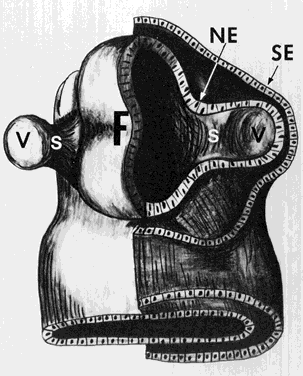
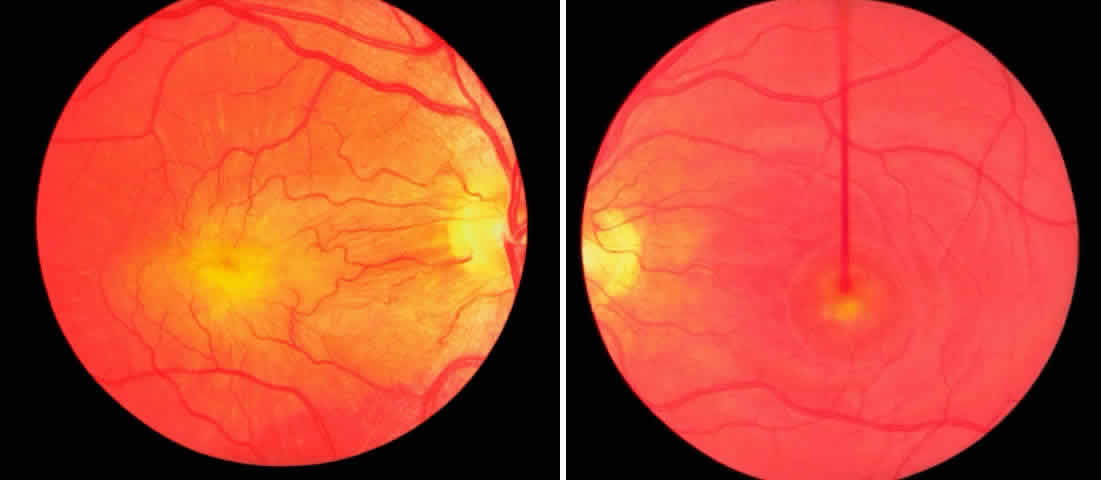
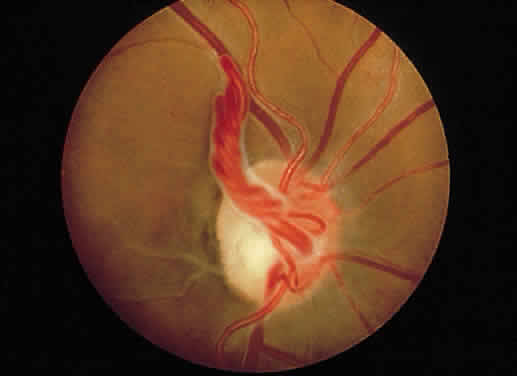
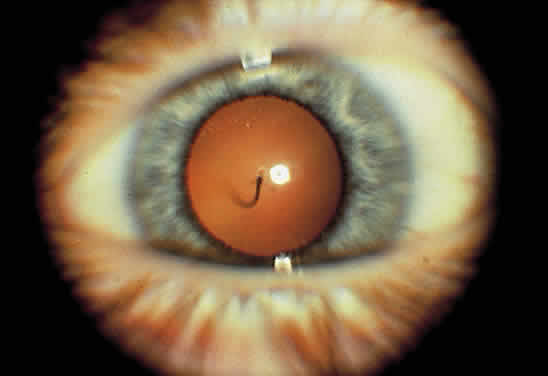
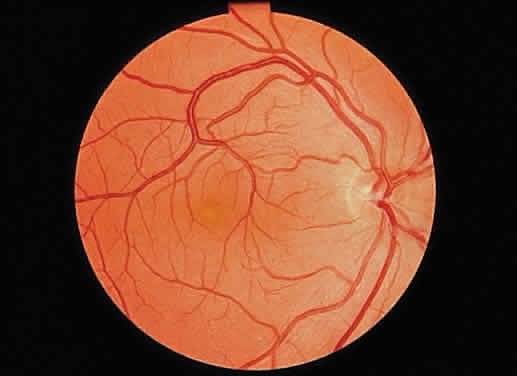
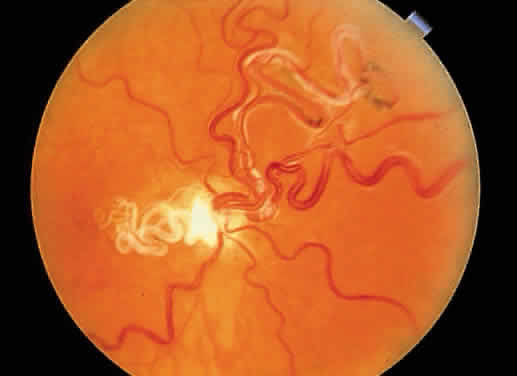
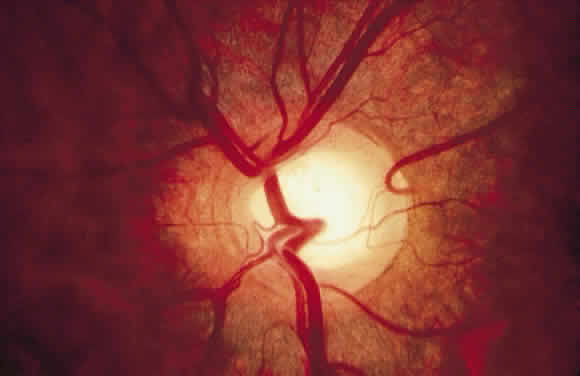
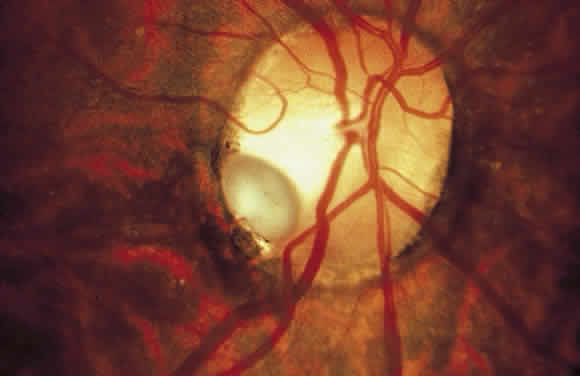
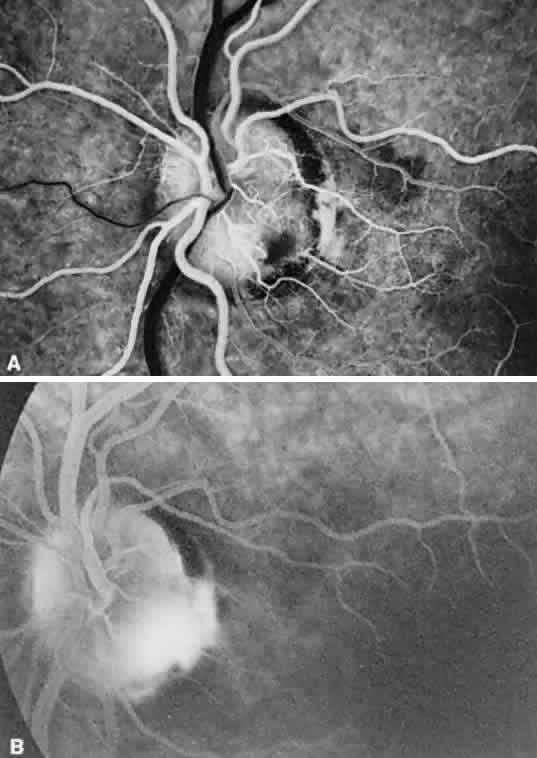
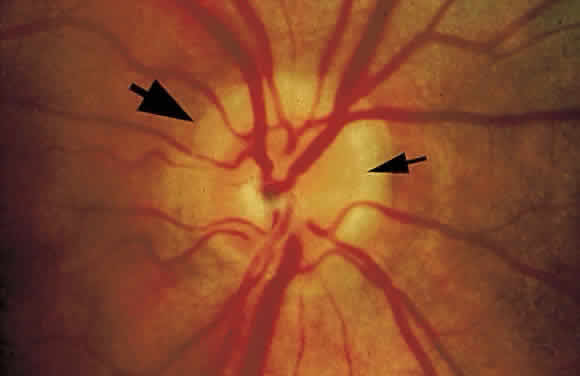
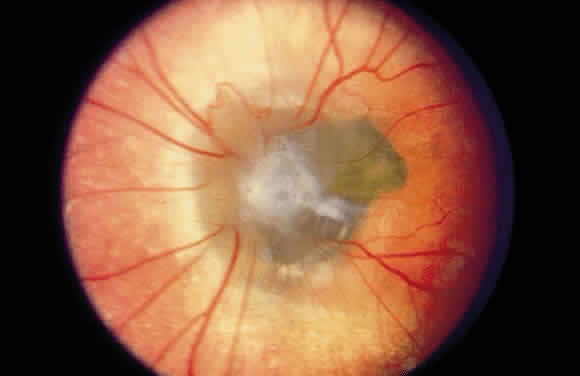
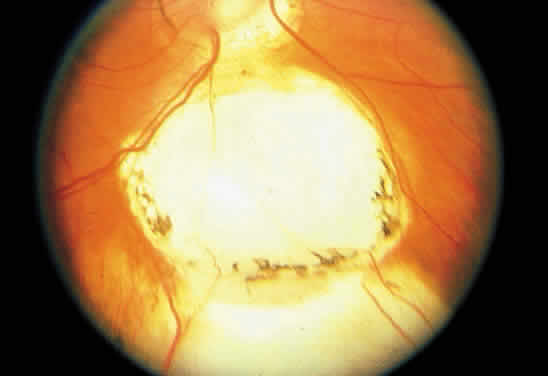
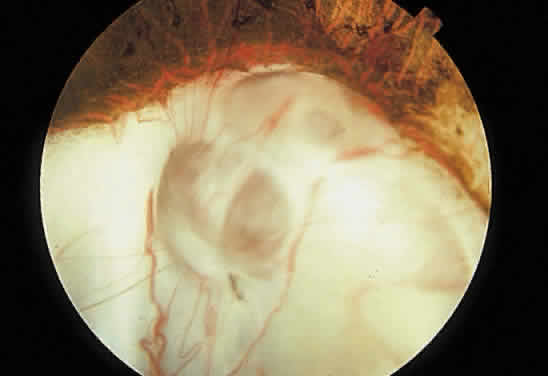
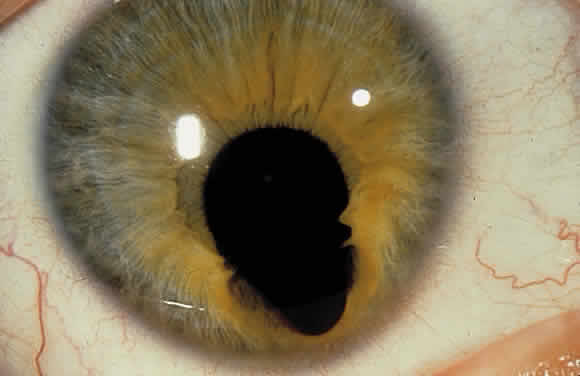
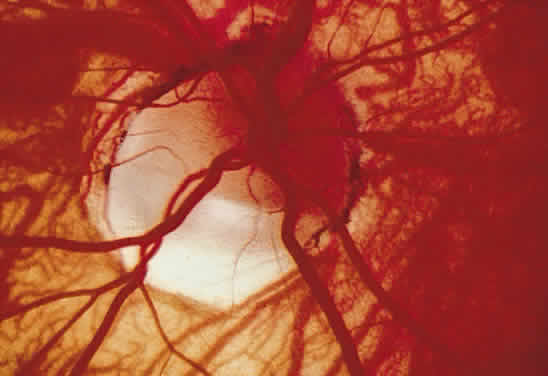
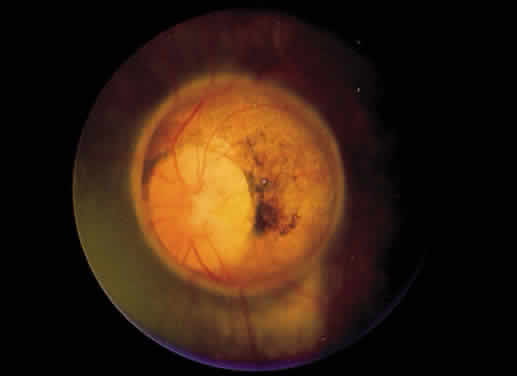
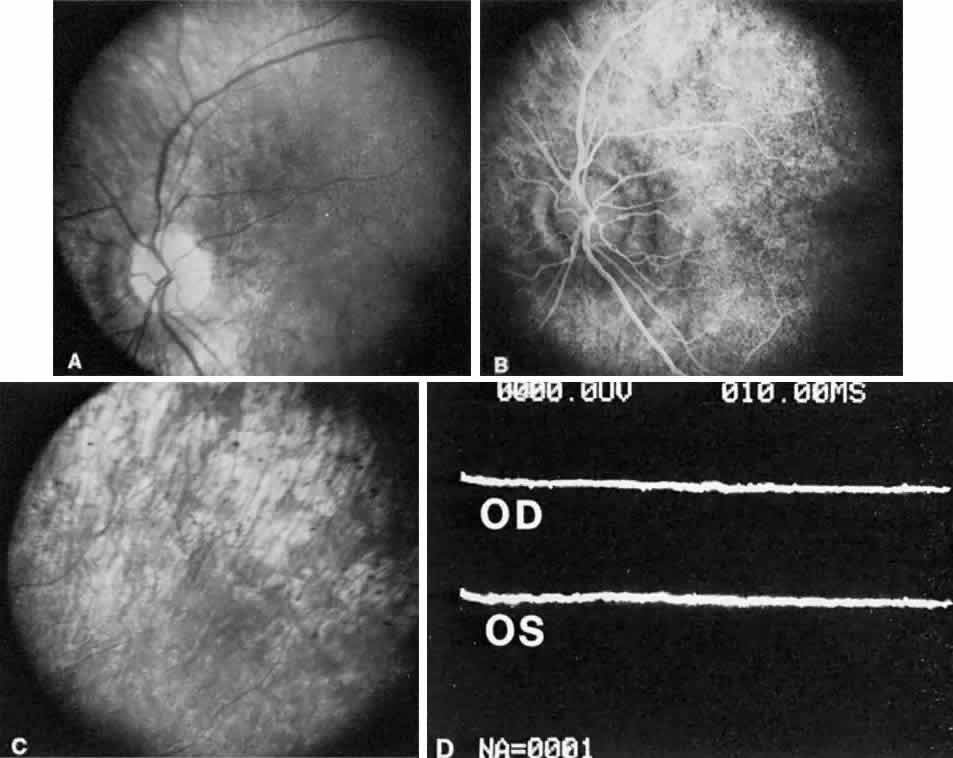
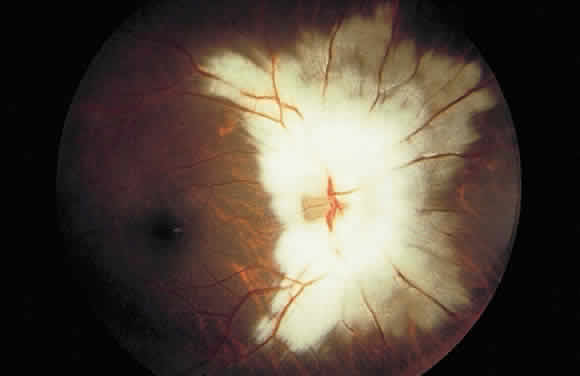
1. Mann I: Development of the Human Eye. New York, Grune & Stratton, 1969 2. Barishak YR: Embryology of the eye and its adnexae. Dev Ophthalmol 24:1, 1992 3. Ashton N: The mode of development of the retinal vessels in man. In Cant
JS (ed): William MacKenzie Centenery Symposium on the Ocular Circulation
in Health and Disease, pp 7–17. St Louis, CV Mosby, 1969 4. Liebrich R: Demonstration of diseases of the eye: Persistent hyaloid artery and vein. Trans Pathol Soc Lond 22:221, 1871 5. Brown GC, Tasman WS: Congenital Anomalies of the Optic Disc. New York, Grune & Stratton, 1983 6. Brown GC, Annesley WH Jr, Magargal LE: Peripheral venous loop. Retina 1:290, 1981 7. Shakin EP, Shields JA, Augsburger JJ, Brown GC: Clinicopathologic correlation of a prepapillary vascular loop. Retina 8:55, 1988 8. Degenhart W, Brown GC, Augsburger JJ, Magargal L: Prepapillary vascular loops. Ophthalmology 88:1126, 1981 9. Mann I: Developmental Abnormalities of the Eye. Philadelphia, JB Lippincott, 1957 10. Awan KJ: Arterial vascular anomalies of the retina. Arch Ophthalmol 95:1197, 1977 11. Oxilia E: Anomalie vascolari della retina: Ansa arteriosa