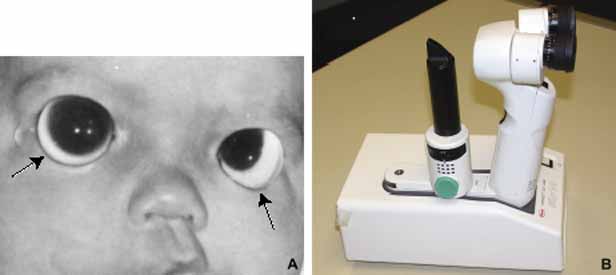
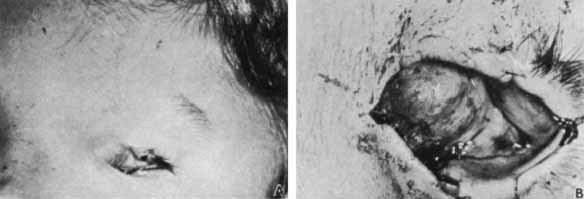
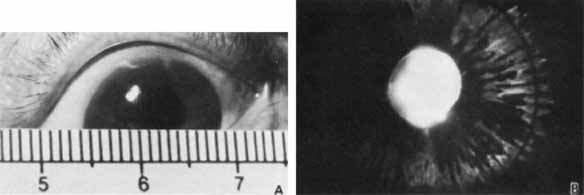
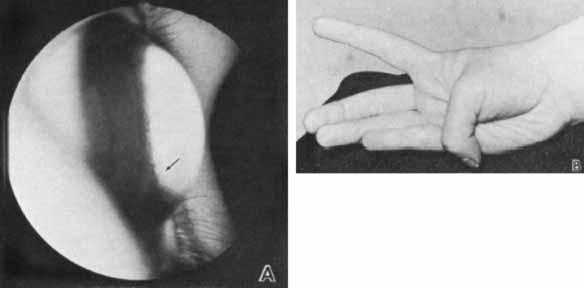
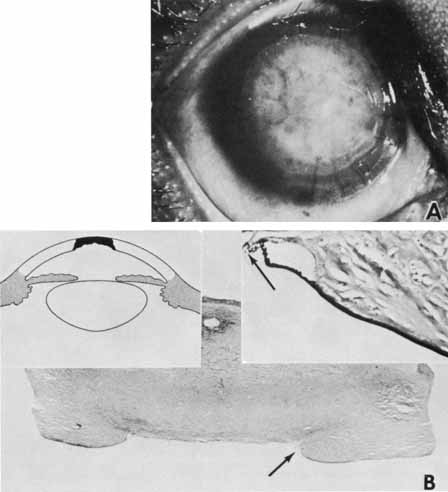
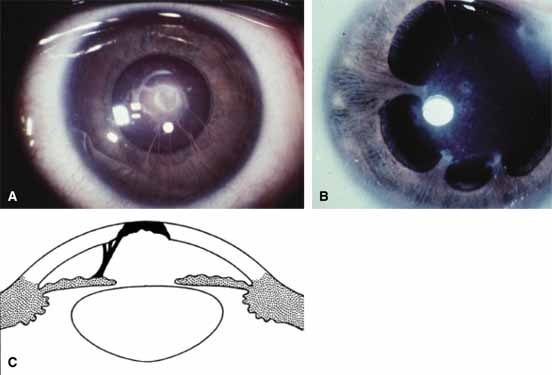
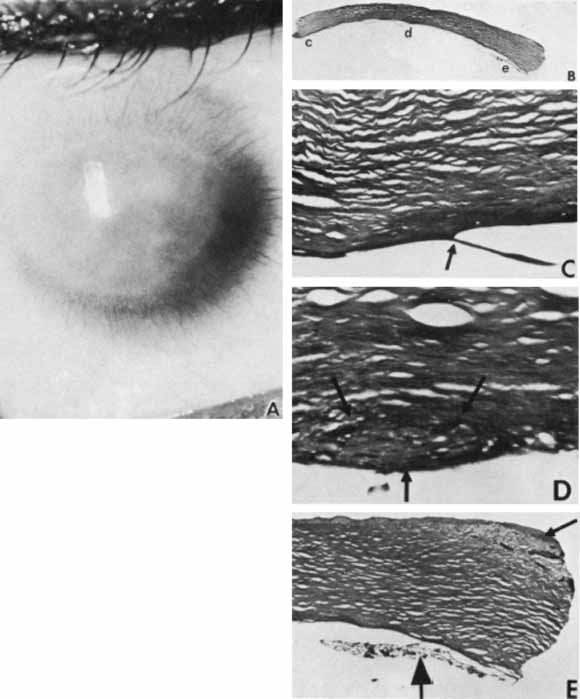
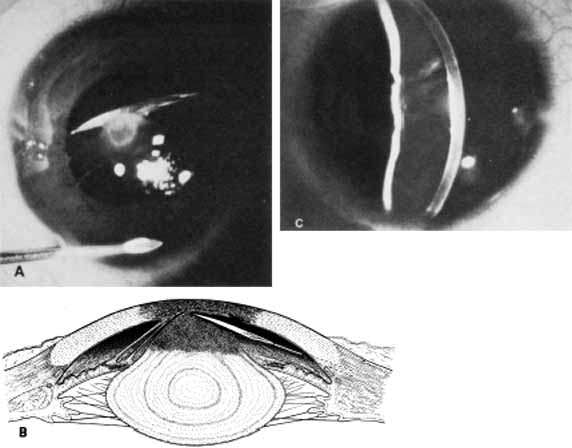
1. Robinson GC, Jan JE, Kinnis C: Congenital ocular blindness in children, 1945 to 1984. Am J Dis Child 141:1321, 1987 2. Foster A, Gilbert C: Epidemiology of childhood blindness. Eye 6:173, 1992 3. Waring GO, Laibson PR: A systematic method of drawing corneal pathologic conditions. Arch Ophthalmol 95:1540, 1977 4. Nischal KK, Naor J, Jay V, MacKeen LD, Rootman DS: Clinicopathological correlation of congenital corneal opacification using
ultrasound biomicroscopy. Br J Ophthalmol 86:62, 2002 5. Duke-Elder S: Normal and Abnormal Development. Congenital Deformities. System of Ophthalmology, Vol 3, Pt 2. St. Louis: C.V. Mosby, 1963 6. Waring GO, Shields JA: Partial unilateral cryptophthalmos with syndactyly, brachycephaly, and
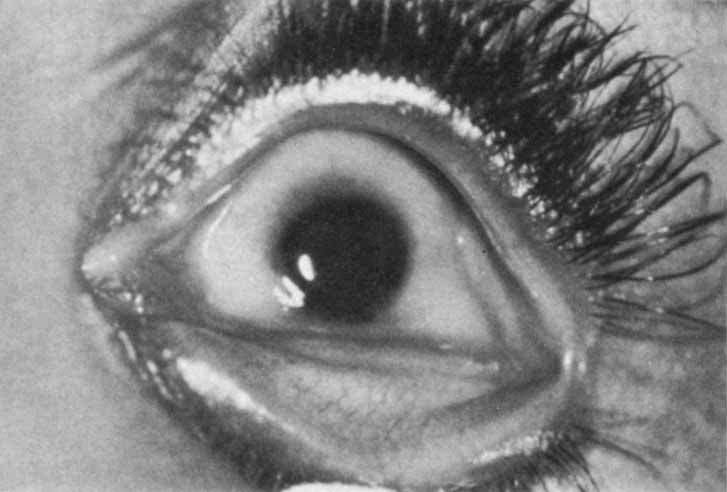
renal anomalies. Am J Ophthalmol 79:437, 1975 7. Sugar HS: The cryptophthalmos-syndactyly syndrome. Am J Ophthalmol 66:897, 1968 8. Thomas IT, Frias JL, Felix V, Sanchez de Leon L, Hernandez RA, Jones MC: Isolated and syndromic cryptophthalmos. Am J Med Genet 25:85, 1986 9. Slavotinek AM, Tifft CJ: Fraser syndrome and cryptophthalmos: review of the diagnostic criteria
and evidence for phenotypic modules in complex malformation syndromes. J Med Genet 39:623, 2002 10. Codere F, Brownstein S, Chen MF: Cryptophthalmos syndrome with bilateral renal agenesis. Am J Ophthalmol 91:737, 1981 11. Francois J: Syndrome malformation avec cryptophthalmie. Acta Genet Med Gemellol 18:18, 1969 12. Ferri M, Harvey JT: Surgical correction for complete cryptophthalmos: case report and review
of the literature. Can J Ophthalmol 34:233, 1999 13. Wilmer HA, Scammon RE: Growth of the components of the human eyeball. Arch Ophthalmol 43:599, 1950 14. Mann I: Developmental Abnormalities of the Eye. Philadelphia: JB Lippincott, 1957:352 15. Wood WJ, Green WR, Marr WG: Megalocornea: a clinico-pathologic clinical case report. Md State Med J 23:57, 1974 16. Rogers GL, Polomeno RC: Autosomal-dominant inheritance of megalocornea associated with Down's
syndrome. Am J Ophthalmol 78:526, 1974 17. Vail DT: Adult hereditary anterior megalophthalmus sine glaucoma: a definite disease
entity. Arch Ophthalmol 6:39, 1931 18. Boles Carenini B: Juvenile familial mosaic degeneration of the cornea associated with megalocornea. Br J Ophthalmol 45:64, 1961 19. Collier M: Hemiatrophie faciale progressive avec megalocornee, micropapille, et dystrophie
nuageuse centrale de la cornee. Acta Ophthalmol 49:946, 1971 20. Dube P, Der Kaloustian VM, Demczuk S, Saabti H, Koenekoop RK: A new association of congenital hydrocephalus, albinism, magalocornea, and
retinal coloboma in a syndromic child: a clinical and genetic study. Ophthalmic Genet 21:211, 2000 21. Kupfer C, Kaiser-Kupfer MI: New hypothesis of developmental anomalies of the anterior chamber associated
