appearance after resolution of exudative detachment in patient with
severe posterior scleritis. Macula was affected and vision much impaired. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5. London, Churchill-Livingstone, 1975)
|
TREATMENT Scleritis is almost always accompanied by very severe pain that prevents
sleep. A response to treatment is heralded by a dramatic relief of pain
even though the condition might appear to be getting worse (Figs. 55 through 59). Treatment may be modified with confidence once the pain has disappeared. 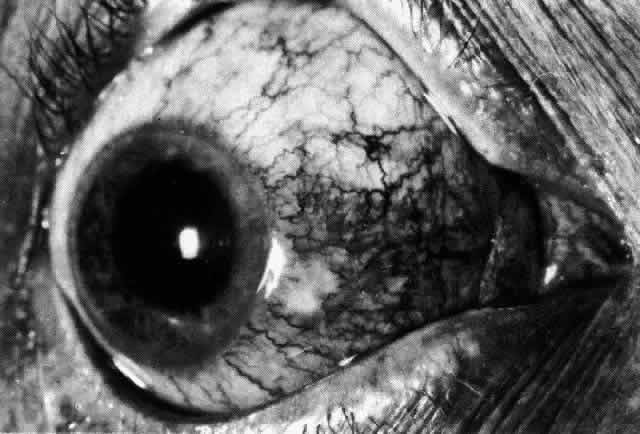 Fig. 55. Necrotizing scleritis 2 days after onset of severe pain in eye and temple. The
eye was red, the sclera edematous, and the overlying episclera
congested. A small paralimbal avascular area was noted. The patient was
treated with 400 mg oxyphenbutazone for 7 days. (Watson PG: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972 Fig. 55. Necrotizing scleritis 2 days after onset of severe pain in eye and temple. The
eye was red, the sclera edematous, and the overlying episclera
congested. A small paralimbal avascular area was noted. The patient was
treated with 400 mg oxyphenbutazone for 7 days. (Watson PG: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972
|
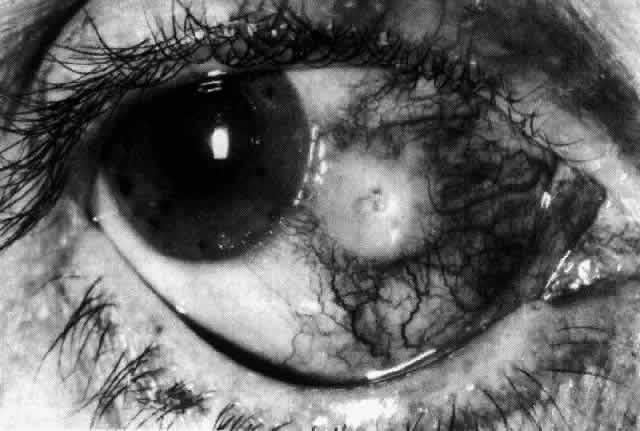 Fig. 56. Appearance of same eye as seen in Figure 55 one week later. The pain persisted, and the avascular patch was much enlarged
with a necrotic center. Treatment was not altered, but the patient
was admitted to the hospital. (Watson PG, Hayreh S, Awdry P: Episcleritis and scleritis. Br J Ophthalmol 52(3): 278–279, 1968) Fig. 56. Appearance of same eye as seen in Figure 55 one week later. The pain persisted, and the avascular patch was much enlarged
with a necrotic center. Treatment was not altered, but the patient
was admitted to the hospital. (Watson PG, Hayreh S, Awdry P: Episcleritis and scleritis. Br J Ophthalmol 52(3): 278–279, 1968)
|
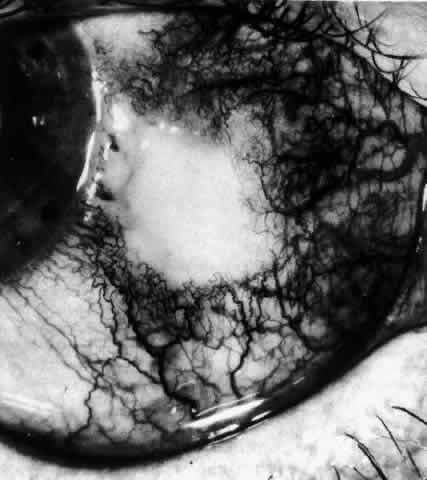 Fig. 57. Appearance of the same eye as seen in Figure 56 five days later. The area has increased in size, and the necrotic center
is beginning to separate. Treatment was changed to 100 mg prednisolone
daily. Within 24 hours, the severe ocular and facial pain had disappeared, but
the appearance of the eye did not change. Fig. 57. Appearance of the same eye as seen in Figure 56 five days later. The area has increased in size, and the necrotic center
is beginning to separate. Treatment was changed to 100 mg prednisolone
daily. Within 24 hours, the severe ocular and facial pain had disappeared, but
the appearance of the eye did not change.
