RETINAL HEMORRHAGES
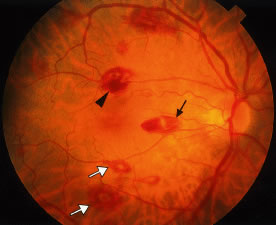
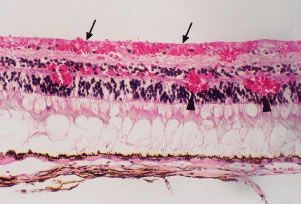
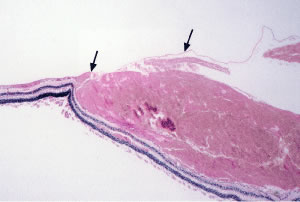
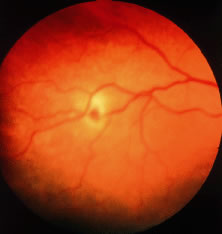
Retinal hemorrhages may take various configurations based on their location within the retina. Flame-shaped and dot-blot hemorrhages are the most commonly encountered intraretinal hemorrhages in blood dyscrasias (Fig. 1). Flame-shaped hemorrhages are located in the nerve fiber layer of the retina, and dot-blot hemorrhages are typically located in the inner nuclear and outer plexiform layers (Fig. 2). Large blot or boat-shaped hemorrhages may be observed and are present beneath the internal limiting membrane (ILM) of the retina (i.e., sub-ILM hemorrhage) (Fig. 3). These large superficial retinal hemorrhages may break through the ILM and extend into the vitreous cavity (Fig. 4). White-centered hemorrhages (see Fig. 1) and, much less commonly, red-centered infiltrates (Fig. 5) are seen in the retinae of patients with a blood dyscrasia. The white center may be associated with a leukemic embolus (Fig. 6) or more commonly a platelet-fibrin thrombus.1 The red center is blood, which may be associated with a cotton wool spot (CWS) or a leukemic retinal infiltrate.
|
|
|
|
|
|
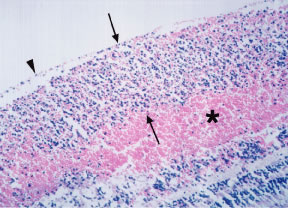
Factors that contribute to the development of retinal hemorrhages in the setting of blood dyscrasias include anemia, thrombocytopenia, elevated white blood cell count, and blood or serum hyperviscosity. In a study of 67 patients with anemia and/or thrombocytopenia, Rubenstein and coworkers2 found that retinal hemorrhage is much more likely to occur when anemia is accompanied by thrombocytopenia than when either is present alone. The same authors postulated that thrombocytopenia is an important causative factor in ocular bleeding in an anemic patient. In a study of 152 patients with blood diseases, Holt and Gordon-Smith3 reported that retinal hemorrhages occurred most often in leukemic patients with significantly more severe anemia and thrombocytopenia and a high percentage of circulating blast cells. In a prospective study of 117 patients with acute leukemia, Guyer and colleagues4 reported that thrombocytopenia is the most important factor in the pathogenesis of intraretinal hemorrhages. On the contrary, Jackson and coworkers found no association between intraretinal hemorrhages and the hemoglobin level or platelet count in 63 newly diagnosed acute leukemia patients.5 The same authors reported a higher median white blood cell count in patients with intraretinal hemorrhages than in those without intraretinal hemorrhages and concluded that a high white blood cell count may be at least as important as anemia and thrombocytopenia in the pathogenesis of retinopathy in acute leukemia.
In a 4-year prospective study of 82 patients with acute leukemia, Jackson and coworkers6 found that patients with a macular hemorrhage were at significantly greater risk for developing intracranial hemorrhage [particularly in the promyelocytic (M3) subtype of acute myeloid leukemia (AML)] within the first 30 days following diagnosis compared with patients without a macular hemorrhage. Based on these findings, the authors recommended that patients with a macular hemorrhage be monitored intensively for the development of intracranial hemorrhage and receive priority in the allocation of platelets when platelets are in short supply. Richards and colleagues7 also reported a high incidence of intraocular hemorrhage in patients with acute promyelocytic leukemia (M3 subtype of AML) and found no consistent detectable abnormalities in the hematologic parameters or coagulation studies predictive of ocular hemorrhage.
