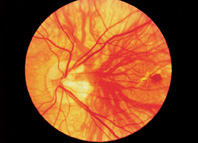
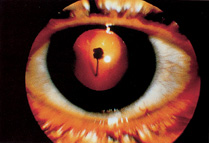
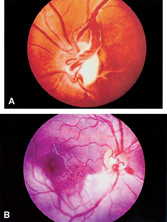
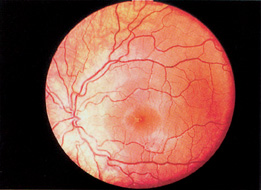
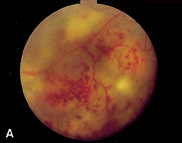
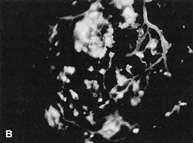
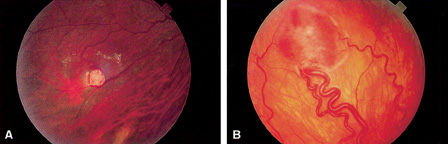
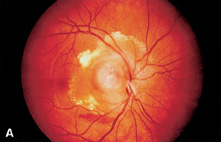
1. Duke-Elder S, Wybar KC: System of ophthalmology. In Duke-Elder S (ed): The Anatomy of the Visual System, vol 2, pp 356–358. St Louis: CV Mosby, 1961 2. Awan KJ: Familial chorioretinal vascular anastomoses and congenital cataracts. J Pediatr Ophthalmol Strab 17:384, 1980 3. Ohno-Matsui K, Morishima N, Ito M et al: Posterior routes of choroidal blood outflow in high myopia. Retina 16:419, 1996 4. Lichter PR, Schmickel RD: Posterior vortex vein and congenital glaucoma in a patient with trisomy 13 syndrome. Am J Ophthalmol 80:939, 1975 5. Mann IC: Developmental Abnormalities of the Eye, pp 138–139. Philadelphia: JB Lippincott, 1957 6. Brown GC, Tasman WS: Congenital Anomalies of the Optic Disc, pp 31–65. New York: Grune & Stratton, 1984 7. Duke-Elder S: System of Ophthalmology, ed 2, vol 3, pp 786–787. St Louis: CV Mosby, 1963 8. Birnholz JC, Farrell EE: Fetal hyaloid artery; Timing of regression with US. Radiology 166:781, 1988 9. Yap EY, Buettner H: Traumatic rupture of a persistent hyaloid artery. Am J Ophthalmol 114:225, 1992 10. Goncalves A, Cruysberg JR, Draaijer RW et al: Vitreous haemorrhage and other ocular complications of a persistent hyaloid
artery. Doc Ophthalmol 92:55, 1996 11. Chen TL, Yarng SS: Vitreous hemorrhage from a persistent hyaloid artery. Retina 13:148, 1993 12. Eller AW, Jabbour NM, Hirose T, Schepens CL: Retinopathy of prematurity: The association of a persistent hyaloid artery. Ophthalmology 94:444, 1987 13. Liebrich R: Demonstrations of diseases of the eye: A persistent hyaloid artery and
vein. Trans Pathol Soc London 22:221, 1871 14. Gass JDM: Stereoscopic Atlas of Macular Diseases; Diagnosis and Treatment, vol 1, pp 208–217, 438–444, 494–502, 844–860, 868–870, St Louis: CV Mosby, 1997 15. Degenhart W, Brown GC, Augsburger JJ et al: Prepapillary vascular loops: A clinical and fluorescein angiographic study. Ophthalmology 88:1126, 1981 16. Shakin EP, Shields JA, Augsburger JJ, Brown GC: Clinico-pathologic correlation of a prepapillary vascular loop, Retina 8:55, 1988 17. Brucker AJ, Michels RG, Fine SL: Congenital arterial loops and retinal arterial occlusion. Am J Ophthalmol 84:220, 1977 18. Brown GC, Margargal LE, Augsburger JJ et al: Preretinal arterial loops and retinal arterial occlusion, Am J Ophthalmol 87:646, 1979 19. Strassman IB, Desai UR: Prepapillary vascular loop and a recurrent vitreous hemorrhage. Retina 17:166, 1997 20. Soltau JB, Oik RJ, Gordon JM: Prepapillary arterial loop associated with vitreous hemorrhage and venous
retinal macrovessel. Retina 16:74, 1996 21. Grossniklaus H, Thall E, Annable W: Familial prepapillary vascular loops, Arch Ophthalmol 104:1755, 1986 22. Lambert HM, Sipperley JO, Shacklett DE: Autosomal dominant preretinal vascular loops, Retina 3:258, 1983 23. Goldberg MF, Pollack IP, Green WR: Familial retinal arte-riolar tortuosity with retinal hemorrhage. Am J Ophthalmol 73:183, 1972 24. Bartlett WJ, Price J: Familial retinal arterial tortuosity with retinal hemorrhage. Am J Ophthalmol 95:556, 1983 25. Wells CG, Kalina RE: Progressive inherited retinal arterio-lar tortuosity with spontaneous retinal