with glaucoma. Trans Ophthal Soc UK 98:213, 1978 22. Pearce WG: Autosomal dominant megalocornea with congenital glaucoma: evidence for
germ-line mosaicism. Can J Ophthalmol 26:21, 1991 23. Mackey DA, Buttery RG, Wise GM, Denton MJ: Description of X-linked megalocornea with identification of the
gene locus. Arch Ophthalmol 109:829, 1991 24. Davis RJ, Shen W, Sandler YI, Heanue TA, Mardon G: Characterization of mouse Dach2, a homologue of Drosophila dachshund. Mech Dev 102:169, 2001 25. Neumann AC: Anterior megalophthalmos and intraocular lens implantation. Am Intra-Ocular Implant Soc J 10:220, 1984 26. Javadi MA, Jafarinasab MR, Mirdehghan SA: Cataract surgery and intraocular lens implantation in anterior megalophthalmos. J Cataract Refract Surg 26:1687, 2000 27. Arkin W: Blue scleras with keratoglobus. Am J Ophthalmol 58:678, 1964 28. Biglan AW, Brown SI, Johnson BL: Keratoglobus and blue sclera. Am J Ophthalmol 83:225, 1977 29. Cameron JA: Corneal abnormalities in Ehlers-Danlos syndrome type VI. Cornea 12:54, 1993 30. Elder MJ: Leber congenital amaurosis and its association with keratoconus and keratoglobus. J Pediatr Ophthalmol Strabismus 31:38, 1994 31. Karabatas CH, Cook SD: Topographic analysis in pellucid marginal corneal degeneration and keratoglobus. Eye 10:451, 1996 32. Jacobs DS, Green WR, Maumenee AE: Acquired keratoglobus. Am J Ophthalmol 77:393, 1974 33. Cameron JA, Al-Rajhi AA, Badr IA: Corneal ectasia in vernal keratoconjunctivitis. Ophthalmol 96:1615, 1989 34. Cameron JA: Keratoglobus. Cornea 12:124, 1993 35. Bertelsen TI: Dysgenesis mesodermalis corneae et sclerae. Acta Ophthalmol 46:486, 1968 36. Macsai MS, Lemley HL, Schwartz T: Management of oculus fragilis in Ehlers-Danlos type VI. Cornea 19:104, 2000 37. Cameron JA, Cotter JB, Risco JM, Alvarez H: Epikeratoplasty for keratoglobus associated with blue sclera. Ophthalmol 98:446, 1991 38. Burk ROW, Joussen AM: Corneoscleroplasty with maintenance of the angle in two cases of extensive
corneoscleral disease. Eye 14:196, 2000 39. Jones DH, Kirkness CM: A new surgical technique for keratoglobus-tectonic lamellar keratoplasty
followed by secondary penetrating keratoplasty. Cornea 20:885, 2001 40. Vajpayee RB, Bhartiya P, Sharma N: Central lamellar keratoplasty with peripheral intralamellar tuck. A new
surgical technique for Keratoglobus. Cornea 21:657, 2002 41. Dinno ND, Lawwill T, Leggett AE, Shearer L, Weisskopf B: Bilateral microcornea, coloboma, short stature and other skeletal anomalies—a
new hereditary syndrome. Birth Defects 8:109, 1976 42. Weiss AH, Kousseff BG, Ross EA, Longbottom J: Complex microphthalmos. Arch Ophthalmol 107:1619, 1989 43. Salmon JF, Wallis CE, Murray ADN: Variable expressivity of autosomal dominant microcornea with cataract. Arch Ophthalmol 106:505, 1988 44. Cross HE, Yoder F: Familial nanophthalmos. Am J Ophthalmol 81:300, 1976 45. Warburg M: Classification of microphthalmos and coloboma. J Med Genet 30:664, 1993 46. Batra DV, Paul SD: Microcornea with myopia. Br J Ophthalmol 51:57, 1967 47. Francois J: Heredity in Ophthalmology. St. Louis: C.V. Mosby, 1961:291 48. Nath K, Nema HV, Shukla BR: Histopathology in a case of unilateral microcornea plana (associated