prepapillare. Ann Ophthalmol Clin Ocul 73:408, 1946 12. Brown GC, Magargal L, Augsburger JJ, Shields JA: Preretinal arterial loops and retinal arterial occlusion. Am J Ophthalmol 87:646, 1979 13. Strassman IB, Desai UR: Prepapillary vascular loop and a recurrent vitreous hemorrhage. Retina 17:166, 1997 14. Soltau JB, Olk RJ, Gordon JM: Prepapillary arterial loop associated with vitreous hemorrhage and venous
retinal macrovessel. Retina 16:74, 1996 15. Wygnanski-Jaffe T, Desatnik H, Treister G, Moisseiev J: Acquired prepapillary vascular loops [letter]. Arch Ophthalmol 115:1329, 1997 16. Bronner A, Risse JF, Flament J: Prepapillary vascular looping and thrombosis of the retinal central vein: Discussion
and observations [in French]. Rev Oto-Neuro-Ophtalmol 48:249, 1976 17. Strek W, Strek P, Nowogrodzka-Zagorska M et al: Hyaloid vessels of the human fetal eye: A scanning electron microscopic
study of corrosion casts. Arch Ophthalmol 111: 1573, 1993 18. Birnholz JC, Farrell EE: Fetal hyaloid artery: Timing of regression with US. Radiology 166:781, 1988 19. Jack RL: Regression of the hyaloid vascular system: An ultrastructural analysis. Am J Ophthalmol 74:261, 1972 20. Pruett RC, Schepens CL: Posterior hyperplastic primary vitreous. Am J Ophthalmol 69:534, 1970 21. Menchini U, Pece A, Alberti M et al: Hyperplastic primary vitreous with persistent hyaloid artery in 2 non-twin
brothers [in French]. J Fr Ophtalmol 10:241, 1987 22. Delaney WV Jr: Prepapillary hemorrhage and persistent hyaloid artery. Am J Ophthalmol 90:419, 1980 23. Thumann G, Bartz-Schmidt KU, Kirchhof B, Heimann K: Branch retinal artery occlusion by diathermy of a persistent hyaloid artery. Am J Ophthalmol 124:415, 1997 24. Eller AW, Jabbour NM, Hirose T, Schepens CL: Retinopathy of prematurity: The association of a persistent hyaloid artery. Ophthalmology 94:444, 1987 25. Nissenkorn I, Kremer I, Ben-Sira