hemorrhages. Ophthalmology 92:1015, 1985 26. Sears J, Oilman J, Sternberg P Jr: Inherited retinal arteriolar tortuosity with retinal hemorrhages. Arch Ophthalmol 116:1185, 1998 27. Connelly BW, Gibson GG: The retinal arterioles in coarcta-tion of the aorta: 14 years' observation
of a case. Am J Ophthalmol 32:1513, 1949 28. Meredith TA: Inherited retinal venous beading, Arch Ophthalmol 105:949, 1987 29. Stewart MW, Gitter KA: Inherited retinal venous beading, Am J Ophthalmol 106:675, 1988 30. Brown GC, Donoso LA, Margargal LE et al: Congenital retinal macrovessels, Arch Ophthalmol 100:1430, 1982 31. Mansour AM, Walsh JB, Henkind P: Arteriovenous anastomoses of the retina. Ophthalmology 94:35, 1987 32. Archer DB, Deutman A, Krill AE: Arteriovenous communications of the retina, Am J Ophthalmol 75:224, 1973 33. Shields JA, Shields CL: Intraocular Tumors: A Text and Atlas, pp 394–418, 514–535, Philadelphia: WB Saunders, 1992 34. Augsburger JJ, Goldberg RE, Shields JA et al: Changing appearance of retinal arteriovenous malformation. Graefes Arch Clin Exp Ophthalmol 215:65, 1980 35. Pauleikhoff D, Wessing A: Arteriovenous communications of the retina during a 17-year follow-up, Retina 11:433, 1991 36. Mansour AM, Wells CG, Jampol LM, Kalina RE: Ocular complications of arteriovenous communications of the retina. Arch Ophthalmol 107:232, 1989 37. Tilanus MD, Hoyng C, Deutman AF et al: Congenital arteriovenous communications and the development of two types
of leaking retinal macroaneurysms, Am J Ophthalmol 112:31, 1991 38. Munoz FJ, Rebolleda G, Cores FJ, Bertrand J: Congenital retinal arteriovenous communication associated with a full-thickness
macular hole, Acta Ophthalmologica 69:117, 1991 39. Schatz H, Chang LF, Ober RR et al: Central retinal vein occlusion associated with retinal arteriovenous malformation. Ophthalmology 100:24, 1993 40. Shah GK, Shields JA, Lanning RC: Branch retinal vein obstruction secondary to retinal arteriovenous communication. Am J Ophthalmol 126:446, 1998 41. Boiling JP, Buettner H: Acquired retinal arteriovenous communications in occlusive disease of the
carotid artery. Ophthalmology 97:1148, 1990 42. Wyburn-Mason R: Arteriovenous aneurysm of midbrain and retina, facial naevi and mental
changes. Brain 66:163, 1943 43. Font RL, Ferry AP: The phakomatoses, Int Ophthalmol Clin 12:1, 1972 44. Bech K, Jensen OA: On the frequency of co-existing racemose haemangiomata of the retina and
brain, Acta Psychi-atr Scand 36:47, 1961 45. Horn G, Rabb MF, Lewickey AO: Retinal telangiectasis of the macula: A review and differential diagnosis, Int Ophthalmol Clin 21:139, 1981 46. Gass JDM, Oyakawa RT: Idiopathic juxtafoveal retinal telangiectasis. Arch Ophthalmol 100:769, 1982 47. Mansour AM, Schachat A: Foveal avascular zone in idiopathic juxtafoveolar telangiectasia. Ophthalmologica 207:9, 1993 48. Chopdar A: Retinal telangiectasis in adults: Fluorescein angiographic findings and
treatment by argon laser. Br J Ophthalmol 62:243, 1978 49. Gass JD, Blodi B A: Idiopathic juxtafoveolar retinal telangiectasis: Update of classification