with coloboma of choroid): First histopathological description. Acta Ophthalmol 42:609, 1964 49. Friedman MW, Wright ES: Hereditary microcornea and cataract in five generations. Am J Ophthalmol 35:1017, 1952 50. Friedman AH, Weingeist S, Brackup A, Marinoff G: Sclero-cornea and defective mesodermal migration. Br J Ophthalmol 59:683, 1975 51. Howard RO, Abrahams IW: Sclerocornea. Am J Ophthalmol 71:1254, 1971 52. Elliott JH, Feman SS, O'Day DM, Garber M: Hereditary sclerocornea. Arch Ophthalmol 103:676, 1985 53. Rodrigues MM, Calhoun J, Weinreb S: Sclerocornea with an unbalanced translocation (17p, 10q). Am J Ophthalmol 78:49, 1974 54. Goldstein JE, Cogan DG: Sclerocornea and associated congenital anomalies. Arch Ophthalmol 67:99, 1962 55. Moriarty AP, Kerr-Muir MG: Sclerocornea and interstitial deletion of the short arm of chromosome 6-(46XY
del [6] [p22p24]). J Pediatr Ophthalmol Strabismus 29:177, 1992 56. Kim T, Cohen EJ, Schnall BM, Affel EL, Eagle RC: Ultrasound biomicroscopy and histopathology of sclerocornea. Cornea 17:443, 1998 57. Wood TO, Kaufman HE: Penetrating keratoplasty in an infant with sclerocornea. Am J Ophthalmol 70:609, 1970 58. Kanai A, Wood TC, Polack FM, Kaufman HE: The fine structure of sclerocornea. Invest Ophthalmol Vis Sci 10:687, 1971 59. Larsen V, Eriksen A: Cornea plana. Acta Ophthalmol 27:275, 1949 60. Sharkey JA, Kervick GN, Jackson AJ, Johnston PB: Cornea plana and sclerocornea in association with recessive epidermolysis
bullosa dystrophica. Case report. Cornea 11:83, 1992 61. Alkemade PPH: Dysgenesis Mesodermalis of the Iris and the Cornea. Assen, Netherlands: Royal Van Gorcum, 1969 62. Nakanishi I, Brown SI: The histopathology and ultrastructure of congenital central corneal opacity (Peter's
anomaly). Am J Ophthalmol 72:801, 1971 63. Redbrake C, Salla S, Becker J, Reim M: A rare case of bilateral congenital corneal malformations. Acta Ophthalmol 71:256, 1993 64. Lesueur L, Arne JL, Mignon-Conte M, Malecaze F: Structural and ultrastructural changes in the developmental process of
premature infants' and children's corneas. Cornea 13:331, 1994 65. Angell LK, Robb RM, Berson FG: Visual prognosis in patients with ruptures in Descemet's membrane
due to forceps injuries. Arch Ophthalmol 99:2137, 1981 66. Costenbader FD, Kwitko ML: Congenital glaucoma: An analysis of seventy-seven consecutive eyes. J Pediatr Ophthalmol 4:9, 1967 67. Waring GO, Laibson PR, Rodrigues M: Clinical and pathologic alterations of Descemet's membrane: with emphasis
on endothelial metaplasia. Surv Ophthalmol 18:325, 1974 68. Pouliquen Y, Saraux H: Ultrastructure de la Cornee, D'un Buphtalme. Arch Ophthalmol (Paris) 27:263, 1967 69. Van Horn DL, Hyndiuk RA, Edelhauser HF, McDonald TO, De Santis LM: Ultrastructural alterations associated with loss of transparency in the
cornea of buphthalmic rabbits. Exp Eye Res 25:171, 1977 70. Mastropasqua L, Carpineto P, Ciancaglini M, Nubile M, Doronzo E: In vivo confocal microscopy in primary congenital glaucoma with megalocornea. J Glaucoma 11:83, 2002 71. Frucht-Pery J, Feldman ST, Brown SI: Transplantation of congenitally opaque corneas from eyes with exaggerated