I: Association of a persistent hyaloid artery and ROP [letter]. Ophthalmology 95:559, 1988 26. Chen TL, Yarng SS: Vitreous hemorrhage from a persistent hyaloid artery. Retina 13:148, 1993 27. Goncalves A, Cruysberg JR, Draaijer RW et al: Vitreous haemorrhage and other ocular complications of a persistent hyaloid
artery. Doc Ophthalmol 92:55, 1996 28. Williamson W, Barac'h D, Poirier L et al: Vitreous hemorrhage associated with persistent hyaloid artery: Apropos
of a case [in French]. J Fr Ophtalmol 17:361, 1994 29. Yap EY, Buettner H: Traumatic rupture of a persistent hyaloid artery [letter]. Am J Ophthalmol 114:225, 1992 30. Jensen VA: Studies on the branchings of retinal blood vessels. Acta Ophthalmol 14:100, 1936 31. Brown GC, Donoso LA, Magargal LE et al: Congenital retinal macrovessels. Arch Ophthalmol 100:1430, 1982 32. Polk TD, Park D, Sindt CW, Heffron ET: Congenital retinal macrovessel. Arch Ophthalmol 115:290, 1997 33. Archer DB, Deutman A, Ernest JT, Krill AE: Arteriovenous communications of the retina. Am J Ophthalmol 75:224, 1973 34. Wyburn-Mason R: Arteriovenous aneurysm of midbrain and retina, facial nevi and mental changes. Brain 66:165, 1943 35. Augsburger JJ, Goldberg RE, Shields JA et al: Changing appearance of retinal arteriovenous malformation. Graefes Arch Clin Exp Ophthalmol 215:65, 1980 36. Gass JDM: Stereoscopic Atlas of Macular Diseases: Diagnosis and Treatment, vol 1, pp 440–441. St Louis, Mosby, 1997 37. Sharma S, Cruess AF: Congenital retinal macrovessel with bilateral pigment epithelial changes. Can J Ophthalmol 31:386, 1996 38. Spraul CW, Lang GE: Congenital retinal macrovessel in German]. Klin Monatsbl Augenheilkd 211:406, 1997 39. Hayreh S: The cilioretinal arteries. Br J Ophthalmol 47:71, 1963 40. Lorentzen SE: Incidence of cilioretinal arteries. Acta Ophthalmol 48:518, 1970 41. Justice J Jr, Lehmann RP: Cilioretinal arteries: A study based on review of stereo fundus photographs