In a pathologic study of the eyes of 76 patients who died of leukemia and allied disorders, Allen and Straatsma8 found that retinal hemorrhage was the most frequent and serious ocular complication and that the most significant hemorrhages occurred in the acute forms of leukemia.
MICROANEURYSMS
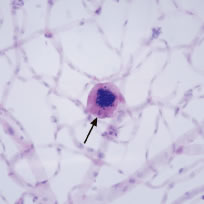
Microaneurysms are small outpouchings in the retinal capillary wall that appear as tiny red dots in the retina on ophthalmoscopic examination. They are classically seen in the posterior fundus in diabetic retinopathy. Microaneurysms have also been reported in leukemia and plasma cell dyscrasias.9–12 Interestingly, the microaneurysms in blood dyscrasias tend to be located in the peripheral retina, in contrast to the location in the posterior retina in diabetic retinopathy. In a pathologic study employing trypsin digestion of flat mounts of the retina, Duke and coworkers10 emphasized the relative preservation of pericytes in patients with chronic leukemia as compared with the marked loss of pericytes in diabetic retinopathy. Figure 7 shows the typical microaneurysms in the retinal periphery in a flat mount preparation with trypsin digestion in a patient with a blood dyscrasia [multiple myeloma (MM)]. Common pathologic features between the microaneurysms of diabetes and blood dyscrasias include the globular shape of the lesion, the location predominantly on the venous side of the capillary, and the presence of intramural and intraluminal periodic acid-Schiff (PAS)-positive deposits.10 Factors that may play a role in the formation of microaneurysms in the setting of blood dyscrasias include increased venous pressure [i.e., central retinal vein occlusion (CRVO)], increased blood viscosity with a secondary increase in venous pressure (i.e., hyperviscosity syndrome associated with plasma cell dyscrasias), and anoxia (i.e., severe anemia).
|
HARD EXUDATES
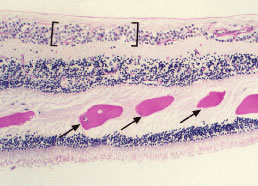
Hard exudates are refractile, yellowish deposits of proteins and lipids that are derived from incompetent or leaky retinal capillaries. Hyperpermeability of the retinal capillaries may be seen in hyperviscosity syndromes, retinal venous congestion, and CRVO secondary to plasma cell dyscrasias.13–15 The hard exudates that result from this capillary hyperpermeability are localized predominantly to the outer plexiform layer of the retina (Fig. 8).