|
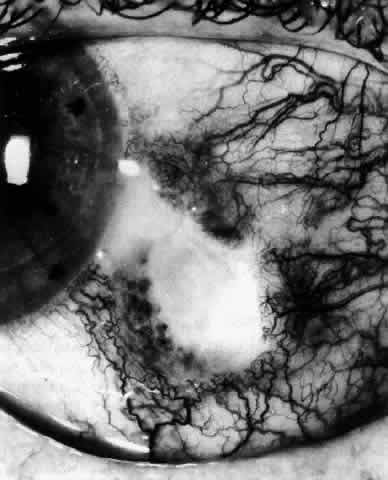 Fig. 58. Appearance of the same eye as in Figure 57. After 5 days' treatment with a high dosage of steroids, new vessels can
be seen growing into the necrotic area both superficially and deep. The
steroids were reduced to a maintenance level of 20 mg and continued
for 6 weeks. (Watson PG: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972) Fig. 58. Appearance of the same eye as in Figure 57. After 5 days' treatment with a high dosage of steroids, new vessels can
be seen growing into the necrotic area both superficially and deep. The
steroids were reduced to a maintenance level of 20 mg and continued
for 6 weeks. (Watson PG: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972)
|
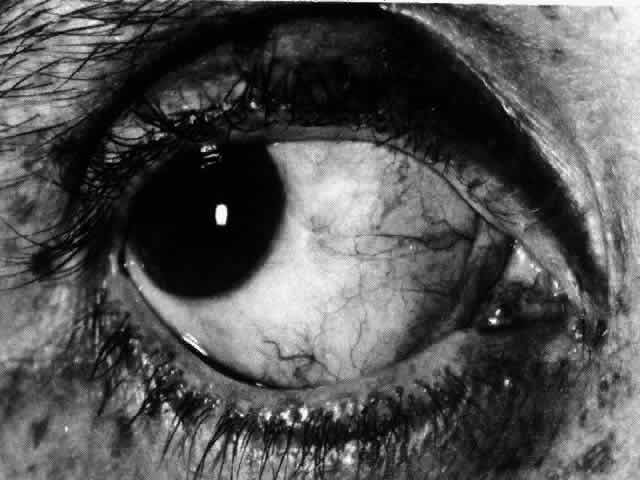 Fig. 59. Same eye as seen in Figures 55 through 58. Four months after the onset of disease, the ulcerated area has completely
healed and filled in with new collagen material, which has assumed
the usual radial pattern of scleral fibers. (Watson PE: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972) Fig. 59. Same eye as seen in Figures 55 through 58. Four months after the onset of disease, the ulcerated area has completely
healed and filled in with new collagen material, which has assumed
the usual radial pattern of scleral fibers. (Watson PE: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972)
|
Local Corticosteroids Local steroid therapy increases the patient's comfort, but it is not
effective in suppressing scleral inflammation. It is occasionally justified
to use local steroid therapy alone when the inflammation is mild, the
pain is slight, and corneal involvement is present, or very occasionally
between attacks in the more severe forms of the disease to
prevent remission. However, local steroids should be used only sparingly, if
at all, in scleral disease because of the high chance of developing
steroid-induced glaucoma or cataract. Systemic Therapy NONSTEROIDAL ANTI-INFLAMMATORY AGENTS. Nonsteroidal anti-inflammatory agents are effective in suppressing the
inflammatory response in the majority of patients with diffuse and nodular
scleritis, especially if they exhibit a high flow pattern on fluorescein
angiography. Dosage levels need to be high initially and, as a
consequence, care must be taken to monitor the patients to ensure that
no toxic side effects occur. Treatment must be continued until the inflammation
subsides, after which it can be stopped abruptly. In assessing the effect of treatment, pain, tenderness, episcleral and
scleral injection, and corneal and intraocular involvement should be used
as parameters of activity of the disease. In a series of double-blind
controlled trials, the effects of different anti-inflammatory and
immunosuppressive agents have been compared. The suggested routines of
treatment are based on the results of these trials. Unfortunately, not
all of the nonsteroidal anti-inflammatory agents are effective in controlling
scleral inflammation. The current practice is to use flurbiprofen (Froben), 100 mg
three times daily, for at least 1 week in all patients
who present with scleritis of whatever type, provided there is
no evidence of vascular closure or scleral destruction on slit lamp examination. The
response, if it is going to occur, is immediate, with