buphthalmos. Am J Ophthalmol 107:655, 1989 72. Spencer WH, Ferguson WJ, Shaffer RN, Fine M: Late degenerative changes in the cornea following breaks in Descemet's
membrane. Trans Am Acad Ophthalmol Otolaryngol 70:973, 1966 73. Toker E, Seitz B, Langenbucher A, Dietrich T, Naumann GOH: Penetratring keratoplasty for endothelial decompensation in eyes with buphthalmos. Cornea 22:198, 2003 74. Kumar P, Tiwari VK: An unusual cleft lip secondary to amniotic bands. Br J Plast Surg 43:492, 1990 75. Nahmias AJ, Visintine AM, Caldwell DR, Wilson LA: Eye infections with herpes simplex viruses in neonates. Survey Ophthalmol 21:100, 1976 76. Whitley, RJ: Herpes simplex virus. In Remington JS, ed. Infectious Diseases of the Fetus and Newborn. Philadelphia: W.B. Saunders, 1990:282–305 77. Liesegang TJ: Herpes simplex virus epidemiology and ocular importance. Cornea 20:1, 2001 78. Isenberg SJ, Apt L, Yoshimori R, Leake RD, Rich R: Povidone-iodine for ophthalmia neonatorum prophylaxis. Am J Ophthalmol 118:701, 1994 79. Wilkie JS, Easterbrook M, Coleman V, Stevens T: Crede prophylaxis and neonatal corneal infection with herpesvirus. Arch Ophthalmol 91:386, 1974 80. el Azazi M, Malm G, Forsgren M: Late ophthalmologic manifestations of neonatal herpes simplex virus infection. Am J Ophthalmol 109:1, 1990 81. Wolff SM: The ocular manifestations of congenital rubella. A prospective study of 328 cases
of congenital rubella. J Pediatric Ophthalmol 10:101, 1973 82. Sears ML: Congenital glaucoma in neonatal rubella. Br J Ophthalmol 51:744, 1967 83. Boniuk V: Systemic and ocular manifestations of the rubella syndrome. Int Ophthalmol Clin 12:67, 1972 84. Givens KT, Lee DA, Jones T, Ilstrup DM: Congenital rubella syndrome: ophthalmic manifestations and associated systemic
disorders. Br J Ophthalmol 77:358, 1993 85. Cruz OA, Sabir SM, Capo H, Alfonso EC: Microbial keratitis in childhood. Ophthalmol 100:192, 1993 86. Burns RP, Rhodes DH: Pseudomonas eye infection as a case of death in premature infants. Arch Ophthalmol 65:517, 1961 87. Davis EA, Dohlman CH: Neurotrpohic keratitis. Int Ophthalmol Clinics 41:1, 2001 88. Vinals AF, Kenyon KR: Corneal manifestations of metabolic diseases. Int Ophthalmol Clinics 38:141, 1998 89. Whitley CB: The mucopolysaccharidoses. In Beighton (ed): McKusick's Heritable Disorders of Connective Tissue, 5th ed. St. Louis: CV Mosby, 1993:367–499 90. Libert J, Kenyon KR: Ocular ultrastructure in inborn lysosomal storages diseases. In Goldberg MA (ed): Genetic and Metabolic Eye Disease, 2nd ed. Boston: Little, Brown & Co, 1986:111–137 91. Newman NJ, Starck T, Kenyon KR, Lessell S, Fish I, Kolodny EH: Corneal surface irregularities and episodic pain in a patient with mucolipidosis
IV. Arch Ophthalmol 108:251, 1990 92. Berliner ML: Lipid keratitis of Hurler's syndrome (gargoylism or dysostosis
multiplex). Clinical and pathologic report. Arch Ophthalmol 22:97, 1939 93. Kenyon KR, Topping TM, Green WR, Maumenee E: Ocular pathology of the Maroteaux-Lamy syndrome (systemic mucopolysaccharidosis