|
RETINAL EDEMA
In addition to the formation of hard exudates, hyperpermeable or leaky retinal capillaries can lead to focal or generalized retinal edema. Mild opacification or graying of the normally transparent retina is noted in areas of retinal edema on ophthalmoscopic examination. Cystic degeneration of the retina may result from long-standing retinal edema. Retinal edema localized to the macular region may manifest as cystoid macular edema (CME). Histologically in CME, the cysts often contain eosinophilic proteinaceous material and are located in the outer plexiform and, to a lesser extent, in the inner nuclear layers of the retina (Fig. 9). Occasionally, aggregates of lipid-laden macrophages are observed in the regions of the cystic degeneration (see Fig. 9). As with hard exudates, retinal edema may be seen in hyperviscosity syndromes induced by the blood dyscrasias. The CME observed in blood dyscrasias may respond to specific treatment for CME (i.e., acetazolamide) and may resolve completely with treatment of the underlying disease process [i.e., after bone marrow transplantation in chronic myeloid leukemia (CML)].16
|
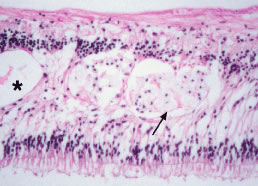
COTTON WOOL SPOTS
CWSs, also known as soft exudates, are microinfarctions in the nerve fiber layer. The normal flow of axoplasm is blocked in response to focal retinal ischemia caused by occlusion of a precapillary arteriole.17 Clinically, CWSs are superficial whitish retinal lesions with irregular feathery borders that may be associated with small retinal hemorrhages. With resolution, the CWSs will fade and an area of retinal depression may develop secondary to inner retinal ischemic atrophy. Histologically, CWSs are characterized by fusiform thickening of the nerve fiber layer with globular cytoid bodies (Fig. 10)—swollen ganglion cell axons with degenerated cellular organelles (i.e., mitochondria, neurofilaments, endoplasmic reticulum). CWSs are often encountered in the retinopathy associated with blood dyscrasias. In one report,18 the presence of a single CWS in each eye of a patient in the absence of diabetes and systemic hypertension led to the diagnosis of MM. In a prospective study of 54 newly diagnosed patients with acute leukemia, Abu El-Asrar and coworkers19 reported that patients with CWSs had significantly lower mean and median survival times than those patients without CWSs. Thus in this study, the presence of CWSs was a poor prognostic sign for survival in acute leukemia.
|
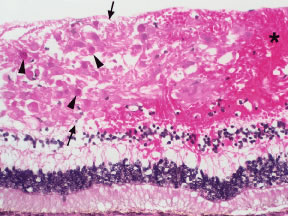
The presence of severe anemia may be a risk factor for the development of CWSs in blood dyscrasias. Holt and Gordon-Smith3 observed CWSs in 14 patients with severe anemia with a mean hemoglobin concentration of 5.6 g/100 ml. In contrast, Guyer and coworkers4 found no statistically significant association between the presence of CWSs and hematologic parameters (including the hematocrit) in their prospective series. CWSs may result from occlusion of the precapillary arterioles by leukemic cells or by platelet-fibrin thrombi.
RETINAL VASCULAR CHANGES
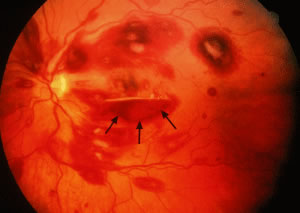
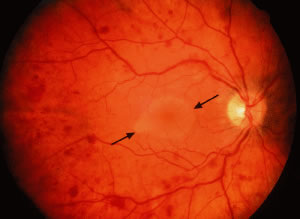
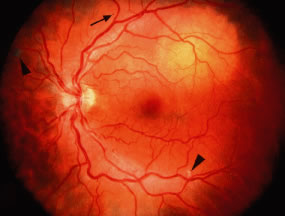
Retinal venous dilatation and tortuosity similar to, and in some cases indistinguishable from, the ophthalmoscopic findings in CRVO may be observed in blood dyscrasias, particularly the plasma cell dyscrasias [i.e., Waldenström's macroglobulinemia (WM), monoclonal gammopathies, and less commonly MM].9,13–15 With venous distention, arteriovenous nicking may become more apparent and give rise to the “sausage link” appearance of the retinal veins. The classical picture of venous stasis retinopathy associated with hyperviscosity is seen in WM (Fig. 11). In a prospective study of 120 patients with newly diagnosed leukemia of all cell types, Schachat and coworkers20 diagnosed a CRVO in 5 patients—all of whom had myeloid leukemia with extremely high white blood cell counts or platelet counts—and attributed the venous occlusion to blood hyperviscosity. The pathologic findings in venous stasis retinopathy associated with blood dyscrasias may be indistinguishable from those found in CRVO (Fig. 12).