the pain disappearing within 48 hours. Within a week, the results of investigations
are known, including those of the angiographic films if
they have been done. If there is a poor response to flurbiprofen and the
angiogram shows a high flow pattern, the drug is changed to another
nonsteroidal anti-inflammatory agent, because there is an individual
susceptibility among the patients. Only if there is no response in the
progression of the disease or if there is evidence of vascular closure
are systemic steroids or other immunosuppressive drugs used. SYSTEMIC STEROID THERAPY. If the scleritis is severe or necrotizing or if areas of vascular closure
are detected with slit lamp examination or fluorescein angiography, then
the use of systemic steroids is mandatory (see Figs. 40 through 44). Prednisone and prednisolone are most commonly used. The principle of treatment with systemic steroids is that a sufficient
amount must be given to suppress the condition; once this has occurred, the
dosage may be rapidly reduced to a maintenance level, which may
have to be continued until a natural remission occurs, or the steroid
may be replaced by a nonsteroidal anti-inflammatory agent. Provided sufficient
amounts are given and the patient can tolerate them, systemic
steroids will control scleritis. The problem is deciding what dosage
is appropriate. The following scheme has been found to be effective. If
the angiogram shows early vascular shutdown and treatment with flurbiprofen
has not been effective, oral prednisolone, 60 or 80 mg, is given
for 2 days and is reduced over 1 week to 20 mg. The dose of prednisolone
is then reduced by 2.5 mg every other day until the pain recurs
or signs of inflammation begin to recur. This maintenance dose is continued
for about 1 month, and then the dose is reduced by 1-mg steps. This
final phase may be aided by the addition of a nonsteroidal anti-inflammatory
drug. Pain relief is by far the most sensitive indicator of
control of the disease. If this course of treatment is not effective, intravenous pulse therapy
of high doses of methylprednisolone, with or without the use of immunosuppressive
therapy, should be considered (Fig. 60). 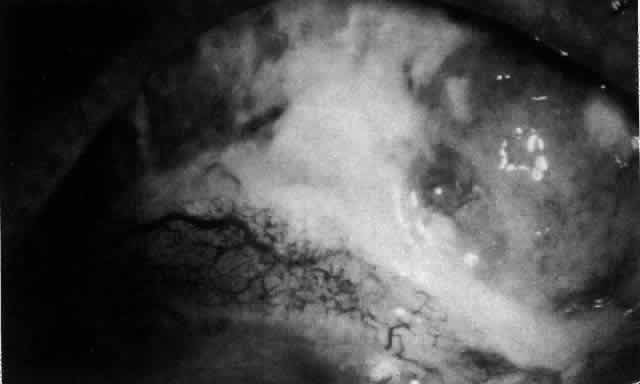 Fig. 60. Severe necrotizing scleritis. The surface of the necrotic sclera is covered
with a thick, sticky, white mucus. The presence of such mucus is
a sensitive indicator that the disease is active. This patient required
two pulses of 1 g of methylprednisolone and 500 mg intravenous cyclophosphamide (5 days
apart) to bring the condition under control. Thereafter, systemic
low-dosage steroid (5 mg daily) and 50 mg of cyclophosphamide
maintained control. The first sign of poor control was the reappearance
of the mucus. Fig. 60. Severe necrotizing scleritis. The surface of the necrotic sclera is covered
with a thick, sticky, white mucus. The presence of such mucus is
a sensitive indicator that the disease is active. This patient required
two pulses of 1 g of methylprednisolone and 500 mg intravenous cyclophosphamide (5 days
apart) to bring the condition under control. Thereafter, systemic
low-dosage steroid (5 mg daily) and 50 mg of cyclophosphamide
maintained control. The first sign of poor control was the reappearance
of the mucus.