and fluorescein angiographic findings. Arch Ophthalmol 94:1355, 1976 42. Randall A: Cilioretinal or aberrant vessels. Trans Am Ophthalmol Soc 4:511, 1887 43. Jackson E: Cilioretinal and other anomalous retinal vessels. Ophthalmol Rev 30:264, 1911 44. Nipken LH, Schmidt D: Incidence, localization, length and course of the cilioretinal artery: Is
there an effect on the course of temporal rental arteries [in
German]? Klin Monatsbl Augenheilkd 208:229, 1996 45. Collier M: Frequence des vaisseaux cillioretiniens, leur rapport avecles ametropies, leur
association avec d'autres anomalies du fond de l'oeil. Bull Soc Ophthalmol Fr 9:598, 1957 46. Limaye SR, Tang RA, Pilkerton AR: Cilioretinal circulation and branch arterial occlusion associated with
preretinal arterial loops. Am J Ophthalmol 89:834, 1980 47. Brown GC, Shields JA: Cilioretinal arteries and retinal arterial occlusion. Arch Ophthalmol 97:84, 1979 48. Lee SS, Schwartz B: Role of the temporal cilioretinal artery in retaining central visual field
in open-angle glaucoma. Ophthalmology 99:696, 1992 49. Perry HD, Mallen FJ: Cilioretinal artery occlusion associated with oral contraceptives. Am J Ophthalmol 84:56, 1977 50. Brown GC, Moffat K, Cruess A et al: Cilioretinal artery obstruction. Retina 3:182, 1983 51. Mizuno K, Sasaoka M, Sasoh M, Uji Y: A case of nonarteritic anterior ischemic optic neuropathy with cilioretinal
artery occlusion [in Japanese]. Nippon Ganka Gakkai Zasshi 99:855, 1995 52. Brazitikos PD, Pournaras CJ, Othenin-Girard P, Borruat FX: Pathogenetic mechanisms in combined cilioretinal artery and retinal vein
occlusion: A reappraisal. Int Ophthalmol 17:235, 1993 53. Schatz H, Fong AC, McDonald HR et al: Cilioretinal artery occlusion in young adults with central retinal vein