|
|
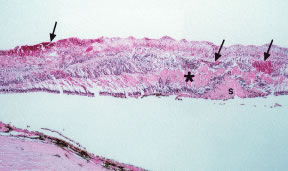
Perivascular sheathing may be observed in the ocular fundus in patients with blood dyscrasias. The apparent opacification or loss of transparency of the retinal blood vessel wall may be due to infiltration by benign inflammatory cells in response to immune complex or other protein (e.g., amyloid, immunoglobulin heavy or light chains) deposition, direct infiltration by leukemic cells, or fibrosis secondary to hypertensive retinal vascular disease. Perivascular sheathing and a clinical picture of retinal vasculitis (with leakage from retinal vessels on fluorescein angiography) have been prominent ocular findings in patients presenting with certain blood dyscrasias, including human T-lymphotropic virus 1 (HTLV-1)-associated adult T-cell leukemia/lymphoma,21,22 cryoglobulinemia,23 hairy cell leukemia,24 and, rarely, MM.25 Kim and coworkers26 described a patient with relapsing acute lymphoblastic leukemia who presented with a retinal vasculopathy resembling frosted branch angiitis and an infiltrative optic neuropathy, which resolved with local radiation and intrathecal chemotherapy.
PALLOR OF THE OCULAR FUNDUS
The normal color of the ocular fundus is derived from the retinal pigment epithelium (RPE), the choroidal melanocytes, and the blood in the retinal and choroidal vasculature. The retina is normally transparent. In patients with severe anemia, the fundus may appear pale and the retinal vessels may be less red than normal.
OPTIC DISC EDEMA
In optic disc edema, nerve fiber swelling in the optic disc and peripapillary retina is present and often associated with flame-shaped hemorrhages, whitish punctate lesions (secondary to obstructed axoplasmic flow), and peripapillary CWSs. On ophthalmoscopy, the optic disc and peripapillary retina appear elevated and the normally well-defined margins of the optic disc are blurred. Optic disc edema is usually bilateral and may be due to elevated intracranial pressure (i.e., papilledema) (Fig. 13) secondary to hemorrhage or a mass effect associated with the blood dyscrasia (i.e., meningeal infiltration, granulocytic sarcoma). The disc edema may also be secondary to serum (blood) hyperviscosity and may resemble that seen in CRVO. Less commonly, direct infiltration of the optic nerve or optic disc may cause marked optic disc edema.
|
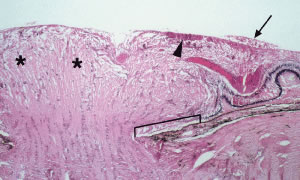
Rosenthal27 emphasized that optic nerve infiltration occurs predominantly in children with acute lymphocytic leukemia (ALL) and must be differentiated clinically from papilledema. Patients with leukemic infiltration of the prelaminar optic nerve typically have marked swelling of the optic disc with a fluffy superficial infiltrate and variable hemorrhage (Fig. 14). The visual acuity may be minimally or severely affected. With retrolaminar optic nerve infiltration, moderate to marked disc elevation and edema with variable hemorrhage may be present and marked vision loss is usually observed. Optic nerve infiltration may occur despite prophylactic brain irradiation in leukemia because of the shields employed to protect the eyes.28 In general, optic nerve infiltration by leukemia responds well to radiotherapy (with or without intrathecal chemotherapy). Brown and coworkers29 reported the case of a patient with acute promyelocytic leukemia and optic disc infiltration who showed complete resolution with oral all trans-retinoic acid alone.