|
Pulsed Therapy With Methylprednisolone. In very severe necrotizing scleritis or posterior scleritis when the vision
is being threatened, it is important to suppress the inflammatory
reaction completely. This is achieved by the use of intravenous methylprednisolone. The
patient must be carefully monitored because this therapy
inhibits all cellular transfer mechanisms, including the heart, and
is therefore potentially dangerous. Methylprednisolone is administered
by intravenous infusion as 500 mg or 1 g given over a period of 1 hour. The
clinical response is observed. If this is unsatisfactory, then
the dose may be repeated at 1- to 3-day intervals for three doses. It
may be necessary to add other immunosuppressives to this regimen. Immunosuppressive Therapy The commonly used immunosuppression drugs used in the control of scleral
disease are cyclophosphamide, cyclosporine, azathioprine, methotrexate
or chlorambucil. Cyclophosphamide is the drug of choice in patients
with systemic vasculitis, periarteritis nodosa, and Wegener's granulomatosis
if they are not of child-bearing age. In severe cases of
these disorders, cyclophosphamide is given with methylprednisolone as
a bolus of 500 mg. A high fluid intake is essential to prevent hemorrhagic
cystitis. Cyclophosphamide is then given orally at a dose of 50 mg
three times per day. This treatment has a profound effect on the lymphocyte
count, which must be monitored at weekly intervals for the first
few months of treatment. Azathioprine can also be used as an adjunct
to steroid therapy in those patients who require an unacceptably high
maintenance dose of steroid (i.e., 15 mg prednisolone or more) to control their disease and in those with
known immune complex disease (Fig. 61). Cyclosporine is helpful sometimes. Considering that pathologically there
is always evidence of T-cell activation, it might be expected that
cyclosporine would be the drug of choice in these diseases. Experience
shows that this is not the case (in sharp contrast to the treatment
of uveitis). Cyclosporine should therefore remain a second-line treatment
and should be reserved for recalcitrant cases. It is strongly recommended
that ophthalmologists always work in close cooperation with internists
or other physicians when undertaking immunosuppressive therapy. 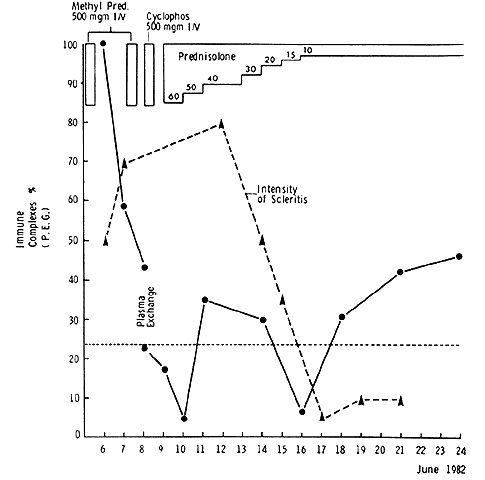 Fig. 61. The course of treatment in a patient who had lost the sight in one eye
from posterior scleritis and who had developed a severe posterior scleritis
in the other eye. She had extremely high circulating immune complexes, but
eliminating these alone was not sufficient to control the scleral
disease. Control was maintained for 3 years after the episode, using 7.5 mg
prednisolone and 50 mg cyclophosphamide daily. Two exacerbations