occlusion. Ophthalmology 98:594, 1991 54. Keyser BJ, Duker JS, Brown GC et al: Combined central retinal vein occlusion and cilioretinal artery occlusion
associated with prolonged retinal arterial filling. Am J Ophthalmol 117:308, 1994 55. Greven CM, Slusher MM, Weaver RG: Retinal arterial occlusions in young adults. Am J Ophthalmol 120:776, 1995 56. Brown GC, Shields JA, Goldberg RE: Congenital pits of the optic nerve head. II. Clinical studies in humans. Ophthalmology 87:51, 1980 57. Petersen HP: Pits or crater-like holes in the optic disc. Acta Ophthalmol 36:435, 1958 58. Rutledge BK, Puliafito CA, Duker JS et al: Optical coherence tomography of macular lesions associated with optic nerve
head pits. Ophthalmology 103:1047, 1996 59. Lincoff H, Lopez R, Kreissig I et al: Retinoschisis associated with optic nerve pits. Arch Ophthalmol 106:61, 1988 60. Theodossiadis GP, Theodossiadis PG: Hemorrhage in macular retinoschisis cavity associated with optic nerve
pit [letter]. Arch Ophthalmol 114:493, 1996 61. Lincoff H, Yannuzzi L, Singerman L et al: Improvement in visual function after displacement of the retinal elevations
emanating from optic pits. Arch Ophthalmol 111:1071, 1993 62. Krivoy D, Gentile R, Liebmann JM et al: Imaging congenital optic disc pits and associated maculopathy using optical
coherence tomography. Arch Ophthalmol 114:165, 1996 63. Brown GC, Shields JA, Patty BE, Goldberg RE: Congenital pits of the optic nerve head. I. Experimental studies in collie
dogs. Arch Ophthalmol 97:1341, 1979 64. Brown GC, Augsburger JJ: Congenital pits of the optic nerve head and retinochoroidal colobomas. Can J Ophthalmol 15:144, 1980 65. Giuffre G: Optic pit syndrome. Doc Ophthalmol 64:187, 1986 66. Theodossiadis GP, Kollia AK, Theodossiadis PG: Cilioretinal arteries in conjunction with a pit of the optic disc. Ophthalmologica 204:115, 1992 67. Kalina RE, Conrad WC: Intrathecal fluorescein for serous macular detachment. Arch Ophthalmol 94:1421, 1976 68. Billi B, Lesnoni G, Giuliano M et al: Post-traumatic macular break associated to congenital optic disc pit and