|
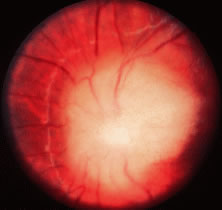
Bilateral optic disc swelling is a common early sign (in up to 73% of cases) of the peripheral neuropathy, organomegaly, endocrinopathy, monoclonal gammopathy, and skin changes (POEMS) syndrome, which typically occurs in middle-aged men in association with a plasma cell dyscrasia.30,31 The optic disc edema may occur in the absence of elevated intracranial pressure and may be mediated by autoantibodies or cytokines.31 Cases of POEMS syndrome—also known as Crow-Fukase syndrome—have been reported worldwide, with the greatest concentration of cases in Japan. Other major features of the syndrome include widespread edema with ascites and pleural effusions, lymphadenopathy, lethargy, weakness, and shortness of breath.30
RETINAL AND OPTIC DISC NEOVASCULARIZATION
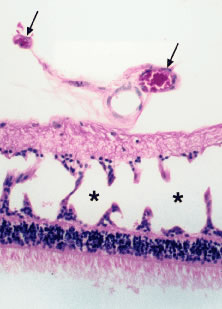
Retinal neovascularization with a sea fan configuration, as seen in sickle cell retinopathy, has been reported in four patients with chronic myelogenous leukemia.27 In most of these reports, the affected patients had extremely high white blood cell counts. Prolonged leukocytosis with or without an increased number of circulating platelets increases the blood viscosity, which may reduce blood flow and cause vascular stagnation.27 The hyperviscosity may lead to microaneurysm formation and retinal capillary nonperfusion or dropout. Retinal ischemia may ensue with the subsequent development of proliferative retinopathy (i.e., neovascularization of the retina and optic disc) (Figs. 15 and 16). More recently, Anderton and coworkers32 reported the case of a patient with chronic myelogenous leukemia who initially presented with a vitreous hemorrhage associated with bilateral retinal and optic disc neovascularization. Wiznia and colleagues33 reported the case of a patient with ALL who developed severe bilateral ischemic retinopathy during chemotherapy with cytosine arabinoside, which progressed to bilateral optic disc and retinal neovascularization despite panretinal photocoagulation. The patient had received low-dose irradiation to the brain within 2 months before presenting with bilateral severe vision loss. The toxic effects of the combination of irradiation and chemotherapy, as well as a hyperviscosity syndrome induced by the leukemia, may have contributed to the acute presentation with proliferative retinopathy.
|
|
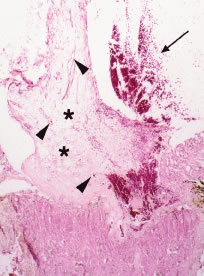
In diabetic patients, the concomitant development of a blood dyscrasia may cause rapid progression from mild background diabetic retinopathy to severe proliferative retinopathy in an unusually short period.34,35 The hyperviscosity syndrome and anemia associated with blood dyscrasias may exacerbate and accelerate diabetic retinopathy. The combination of anemia and diabetes results in both reduced blood oxygen-carrying capacity and a reduced dissociation of oxygen from the blood causing greater tissue hypoxia (i.e., retinal ischemia).34
VITREOUS HEMORRHAGE AND INFLAMMATION
Vitreous hemorrhage may be an ophthalmic manifestation of blood dyscrasias, particularly in patients with the combination of anemia and thrombocytopenia. Vitreous hemorrhage may occur in 10% to 15% of patients with aplastic anemia.36 Schachat and coworkers20 reported 3 patients with vitreous hemorrhage in a prospective series of 120 patients with newly diagnosed leukemia of all cell types. Vitreous hemorrhage may occur more often in acute promyelocytic leukemia (M3 subtype of AML), which is characterized by severe hemorrhagic manifestations either at presentation or following commencement of cytotoxic therapy.7 In addition, patients with acute promyelocytic leukemia may develop Terson's syndrome (vitreous hemorrhage associated with intracranial bleeding) during induction therapy with all trans-retinoic acid.37 The vitreous hemorrhage may also occur as a complication of proliferative retinopathy32 from neovascularization of the retina (see Fig. 15) or disc (see Fig. 16).
Apparent vitreous inflammation may be caused by the presence of leukemic cells within the vitreous.21,38,39 Swartz and Schumann38 reported the case of a patient with acute lymphoblastic leukemia in apparent remission who presented with vitreous infiltration diagnosed by cytopathologic evaluation of a vitreous aspirate. The apparent vitreous inflammation was the first clinical sign of central nervous system involvement. Prompt institution of radiotherapy and chemotherapy produced a rapid reduction of cells in the vitreous and clearing of cells from the cerebrospinal fluid.