required treatment with short 7-day courses of oral prednisolone. (Watson PG: The nature and treatment of scleral inflammation [Doyne
Memorial Lecture]. Trans Ophthalmol Soc UK 102:257–281, 1982) Fig. 61. The course of treatment in a patient who had lost the sight in one eye
from posterior scleritis and who had developed a severe posterior scleritis
in the other eye. She had extremely high circulating immune complexes, but
eliminating these alone was not sufficient to control the scleral
disease. Control was maintained for 3 years after the episode, using 7.5 mg
prednisolone and 50 mg cyclophosphamide daily. Two exacerbations
required treatment with short 7-day courses of oral prednisolone. (Watson PG: The nature and treatment of scleral inflammation [Doyne
Memorial Lecture]. Trans Ophthalmol Soc UK 102:257–281, 1982)
|
SUBCONJUNCTIVAL AND ORBITAL FLOOR STEROIDS. Treatment with subconjunctival steroids is contraindicated in scleritis. Perforation can occur at the site of subconjunctival injections (Fig. 62). Depot steroids should not be used because the particulate matter may
induce or perpetuate the inflammatory reaction in the sclera. Orbital
floor steroids are occasionally helpful in patients with necrotizing
disease who are unable to take systemic steroids. The effects unfortunately
tend to be transient, and the injections often need to be repeated
at 7- to 10-day intervals. In this situation, intravenous pulse therapy
should be considered as an alternative. 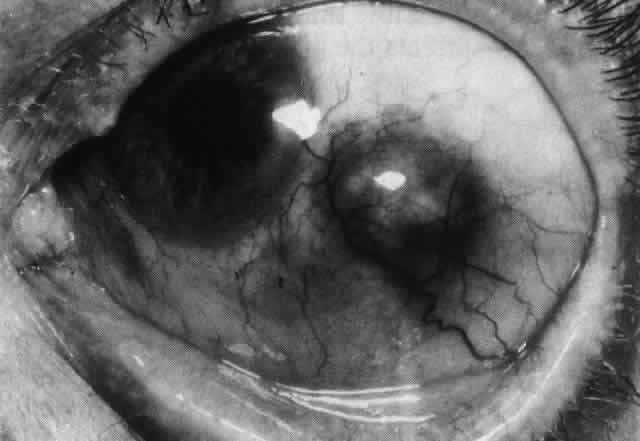 Fig. 62. Extreme scleral thinning in nasal and inferior quadrants surrounding a
deposit of hyodrocortisone given for suppression of scleral disease. All
sclera in the area later disappeared, and the eye threatened to perforate. Fig. 62. Extreme scleral thinning in nasal and inferior quadrants surrounding a
deposit of hyodrocortisone given for suppression of scleral disease. All
sclera in the area later disappeared, and the eye threatened to perforate.
|
Surgery Surgical treatment for defects in the sclera is rarely necessary. Adequate
medical therapy allows the base of all small scleral defects to be
covered by newly formed collagen, rendering them safe from perforation (see Fig. 59). However, very large defects may have to be covered with sclera or cornea. Provided
these grafts can be covered by conjunctiva, they usually remain
in place, apparently viable. Scleral grafts ensure the comfort of
the patient but do not prevent progressive necrotizing disease in the
host sclera or even the graft (Figs. 63 and 64). Scleral replacement should be performed with the use of cornea rather
than scleral tissue. Sclera rapidly resorbs, whereas corneal tissue
is attacked only if there is a recurrence of the original disease. 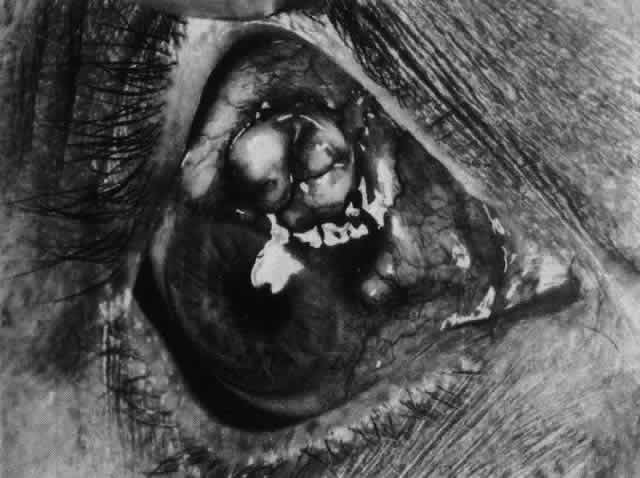 Fig. 63. Severe necrotizing scleritis. Fig. 63. Severe necrotizing scleritis.