pre-existing sensory macular detachment. Int Ophthalmol 20:269, 1996 69. Bartz-Schmidt KU, Heimann K, Esser P: Vitrectomy for macular detachment associated with optic nerve pits. Int Ophthalmol 19:323, 1995 70. Theodossiadis GP: Treatment of maculopathy associated with optic disk pit by sponge explant. Am J Ophthalmol 121:630, 1996 71. Theodossiadis GP, Panopoulos M, Kollia AK, Georgopoulos G: Long-term study of patients with congenital pit of the optic nerve and
persistent macular detachment. Acta Ophthalmol 70:495, 1992 72. Sobol WM, Blodi CF, Folk JC, Weingeist TA: Long-term visual outcome in patients with optic nerve pit and serous retinal
detachment of the macula. Ophthalmology 97: 1539, 1990 73. Brockhurst RJ: Optic pits and posterior retinal detachment. Trans Am Ophthalmol Soc 73:264, 1976 74. Theodossiadis G: Evolution of congenital pit of the optic disk with macular detachment in
photocoagulated and nonphotocoagulated eyes. Am J Ophthalmol 84:620, 1977 75. McDonald HR, Schatz H, Johnson RN: Treatment of retinal detachment associated with optic pits. Int Ophthalmol Clin 32:35, 1992 76. Annesley W, Brown G, Bolling J et al: Treatment of retinal detachment with congenital optic pit by krypton laser
photocoagulation. Graefes Arch Clin Exp Ophthalmol 225: 311, 1987 77. Snead MP, James N, Jacobs PM: Vitrectomy, argon laser, and gas tamponade for serous retinal detachment
associated with an optic disc pit: A case report. Br J Ophthalmol 75:381, 1991 78. Lee KJ, Peyman GA: Surgical management of retinal detachment associated with optic nerve pit. Int Ophthalmol 17:105, 1993 79. Schatz H, McDonald HR: Treatment of sensory retinal detachment associated with optic nerve pit