and follow-up study. Ophthalmology 100:1536, 1993 50. Green WR, Quigley HA, De La Cruz Z et al: Parafoveal retinal telangiectasis: Light and electron microscopy studies, Trans Ophthalmol Soc UK 100:162, 1980 51. Berger AS, McCuen BW, Brown GC, Brownlow RL Jr: Surgical removal of subfoveal neovascularization in idio-pathic juxtafoveolar
retinal telangiectasis, Retina 17:94, 1997 52. Asdourian G, Nagpal K, Busse B et al: Macular and perima-cular vascular remodeling in sickling hemoglobinopathies. Br J Ophthalmol 60:431, 1976 53. Campo RV, Reeser FH: Retinal telangiectasia secondary to bilateral carotid artery occlusion. Arch Ophthalmol 101:1211, 1983 54. Deutman AF, Pinckers AJ, Aan de Kerk A: Dominantly inherited cystoid macular edema, Am J Ophthalmol 82: 540, 1976 55. Gurwin EB, Fitzsimons RB, Sehmi KS et al: Retinal telangiectasis in fascioscapulohumeral muscular dystrophy with
deafness. Arch Ophthalmol 102:1695, 1985 56. Chew EY, Murphy RD, Newsome DA et al: Parafoveal telangiectasis and diabetic retinopathy, Arch Ophthalmol 104:71, 1986 57. Milay RH, Klein ML, Handelman IL, Watzke RC: Abnormal glucose metabolism and parafoveal telangiectasia, Am J Ophthalmol 102:363, 1986 58. Coats G: Forms of retinal disease with massive exudation, R Lond Ophthalmol Hosp Rep 17:440, 1908 59. Henkind P, Morgan G: Peripheral retinal angioma with exudative retinopathy in adults (Coat's
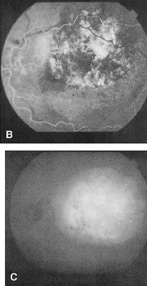
lesion). Br J Ophthalmol 50:2, 1966 60. Reese AB: Telangiectasis of the retina and Coats' disease, Am J Ophthalmol 42:1, 1956 61. Goel SD, Augsburger JJ: Hemorrhagic retinal macrocysts in advanced Coats' disease. Retina 11:437, 1991 62. Senft SH, Hidayat AA, Cavender JC: Atypical presentation of Coats' disease, Retina 14:36, 1994 63. Noble KG, Carr RE: Retinal telangiectasia. Ophthalmology 91:999, 1981 64. Ridley ME, Shields JA, Brown GC et al: Coats' disease: Evaluation of management. Ophthalmology 89:1381, 1982 65. Silodor SW, Augsburger JJ, Shields JA, Tasman WS: Natural history and management of advanced Coats' disease, Ophthalmic Surg 19:89, 1988 66. Yoshizumi MO, Kreiger AE, Lewis H et al: Vitrectomy techniques in late-stage Coats'-like exudative retinal
detachment. Doc Ophthalmol 90:387, 1995 67. Small RG: Coats' disease and muscular dystrophy, Trans Am Acad Ophthalmol Otolaryngol 72:225, 1969 68. Tasman WS: Exudative retinal detachment in retrolental flbroplasia. Trans Am Acad Ophthalmol Otolaryngol 81: 535, 1976 69. Scimeca G, Magargal LE, Augsburger JJ: Chronic exudative ischemic superior temporal branch retinal vein obstruction
simulation Coats' disease, Ann Ophthalmol 18:118, 1986 70. Khan JA, Ide CH, Strickland MP: Coats'-type retinitis pig-mentosa-check title. Surv Ophthalmol 32:317, 1988 71. Chen PP, Chong LP: Coats'-like response in a patient with pars planitis. Br J Ophthalmol 80:675, 1996 72. von Hippel E: Concerning an unusual disorder of the retina. Graefes Arch Clin Exp Ophthalmol 59:199, 1904 73. Welch RB: von Hippel-Lindau disease: The recognition and treatment of early angiomatosis
retinae and the use of cryosurgery as an adjunct to therapy. Trans Am Ophthalmol Soc 68:367, 1970 74. Lindau A: Studies on cysts of the posterior fossa: Structure, pathogenesis, and relationship