|
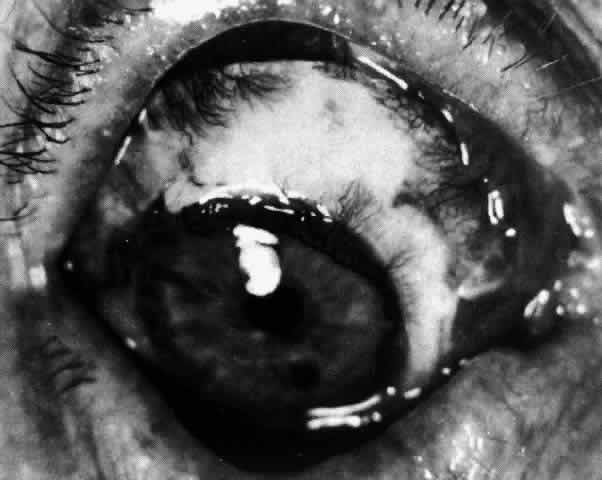 Fig. 64. Scleral graft in eye shown in Figure 63. Three months later the scleritis was still being treated with steroids. New
vessels invaded the graft, which later took on the appearance of
normal sclera. Fig. 64. Scleral graft in eye shown in Figure 63. Three months later the scleritis was still being treated with steroids. New
vessels invaded the graft, which later took on the appearance of
normal sclera.
|
Keratolysis or progressive peripheral corneal thinning sometimes requires
lamellar corneal grafting (Figs. 65 and 66). Penetrating keratoplasty should be avoided if possible. The endothelium
always remains normal in sclerokeratitis, and because of the proximity
of the grafts to the limbus, larger penetrating grafts do less well. Under
no circumstances should surgery be attempted until the systemic
disease and ocular inflammation are brought under control. If necessary, cyanacrylate
glue can be used to seal a perforation until immunosuppression
is achieved. Methylprednisolone 500 mg should be given during
the operation and, if required, after surgery. 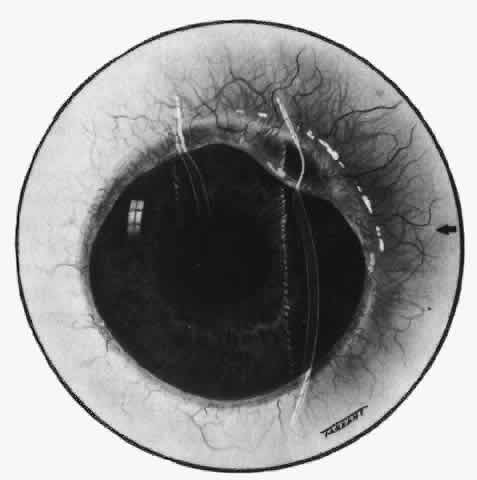 Fig. 65. Sclerokeratitis. Terrien-like ulcer at site of long-standing scleritis. The
eye began to expand in this area after 6 years. (Watson PG: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972) Fig. 65. Sclerokeratitis. Terrien-like ulcer at site of long-standing scleritis. The
eye began to expand in this area after 6 years. (Watson PG: Contemporary Ophthalmology. Baltimore, Williams & Wilkins, 1972)
|
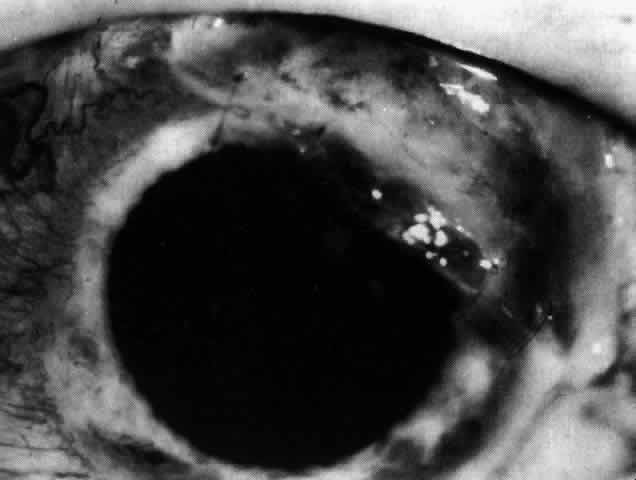 Fig. 66. The affected area of cornea seen in Figure 65 covered by a corneal graft. The scleritis has since settled and no longer
requires treament. Fig. 66. The affected area of cornea seen in Figure 65 covered by a corneal graft. The scleritis has since settled and no longer
requires treament.