or coloboma. Ophthalmology 95:178, 1988 80. Alexander TA, Billson FA: Vitrectomy and photocoagulation in the management of serous detachment
associated with optic nerve pits. Aust J Ophthalmol 12:139, 1984 81. Monin C, Le Guen Y, Morel C, Haut J: Treatment of coloboma pits of the optic nerve complicated by serous detachment
of the neuroepithelium [in French]. J Fr Ophtalmol 17:574, 1994 82. Taiel-Sartral M, Mimoun G, Glacet-Bernard A et al: Vitrectomy-laser-gas for treating optic disk pits complicated by serous
macular detachment [in French]. J Fr Ophtalmol 19:603, 1996 83. Van Nouhuys J, Bruyn G: Nasopharyngeal transsphenoidal encephalocele, craterlike hole in the optic
disc and agenesis of the corpus callosum: Pneumoencephalographic visualization
in a case. Psychiatr Neurol Neurochir 67:243, 1964 84. Corbett JJ, Savino PJ, Schatz NJ, Orr LS: Cavitary developmental defects of the optic disc: Visual loss associated
with optic pits and colobomas. Arch Neurol 37:210, 1980 85. Jonas JB, Freisler KA: Bilateral congenital optic nerve head pits in monozygotic siblings. Am J Ophthalmol 124:844, 1997 86. Ragge NK, Ravine D, Wilkie AO: Dominant inheritance of optic pits. Am J Ophthalmol 125:124, 1998 87. Veugelen D, Leys A: Autosomal-dominant heredity of optic pits: A report of two families. Bull Soc Belge Ophtalmol 223:75, 1987 88. Mosier MA, Lieberman MF, Green WR, Knox DL: Hypoplasia of the optic nerve. Arch Ophthalmol 96:1437, 1978 89. Hotchkiss ML, Green WR: Optic nerve aplasia and hypoplasia. J Pediatr Ophthalmol Strabismus 16:225, 1979 90. Jacobson L, Hellstrom A, Flodmark O: Large cups in normal-sized optic discs: A variant of optic nerve hypoplasia
in children with periventricular leukomalacia. Arch Ophthalmol 115:1263, 1997 91. Walton DS, Robb RM: Optic nerve hypoplasia: A report of 20 cases. Arch Ophthalmol 84:572, 1970 92. Edwards WC, Layden WE: Optic nerve hypoplasia. Am J Ophthalmol 70:950, 1970 93. Gardner HB, Irvine AR: Optic nerve hypoplasia with good visual acuity. Arch Ophthalmol 88:255, 1972 94. Petersen RA, Walton DS: Optic nerve hypoplasia with good visual acuity and visual field defects: A