Vitreous deposits of amyloid are rarely observed in primary systemic nonfamilial amyloidosis.40 This systemic disorder is characterized by an aberrant deposition in various organ systems of insoluble polypeptides derived from a portion of the light chain of immunoglobulins.
RETINAL AND RETINAL PIGMENT EPITHELIUM DETACHMENT
Retinal and retinal pigment epithelium (RPE) detachments are not infrequently observed in blood dyscrasias. A clinical picture with fluorescein angiography resembling central serous retinopathy (i.e., serous retinal and RPE detachments) has been described in patients with plasma cell dyscrasias, including monoclonal gammopathies and cryoglobulinemia.41,42 Rarely, apparent serous macular detachments can occur, and fluorescein angiography shows macular hypofluorescence and no evidence of retinal vascular or RPE leakage associated with the macular elevation. Ho and coworkers43 reported three such cases in patients with MM, WM, and benign polyclonal gammopathy. The same authors hypothesized that the macular detachments were transudative rather than exudative with subretinal precipitates of immunoglobulin or other serum proteins. Ogata and colleagues44 described a patient with WM who presented with an apparent serous macular detachment with no abnormal hyperfluorescence on fluorescein angiography; however, with optical coherence tomography (OCT), a large occult RPE detachment was noted beneath the serous retinal detachment. In plasma cell dyscrasias, the choriocapillaris may be partly obstructed by light chain or other immunoglobulin deposits, and this may play an important role in the pathogenesis of the serous retinal and RPE detachments observed in these disorders.12,45
Rarely, visual impairment secondary to a retinal or RPE detachment may be the first clinical sign of a blood dyscrasia or a leukemic relapse during apparent remission.46–49 In leukemia, the serous retinal and RPE detachments may occur secondary to leukemic infiltration of the choroid.11,27,50 Solid detachments of the retina by leukemic cell infiltrates in the subretinal space have also been reported.11,51 In one such case, a hypopyon-like configuration of the leukemic cells was observed in the subretinal space.51
Serous retinal and RPE detachments may also be seen in platelet disorders and coagulopathies such as thrombotic thrombocytopenic purpura (TTP) and disseminated intravascular coagulation (DIC).52–55 The mechanism of retinal and RPE detachment in these disorders is associated with choriocapillary occlusion by platelet-fibrin thrombi (Fig. 17).
|
RETINAL AND CHOROIDAL INFILTRATES
Retinal and choroidal infiltrates are often observed in patients with leukemia. A retinal leukemic infiltrate may appear as an elevated yellowish white mass of variable size with or without associated hemorrhage or vitreous inflammation and is most often located in the posterior pole or peripapillary region. Leukemic infiltration of the retina and/or choroid may simulate an infectious chorioretinitis.21,22,56 Gordon and coworkers56 emphasized the importance of distinguishing between infectious and neoplastic retinal infiltrates in patients with a history of leukemia. The same authors found that neoplastic (or leukemic) retinal infiltrates occurred in patients who had newly diagnosed leukemia and those patients in blast crisis. In contrast, two patients who were in complete remission after bone marrow transplantation had infectious retinal infiltrates. Thus the authors concluded that the systemic status (i.e., newly diagnosed leukemia vs. immunosuppression after bone marrow transplantation) ofthe patient is highly informative in determining whether infection or neoplasia is responsible for the retinal infiltration.