to angiomatosis retinae, Ada Pathol Microbiol Scand 1:1, 1926 75. Augsburger JJ, Shields JA, Goldberg RE: Classification and management of hereditary retinal angiomas, Jnt Ophthalmol 4:93, 1981 76. Hardwig T, Robertson DM: von Hippel-Lindau disease; A familial, often lethal, multisystem phakomatosis. Ophthalmology 91:263, 1981 77. Maher ER, Yates JR, Harries R et al: Clinical features and natural history of von Hippel-Lindau disease, Q J Med 77:1151, 1990 78. Decker HJ, Weidt EJ, Brieger J: The von Hippel-Lindau tumor suppressor gene: A rare and intriguing disease
opening new insight into basic mechanisms of carcinogenesis. Cancer Genetic Cytogenet 93:74, 1997 79. Destro M, D'Amico DJ, Gragoudas ES et al: Retinal manifestations of neurofibromatosis; Diagnosis and management. Arch Ophthalmol 109:662, 1991 80. Shields JA, Shields CL, Deglin E: Retinal capillary heman-gioma in Marshall-Stickler syndrome, Am J Ophthalmol 124:120, 1997 81. Brown GC, Shields JA: Tumors of the optic nerve head, Surv Ophthalmol 29:239, 1985 82. Schmidt D, Neumann HP: Retinal vascular hamartoma in von Hippel-Lindau disease, Arch Ophthalmol 113:1163, 1995 83. Grossniklaus HE, Thomas JW, Vigneswaran N, Jarrett WHD: Retinal hemangioblastoma; A histologic, immuno-histochemical, and ultrastructural
evaluation. Ophthalmology 99:140, 1992 84. Schwartz PL, Fastenberg DM, Shakin JL: Management of macular puckers associated with retinal angiomas, Ophthalmic Surg 21:550, 1990 85. Johnson MW, Flynn HW Jr, Gass JD: Pars plana vitrectomy and direct diathermy for complications of multiple
retinal angiomas. Ophthalmic Surg 23:47, 1992 86. Gass JD, Braunstein R: Sessile and exophytic capillary hem-angiomas of the juxtapapillary retina
and optic nerve head. Arch Ophthalmol 98:1790, 1980 87. Darr JL, Hughes RP, McNair JN: Bilateral peripapillary retinal hemangiomas; A case report. Arch Ophthalmol 75:77, 1966 88. Goldberg MF, Koenig S: Argon laser treatment of von Hippel-Lindau retinal angiomas, I. Clinical
and angio-graphic findings. Arch Ophthalmol 92:121, 1974 89. Amoils SP, Smith TR: Cryotherapy of angiomatosis retinae. Arch Ophthalmol 81:689, 1969 90. Cardoso RD, Brockhurst RJ: Perforating diathermy coagulation for retinal angiomas. Arch Ophthalmol 94:1702, 1976 91. Rosa RH Jr, Goldberg MF, Green WR: Clinicopathologic correlation of argon laser photocoagulation of retinal
angiomas in a patient with von Hippel-Lindau disease followed for more
than 20 years. Retina 16:145, 1996 92. Augsburger JJ, Freire J, Brady LW: Radiation therapy for choroidal and retinal hemangiomas. Front Radiat Ther Ocular Dis 30:265, 1997 93. McDonald HR, Schatz H, Johnson RN et al: Vitrectomy in eyes with peripheral retinal angioma associated with traction
macular detachment. Ophthalmology 103:329, 1996 94. Peyman GA, Rednam KR, Mottow-Lippa L et al: Treatment of large von Hippel tumors by eye wall resection. Ophthalmology 90:840, 1983 95. Annesley WH, Leonard BC, Shields JA et al: Fifteen year review of treated cases of retinal angiomatosis, Trans Am Acad Ophthalmol Otolaryngol 83:446, 1977 96. Shields JA, Decker WL, Sanborn GE et al: Presumed acquired retinal hemangiomas, Ophthalmology 90:1292, 1983 97. Campochiaro PA, Conway BP: Hemangioma-like masses of the retina. Arch Ophthalmol 106:1409, 1988 98. Laqua H, Wessing A: Peripheral retinal telangiectasis in adults simulating a vascular tumor