type VI). Histologic and ultrastructural report
of two cases. Am J Ophthalmol 73:718, 1972 94. Weingeist TA, Blodi FC: Fabry's disease: ocular findings in a female carrier—a light
and electron microscopic study. Arch Ophthalmol 85:169, 1971 95. Rodrigues MM, Calhoun J, Harley RD: Corneal clouding with increased acid mucopolysaccharide accumulation in
Bowman's membrane. Am J Ophthalmol 79:916, 1975 96. Heathcote JG, Sholdice J, Walton JC, Willis NR, Sergovich FR: Anterior segment mesenchymal dysgenesis associated with partial duplication
of the short arm of chromosome 2. Can J Ophthalmol 26:35, 1991 97. Burns RP: Soluble tyrosine aminotransferase deficiency: an unusual case of corneal
ulcers. Am J Ophthalmol 73:400, 1972 98. Macsai MS, Schwartz TL, Hinkle D, Hummel MB, Mulhern MG, Rottman D: Tyrosinemia type II:. nine cases of ocular signs and symptoms. Am J Ophthalmol 132:522, 2001 99. Goldsmith LA, Kang E, Bienfang DC, Jimbow K, Gerald P, Baden HP: Tyrosinemia with plantar and palmar keratosis and keratitis. J Pediatr 83:789, 1973 100. Chitayat D, Balbul A, Hani V, et al: Hereditary tyrosinemia type II in a consanguineous Ashkenazi Jewish family: intrafamilial
variation in phenotype; absence of parental phenotype
effects on the fetus. J Inher Metab Dis 15:198, 1992 101. Gipson IK, Anderson RA: Response of the lysosomal system of the corneal epithelium to tyrosine-induced
cell injury. J Histochem Cytochem 125:1351, 1977 102. Ahmad S, Teckman JH, Lueder GT: Corneal opacities associated with NTBC treatment. Am J Opthtalmol 134:266, 2002 103. Mungan N, Nischal KK, Heon E, MacKeen L, Balfe JW, Levin AV: Ultrasound biomicroscopy of the eye in cystinosis. Arch Ophthalmol 118:1329, 2000 104. Kaiser-Kupfer MI, Gazzo MA, Datiles MB, Caruso RC, Kuehl EM, Gahl WA: A randomized placebo-controlled trial of cysteamine eye drops in
nephropathic cystinosis. Arch Ophthalmol 108:689, 1990 105. Katz B, Melles RB: Crystal deposition following keratoplasty in nephropathic cystinosis. Arch Ophthalmol 107:1727, 1989 106. Tsilou ET, Rubin BI, Reed GF, Iwata F, Gahl W, Kaiser-Kupfer MI: Age-related prevalence of anterior segment complications in patients
with infantile nephropathic cystinosis. Cornea 21:173, 2002 107. Reese AB, Ellsworth RM: The anterior chamber cleavage syndrome. Arch Ophthalmol 75:307, 1966 108. Waring GO, Rodrigues MM, Laibson PR: Anterior chamber cleavage syndrome. A stepladder classification. Surv Ophthalmol 20:3, 1975 109. Rao SK, Padmanabhan P: Posterior keratoconus. An expanded classification scheme based on corneal
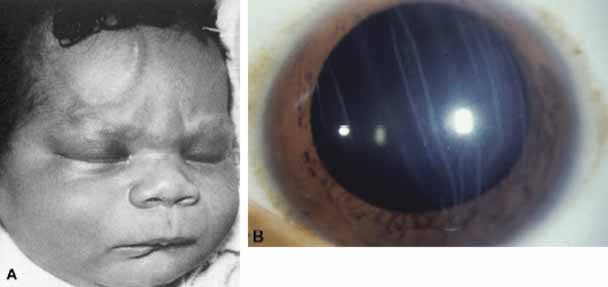
topography. Ophthalmology 105:1206, 1998 110. Streeten BW, Karpik AG, Spitzer KH: Posterior keratoconus associated with systemic abnormalities. Arch Ophthalmol 101:616, 1983 111. Williams R: Acquired posterior keratoconus. Br J Ophthalmol 71:16, 1987 112. Cote MA, Gaster RN: Keratohematoma leading to acquired posterior keratoconus. Cornea 13:534, 1994 113. Haney WP, Falls HF: The occurrence of congenital keratoconus posticus circumscriptus. Am J Ophthalmol 52:53, 1961 114. Krachmer JH, Rodrigues MM: Posterior keratoconus. Arch Ophthalmol 96:1867, 1978 115. Kupfer C, Kuwabara T, Stark WJ: The histopathology of Peters' anomaly. Am J Ophthalmol 80:653, 1975 116. Stone DL, Kenyon KR, Green WR, Ryan SJ: Congenital corneal leukoma (Peters' anomaly). Am J Ophthalmol 81:173, 1976 117. Townsend WM, Font RL, Zimmerman LE: Congenital corneal leukomas. II. Histopathological findings in 19 eyes