|
COMPLICATIONS Complications occur late in the disease and vary with the severity of inflammation. They
occur most frequently in posterior scleritis and in
severe necrotizing disease, particularly when the condition has become
circumferential and when the inflammation is so severe as to produce
secondary intraocular inflammation. Visual Acuity The object of early diagnosis and treatment is to prevent a decrease in
visual acuity. The treatment must not produce iatrogenic changes that
cause decreased acuity. Over a 3-year period, approximately 27% of the patients who develop this
disease will experience a decrease in visual acuity of two or more lines, which
can be the result of cataracts and keratitis developing in
patients with severe diffuse anterior scleritis. However, over a 25-year
period, only 3% have lost useful vision. Increased Scleral Transparency and Thinning Alteration in the collagen and ground substance results in increased scleral
transparency. Scleral thinning occurred, particularly in necrotizing
disease or scleromalacia perforans. Of these patients, 22% showed
increased scleral transparency after the first attack; however, only 6% developed
a scleral defect. If scleral defects are small, they will
refill with new collagen after treatment; but if they are very large, they
may have to be covered with a graft (see Figs. 63 and 64). Uveitis Although roughly 35% of patients with scleral disease show some evidence
of cellular activity in either the anterior or the posterior segment, a
severe uveitis with a marked flare and heavy cellular response is
very unusual. If it does occur, it is a serious sign, and intensive treatment
must be instituted at once with systemic steroids. In posterior
scleritis, if the granuloma is behind the equator, there may be little
or no intravitreal cellular reaction, even though there is a visible
granuloma and a retinal detachment. Scleritis occurring between the
pars plana and the equator affects the ciliary body, so some inflammatory
response occurs. Unless patients with this form of inflammation are
treated rapidly, the intraocular pressure sometimes rises disastrously. Most
patients with posterior scleritis have high intraocular pressures
at some stage in the disease. As the scleral disease is brought under control, the uveitis resolves, leaving
anterior and posterior synechiae unless care is taken to prevent
them. The inflammation of the pars plana sometimes leads to massive
pigment migration at the retinal periphery, leaving a reaction rather
like a diathermy or cryotherapy reaction in retinal detachment surgery (see Fig. 52). Glaucoma The intraocular pressure may become raised at any stage of the disease
because of an acute congestion of the outflow channels,27 raised episcleral venous pressure, angle closure, or a steroid-induced
rise. Therefore, it is important that the intraocular pressure be monitored; 13.5% of
all patients with nodular or necrotizing scleritis had
a pressure rise, albeit transient, during the course of the disease. Permanent
field changes occurred in 5%. Patients with posterior scleritis
are particularly prone to develop rises of intraocular pressure. The treatment of the glaucoma is the treatment of the scleritis. Once the
scleritis is controlled, the pressure will fall to normal. While the
eye is inflamed, particularly if there is a limbitis, acetazolamide
should be used to control the intraocular pressure. Should the pressure
remain high after the attack, topical timolol can often help to control
the intraocular pressure. If control fails, trabeculectomy can be