or melanoma, Ophthla-mology 90:1284, 1983 99. Laatikainen L, Immonen I, Surnmanen P: Peripheral retinal angioma-like lesion and macular pucker, Am j Ophthalmol 108:563, 1989 100. Gray RH, Gregor ZJ: Acquired peripheral retinal telangiec-tasia after retinal surgery. Retina 14:10, 1994 101. Hanssens M, De Laey JJ: Haemangioma of the optic disc or hyperplasia of the pigment epithelium: A
clinicopatho-logical correlation, Bull Soc Beige Ophthalmol 224:61, 1987 102. Shields CL, Shields JA, Barrett J, De Potter P: Vasoprolifer-ative tumors of the ocular fundus: Classification and clinical
manifestations in 103 patients. Arch Ophthalmol 113:615, 1995 103. Smeets MH, Mooy CM, Baarsma GS et al: Histopathology of a vasoproliferative tumor of the ocular fundus, Retina 18:470, 1998 104. McCabe CM, Mieler WF: Six-year follow-up of an idio-pathic retinal vasoproliferative tumor [letter]. Arch Ophthalmol 114:617, 1996 105. Gass JDM: Cavernous hemangioma of the retina: A neuro-oculocutaneous syndrome, Am J Ophthalmol 71:799, 1971 106. Kushner MS, Jampol LM, Haller JA: Cavernous hemangioma of the optic nerve. Retina 14:359, 1994 107. Messmer E, Font RL, Laqua H et al: Cavernous hemangioma of the retina: Immunohistochemical and ultrastructura! observations. Arch Ophthalmol 102:413, 1984 108. Colvard DM, Robertson DM, Trautman JC: Cavernous hemangioma of the retina. Arch Ophthalmol 96:2042, 1978 109. Goldberg RE, Pheasant TR, Shields JA: Cavernous hemangioma of the retina: A four-generation pedigree with neuro-oculocutaneous
involvement and an example of bilateral involvement. Arch Ophthalmol 97:2321, 1979 110. Pancurak J, Goldberg MF, Frenkel M, Crowell RM: Cavernous hemangioma of the retina: Genetic and central nervous system
involvement, Retina 5:215, 1985 111. Roberson GH, Kase CS, Wolpow ER: Telangiectasis and cavernous angiomas of the brain stem: “Cryptic” vascular
malformations; Report of a case, Neuroradiology 8:83, 1974 112. Sanborn GE, Augsburger JJ, Shields JA: Treatment of circumscribed choroidal hemangiomas. Ophthalmology 89:1374, 1982 113. Witschel H, Font R: Hemangioma of the choroid: A clinico-pathologic study of 71 cases and review
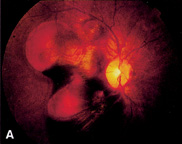
of the literature, Surv Ophthalmol 20:415, 1976 114. Anand R, Augsburger JJ, Shields JA: Circumscribed choroidal hemangiomas. Arch Ophthalmol 107:1338, 1989 115. Leys AM, Bonnet S: Case report: Associated retinal neo-vascularization and choroidal hemangioma. Retina 13:22, 1993 116. Ruby AJ, Jampol LM, Goldberg MF et al: Choroidal neo-vascularization associated with choroidal hemangiomas. Arch Ophthalmol 110:658, 1992 117. Medlock RD, Augsburger JJ, Wilkinson CP et al: Enlargement of circumscribed choroidal hemangiomas, Retina 11:385, 1991 118. Piccolino FC, Borgia L, Zinicola E: Indocyanine green angi-ography of circumscribed choroidal hemangiomas. Retina 16:19, 1996 119. Shields CL, Shields JA, De Potter P: Patterns of indocya-nine green videoangiography of choroidal tumours, Br J Ophthalmol 79:237, 1995 120. Griffiths PD, Boodram MB, Blaser S et al: Abnormal ocular enhancement in Sturge-Weber syndrome; Correlation of ocular