with central defect in Descemet's membrane. Am J Ophthalmol 77:192, 1974 118. Townsend WM, Font RL, Zimmerman LE: Congenital corneal leukomas. III. Histopathological findings in 13 eyes
with noncentral defect in Descemet's membrane. Am J Ophthalmol 77:400, 1974 119. Lee CF, Yue BYJT, Robin J, Sawaguchi S, Sugar J: Immunohistochemical studies of Peters' anomaly. Ophthalmology 96:958, 1989 120. Hagedoorn A, Velzeboer CMJ: Postnatal partial spontaneous correction of a severe congenital anomaly
of the anterior segment of an eye. Arch Ophthalmol 62:685, 1959 121. Waring GO, Parks MM: Successful lens removal in congenital corneolenticular adhesion (Peters' anomaly). Am J Ophthalmol 83:526, 1977 122. Myles WM, Flanders ME, Chitayat D, Brownstein S: Peters' anomaly: a clinicopathologic study. J Pediatr Ophthalmol Strabismus 29:374, 1992 123. Traboulsi EI, Maumenee IH: Peters' anomaly and associated congenital malformations. Arch Ophthalmol 110:1739, 1992 124. Heon E, Barsoum-Homsy M, Cevrette L, et al: Peters' anomaly. The spectrum of associated ocular and systemic malformations. Ophthalmic Paediatr Genet 13:137, 1992 125. Cross H: Penetrance of variability in anterior chamber malformations. Birth Defects 15:131, 1979 126. Holmstrom GE, Reardon WP, Baraitser M, Elston JS, Taylor DS: Heterogeneity in dominant anterior segment malformations. Br J Ophthalmol 75:591, 1991 127. Halder G, Callaerts P, Gehring WJ: Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 267:1788, 1995 128. Hanson IM, Fletcher JM, Jordan T, et al: Mutations at the PAX6 locus are found in heterogenous anterior segment malformations including
Peters' anomaly. Nat Genet 6:168, 1994 129. Doward W, Perveen R, Lloyd IC, et al: A mutation in the RIEG1 gene associated with Peters' anomaly. J Med Genet 36:152, 1999 130. Honkanen RA, Nishimura DY, Swiderski RE, et al: A family with Axenfeld-Rieger syndrome and Peters anomaly caused
by a point mutation (Phe112Ser) in the FOXC1 gene. Am J Ophthalmol 135:368, 2003 131. Vincent A, Billinsgly G, Priston M, et al: Phenotypic heterogeneity of CYP1B1: mutations in a patient with Peters' anomaly. J Med Genet 38:324, 2001 132. Jotterand V, Boisjoly HM, Harnois C, Bigonesse P, Laframboise R, Gagne R, St-Pierre A: 11p13 deletion, Wilms' tumor, and aniridia: unusual genetic, non-ocular
and ocular features of three cases. Br J Ophthalmol 74:568, 1990 133. Dichlt A, Jonas JB, Naumann GOH: Atypical Peters' anomaly associated with partial trosomy 5p. Am J Ophthalmol 120:541, 1995 134. Schanzlin DJ, Robin JB, Erickson G, Lingua R, Minckler D, Pickford M: Histopathologic and ultrastructural analysis of congenital corneal staphyloma. Am J Ophthalmol 95:506, 1983 135. Smith HC: Keloid tumors of the cornea. Trans Am Ophthalmol Soc 38:519, 1940 136. Leff SR, Shields JA, Augsburger JJ, Sakowski AD, Blair CJ: Congenital corneal staphyloma: clinical, radiological, and pathological