performed successfully in an area of normal sclera and conjunctiva. Cataract Involutional changes that are already present will be increased by the
presence of a severe inflammation. However, there is no doubt that the
transparency of the lens can be affected directly in patients who have
had previously normal lenses and who have developed severe necrotizing
scleral disease. If a cataract advances to the extent that it requires removal, the extraction
can be performed with use of a corneal section in spite of the
presence of scleritis. Healing is a little delayed in some cases, but
no operative or postoperative complications have occurred. Cataract extraction and, for that matter, any other surgical procedure
can precipitate scleral inflammation in a patient who is predisposed, usually
because of circulating immune complex disease. These patients
usually have necrotizing scleritis and require vigorous therapy (Fig. 67).23–25,30–33 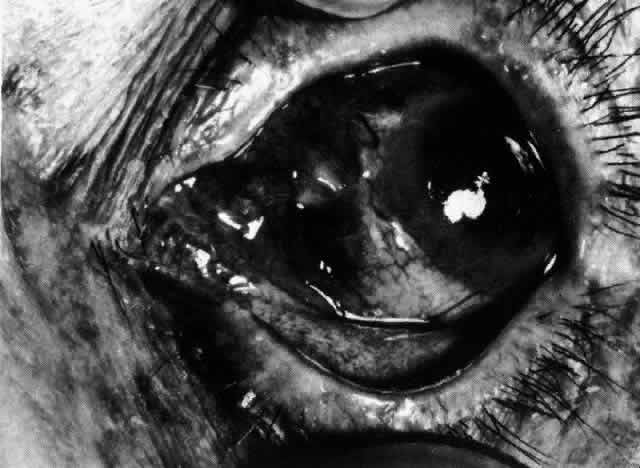 Fig. 67. Severe necrotizing scleritis that started in the wound edges 3 weeks after
cataract extraction. The patient had no previous history of eye disease
other than involutional cataract. Ill-advised subconjunctival depot
steroid led to tissue loss at 8 o'clock. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5. London, Churchill-Livingstone, 1975) Fig. 67. Severe necrotizing scleritis that started in the wound edges 3 weeks after
cataract extraction. The patient had no previous history of eye disease
other than involutional cataract. Ill-advised subconjunctival depot
steroid led to tissue loss at 8 o'clock. (Watson PG: Management of scleritis. In: Recent Advances in Ophthalmology, Vol 5. London, Churchill-Livingstone, 1975)
|
Retinal Detachment Exudative retinal detachment occurs in patients who have posterior scleritis, and
it may, indeed, be the only sign in a very painful eye. The
detachment is poorly mobile. A pale gray granuloma can be seen extending
from the choroid beneath the retina and is accompanied by a poorly
mobile serous detachment that may become total. The scleral granuloma
sometimes leaves a permanent, inward indentation of the retina and a
subretinal mass, although this does not always occur. An increasing hypermetropia
has also been noted; it is of rapid onset (over a period of 1 week) and
is caused by the diffuse scleral edema in the early stages
of the disease before the detachment of the retina occurs. The exudative detachment usually resolves completely with treatment of
the scleritis. However, if the inflammatory changes have affected the
macular area, vision will be severely and permanently affected. After
resolution, the retina shows a diffuse, heavy pigmentation of the affected
area with a “high-water mark” at the edge (see Fig. 52). Patchy changes outside this area do not seem to occur. Surgery is not
indicated. Optic Nerve Swelling Granulomatous processes inside the muscle cone or affecting the optic nerve
sheaths may be accompanied by edema of the optic nerve (see Fig. 51). Although it is not possible to make a diagnosis of posterior scleritis
on the basis of this sign alone, should there be severe pain, proptosis, limitation
of movement, and a retinal detachment, a presumptive
diagnosis is permissible; however, it can be confirmed only if the anterior
sclera becomes involved later in the disease. B-scan ultrasonography
is very helpful in defining granulomas involving the sclera and the
optic nerve. Swelling of the disc in patients who have presented with
anterior scleritis is unusual, but it has occurred in patients in whom
it was known that the process had advanced to involve the posterior
segment. |