MR and CT findings with clinical and intracranial imaging findings. Am J Neuroradiol 17:749, 1996 121. Lindsey PS, Shields JA, Goldberg RE et al: Bilateral choroidal hemangiomas and facial nevus flammeus, Retina 1:88, 1981 122. Sullivan TJ, Clarke MP, Morin JD: The ocular manifestations of the Sturge-Weber syndrome. J Pediatr Ophthalmol Strab 29:349, 1992 123. Lanzetta P, Virgili G, Ferrari E, Menchini U: Diode laser photocoagulation of choroidal hemangioma. Int Ophthalmol 19:239, 1995 124. Zografos L, Bercher L, Chamot L et al: Cobalt-60 treatment of choroidal hemangiomas. Am J Ophthalmol 121:190, 1996 125. Schilling H, Sauerwein W, Lommatzsch A et al: Long-term results after low dose ocular irradiation for choroidal hae-mangiomas. Br J Ophthalmol 81:267, 1997 126. Yannuzzi LA, Sorenson JA, Spaide RF, Lipson B: Jdio-pathic polypoidal choroidal vasculopathy. Retina 10:1, 1990 127. Stern RM, Zakov ZN, Zegarra H, Gutman FA: Multiple recurrent serosanguineous retinal pigment epithelial detachments
in black women. Am J Ophthlamol 100:560, 1985 128. Kleiner RC, Brucker AJ, Johnston RL: The posterior uveal bleeding syndrome. Retina 10:9, 1990 129. Ross RD, Gitter KA, Cohen G, Schomaker KS: Idiopathic polypoidal choroidal vasculopathy associated with retinal arterial
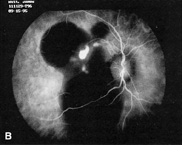
macroanurysm and hypertensive retinopathy. Retina 16:105, 1996 130. Yannuzzi LA, Ciardella A, Spaide RF et al: The expanding clinical spectrum of idiopathic polypoidal choroidal vasculopathy, Arch Ophthalmol 115:478, 1997 131. Moorthy RS, Lyon AT, Rabb MF et al: Idiopathic polypoidal choroidal vasculopathy of the macula. Ophthalmology 105:1380, 1998 132. Spaide RF, Yannuzzi LA, Slakter JS et al: Indocyanine green videoangiography of idiopathic polypoidal choroidal vasculopathy. Retina 15:100, 1995 133. Chang TS, Aylward W, Davis JL et al: Idiopathic retinal vasculitis, aneurysms, and neuro-retinitis. Ophthalmology 102:1089, 1995 134. Kincaid J, Schatz H: Bilateral retinal arteritis with multiple aneurysmal dilatations. Retina 3:171, 1983 135. Karel I, Peleska M, Divisova G: Fluorescence angiography in retinal vasculitis in children's uveitis. Ophthalmologica 166:251, 1973 136. Malan P, Ciurana AJ, Boudet C: Arterite retinienne bilater-ale avec dilatations aneurysmales multiples, J Fr Ophtalmol 9:23, 1986 137. Owens SL, Gregor ZJ: Vanishing retinal arterial aneurysms; A case report, Br J Ophthalmol 76:637, 1992 138. Gunduz K, Shields CL, Shields JA: Varix of the vortex vein ampulla simulating choroidal melanoma; Report
of four cases. Retina 18:343, 1998 139. Buettner H: Varix of the vortex ampulla simulating a choroidal melanoma, Am J Ophthalmol 109:607, 1990 140. da Cruz L, James B, Gray R, Elston J: Multiple vortex vein varices masquerading as choroidal secondaries, Br J Ophthalmol 78:800, 1994 141. Osher RH, Abrams GW, Yarian D, Armao D: Varix of the vortex vein ampulla. Am J Ophthalmol 92:653, 1981 142. Hunter JE: Vortex vein varix. Am J Optom Phys Optics 60:995, 1983 143. Singh AD, DePotter P, Shields CL, Shields JA: Indocyanine green angiography and ultrasonography of a varix of the vortex
vein, Arch Ophthalmol 111:1283, 1993 144. Lieb WE, Merton DA, Shields JA et al: Color Doppler imaging in the demonstration of an orbital varix, Br J Ophthalmol 74:305, 1990 |