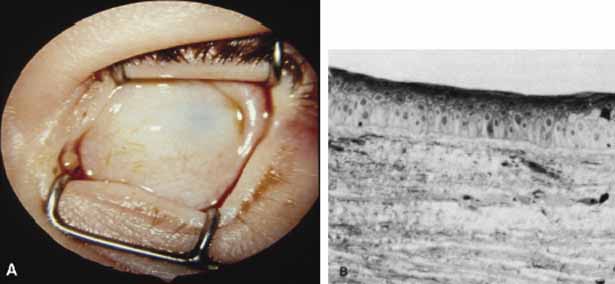
correlation. Br J Ophthalmol 70:427, 1986 137. Townsend WM: Congenital corneal leukomas. I. Central defect in Descemet's membrane. Am J Ophthalmol 77:80, 1974 138. BenEzra D, Sela M, Peer J: Bilateral anophthalmia and unilateral microphthalmia in two siblings. Ophthalmologica 198:140, 1989 139. Waring GO, Rodrigues MM, Laibson PR: Corneal dystrophies. I. Dystrophies of the epithelium, Bowman's layer
and stroma. Surv Ophthalmol 23:71, 1978 140. Waring GO, Rodrigues MM, Laibson PR: Corneal dystrophies. II. Endothelial dystrophies. Surv Ophthalmol 23:147, 1978 141. Toma NMG, Ebenezer ND, Inglehearn CF, Plant C, Ficker LA, Bhattacharya SS: Linkage of congenital hereditary endothelial dystrophy to chromosome 20. Hum Mol Genet 4:2395, 1995 142. Hand CK, Harmon DL, Kennedy SM, FitzSimon JS, Collum LMT, Parfrey NA: Localization of the gene for autosomal recessive congenital hereditary
endothelial dystrophy (CHED2) to chromosome 20 by homozygosity
mapping. Genomics 61:1, 1999 143. Kenyon KR, Maumenee AE: Further studies of congenital hereditary endothelial dystrophy of the cornea. Am J Ophthalmol 76:419, 1973 144. Schaumberg DA, Moyes AL, Gomes JAP, Dana MR: Corneal transplantation in young children with congenital hereditary endothelial
dystrophy. Am J Ophthalmol 127:373, 1999 145. Cockerham GC, Laver NV, Hidayat AA, McCoy DL: An immunohistochemical analysis and comparison of posterior polymorphous
dystrophy with congenital hereditary endothelial dystrophy. Cornea 21:787, 2002 146. Cibis GW, Krachmer JA, Phelps CD, Weingeist TA: The clinical spectrum of posterior polymorphous dystrophy. Arch Ophthalmol 95:1529, 1977 147. Levy SG, Moss J, Nobel BA, McCartney ACE: Early onset posterior polymorphous dystrophy. Arch Ophthalmol 114:1265, 1996 148. Heon E, Greenberg A, Kopp KK, et al: VSX1: A gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet 11:1029, 2002 149. Biswas S, Munier FL, Yardley J, et al: Missense mutations in COL8A2, the gene encoding the α2 chain of type VIII collagen, causing two
forms of corneal endothelial dystrophy. Hum Mol Genet 10:2415, 2001 150. Rodrigues MM, Waring GO, Laibson PR, Weinreb S: Endothelial alterations in congenital corneal dystrophies. Am J Ophthalmol 80:678, 1975 151. Sekundo W, Lee WR, Kirkness CM, Aitken DA, Fleck B: An ultrastructural investigation of an early manifestation of the posterior
polymorphous dystrophy of the cornea. Ophthalmology 101:1422, 1994 152. Ross JR, Foulks GN, Sanfilippo FP, Howell DN: Immunohistochemical analysis of the pathogenesis of posterior polymorphous
dystrophy. Arch Ophthalmol 113:340, 1995 153. Brooks AMV, Grant G, Gillies WE: Differentiation of posterior polymorphous dystrophy from other posterior