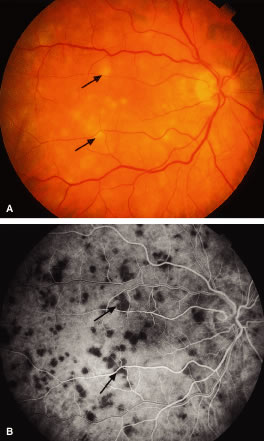
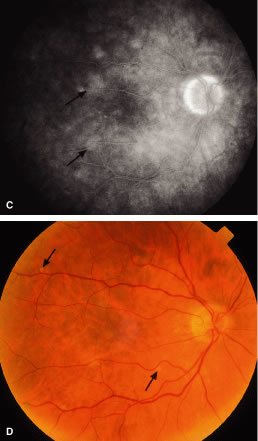
Retinal detachment may occur in the region of a choroidal leukemic infiltrate.8,11,27,49 RPE changes, including pigment clumping and pigment mottling, may be observed overlying the choroidal infiltrate.57 A choroidal leukemic infiltrate typically appears as an elevated, creamy, yellowish-white lesion (Fig. 18). Fluorescein angiography may reveal early multifocal punctate hyperfluorescence over the choroidal infiltrate with late pooling of fluorescein in the subretinal space resembling Harada's disease.49 Ophthalmoscopic and fluorescein angiographic findings resembling acute posterior multifocal placoid pigment epitheliopathy (APMPPE) have been described in patients with leukemia.58,59 The APMPPE-like features include deep, whitish, well-circumscribed fundus lesions that exhibit early blockage and late leakage on fluorescein angiography (Fig. 19).
|
|
McManaway and Neely60 reported the case of a patient with acute lymphoblastic leukemia who presented with leukocoria and proptosis resulting from an extensive intraocular and orbital tumor mimicking advanced retinoblastoma. An elevated leukocyte blood count, blast forms on the peripheral blood smear, rubbery suboccipital nodules, and subcutaneous scalp masses all suggested leukemia, although the fundus picture was atypical and caused diagnostic confusion. No intratumoral calcification was observed on a computed tomography (CT) scan. The ocular and orbital infiltrates responded rapidly to chemotherapy and low-dose orbital irradiation.
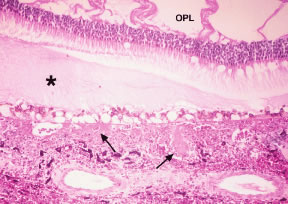
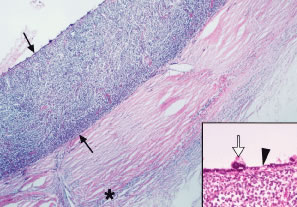
The frequency of these infiltrates, particularly choroidal infiltrates, is much higher in pathologic studies (compared with clinical studies) of eyes from patients with acute and chronic leukemia and ranges between 28% and 80%.8,11,61 In a prospective study of 120 patients with newly diagnosed leukemia (all cell types), Schachat and coworkers20 found 4 patients (3%) with leukemic retinal infiltrates and none with choroidal infiltrates. Leukemic choroidal infiltrates may be difficult to diagnose clinically without special ancillary tests (i.e., fluorescein angiography, A- and B-scan ultrasonography). In a pathologic study of 135 autopsy eyes in patients with leukemia, Leonardy and coworkers61 reported choroidal involvement in 93% of eyes with leukemic infiltrates. Allen and Straatsma8 found a much higher incidence of ocular involvement, including choroidal infiltration, in eyes of patients with acute leukemia (80%) compared with chronic leukemia (29%). The same authors emphasized the diffuse pattern of leukemic infiltration of the posterior choroid (Fig. 20) and infrequent association with hemorrhage as opposed to the focal leukemic infiltration observed in the retina with associated hemorrhage in nearly every case.
|
Diffuse bilateral chorioretinal abnormalities including deep hemorrhages and pigment mottling in the posterior pole, diffuse areas of hypofluorescence on fluorescein and indocyanine green angiography in the posterior pole, and hypofluorescent lines or streaks in the midperipheral fundus were observed by Pece and coworkers62 in a patient with primary systemic nonfamilial amyloidosis. These authors postulated that the areas of hypofluorescence may represent either choroidal vascular occlusion or intravascular and/or perivascular amyloid deposits.