corneal opacities by specular microscopy. Ophthalmol 96:1639, 1989 154. Hirst LW, Waring GO: Clinical specular microscopy of posterior polymorphous endothelial dystrophy. Am J Ophthalmol 95:143, 1983 155. Grupcheva CN, Chew GSM, Edwards M, Craig JP, McGhee CNJ: Imaging posterior polymorphous corneal dystrophy by in vivo confocal microscopy. Clinical and Exp Ophthalmol 29:256, 2001 156. Witschel H, Fine BS, Grutzner P, McTigue JW: Congenital hereditary stromal dystrophy of the cornea. Arch Ophthalmol 96:1043, 1978 157. Elsas FJ, Green WR: Epibulbar tumors in childhood. Am J Ophthalmol 79:1001, 1975 158. Baum JL, Feingold M: Ocular aspects of Goldenhar's syndrome. Am J Ophthalmol 75:250, 1973 159. Shields JA, Laibson PR, Augsburger JJ, Michon CA: Cerntal corneal dermoid: a clinicopathologic correlation and review of
the literature. Can J Ophthalmol 21:23, 1986 160. Hayasaka S, Sekimoto M, Setogawa T: Epibulbar complex choristoma involving the bulbar conjunctiva and cornea. J Pediatr Ophthalmol Strabismus 26:251, 1989 161. Mann I: Developmental Abnormalities of the Eye. Philadelphia: JB Lippincott, 1957:357–361 162. Henkind P, Marinoff G, Manas A, Friedman A: Bilateral corneal dermoids. Am J Ophthalmol 76:972, 1973 163. Nsiaye PA, Ndiaye MR, Ba EA, et al: Dermoides de la cornee: A propos de deux observations du 2edegre de Ida Mann. J Fr Ophthalmol 13:255, 1990 164. Murata T, Ishibashi T, Ohnishi Y, Inomata H: Corneal choristoma with microphthalmos. Arch Ophthalmol 109:1130, 1991 165. Waring GO, Laibson PR: Keratoplasty in infants and children. Trans Am Acad Ophthalmol Otolaryngol 83:283, 1977 166. Stulting RD, Sumers KD, Cavanagh HD, Waring GO, Gammon JA: Penetrating keratoplasty in children. Ophthalmology 91:1222, 1984 167. Gollamudi SR, Traboulsi EI, Chamon W, et al: Visual outcome after surgery for Peters' anomaly. Ophthalmic Genet 15:31, 1994 168. Dana MR, Schaumberg DA, Moyes AL, Gomes JAP: Corneal transplantation in children with Peters anomaly and mesenchymal
dysgenesis. Ophthalmology 104:1580, 1997 169. Frueh BE, Brown SI: Transplantation of congenitally opaque corneas. Br J Ophthalmol 81:1064, 1997 170. Yang LLH, Lambert SR, Lynn MJ, Stulting RD: Long-term results of corneal graft survival in infants and children
with Peters' anomaly. Ophthalmology 106:833, 1999 171. Aasuri MK, Garg P, Gokhle N, Gupta S: Penetrating keratoplasty in children. Cornea 19:140, 2000 172. Cameron JA: Good visual result following early penetrating keratoplasty for Peters' anomaly. J Pediatr Ophthalmol Strabismus 30:109, 1993 173. Comer RM, Daya SM, O'Keefe M: Penetrating keratoplasty in infants. J AAPOS 5:285, 2001 174. Beauchamp GR: Pediatric keratoplasty: problems in management. J Pediatr Ophthalmol Strabismus 16:388, 1979 175. Schanzlin DJ, Goldberg DB, Brown SI: Transplantation of congenitally opaque corneas. Ophthalmology 87:1253, 1980 176. Parmley VC, Stonecipher KG, Rowsey JJ: Peters' anomaly: a review of 26 penetrating keratoplasties in infants. Ophthalmic Surg 24:31, 1993 177. Dana MR, Moyes AL, Gomes JAP, Rosheim KM, Schaumberg DA, Laibson PR, Holland EJ, Sugar A, Sugar J: The indications for and outcome in pediatric keratoplasty. A multicenter
study. Ophthalmology 102:1129, 1995 178. Schaumberg DA, Moyes AL, Gomes JAP, Dana MR: Corneal transplantation in young children with congenital hereditary endothelial
dystrophy. Am J Ophthalmol 127:373, 1999 |