study of children of diabetic mothers. Arch Ophthalmol 95:254, 1977 95. Zeki SM: Optic nerve hypoplasia and astigmatism: A new association. Br J Ophthalmol 74:297, 1990 96. Zeki SM, Hollman AS, Dutton GN: Neuroradiological features of patients with optic nerve hypoplasia. J Pediatr Ophthalmol Strabismus 29:107, 1992 97. Sprague JB, Wilson WB: Electrophysiologic findings in bilateral optic nerve hypoplasia. Arch Ophthalmol 99:1028, 1981 98. Wakakura M, Alvarez E: A simple clinical method of assessing patients with optic nerve hypoplasia: The
disc-macula distance to disc diameter ratio (DM/DD). Acta Ophthalmol 65:612, 1987 99. Alvarez E, Wakakura M, Khan Z, Dutton GN: The disc-macula distance to disc diameter ratio: A new test for confirming
optic nerve hypoplasia in young children. J Pediatr Ophthalmol Strabismus 25:151, 1988 100. Brodsky MC, Glasier CM, Pollock SC, Angtuago EJ: Optic nerve hypoplasia: Identification by magnetic resonance imaging. Arch Ophthalmol 108:1562, 1990 101. Boynton JR, Pheasant TR, Levine MR: Hypoplastic optic nerves studied with B-scan ultrasonography and axial
tomography of the optic canals. Can J Ophthalmol 10:473, 1975 102. Acers TE. Optic nerve hypoplasia: Septo-optic-pituitary dysplasia syndrome. Trans Am Ophthalmol Soc 79:425, 1981 103. DeMorsier G: Etuds sur les dysraphies cranioencephaliques. III. Agenesie du septum lucidum
avec malformation du tractus optique: La dysplasie septa optique. Schweiz Arch Neurol Psychiatr 77:267, 1956 104. Hoyt WF, Kaplan SL, Grumbach MM, Glaser JS: Septo-optic dysplasia and pituitary
dwarfism. Lancet i:893, 1970 105. Sheridan SJ, Robb RM: Optic nerve hypoplasia with diabetes insipidus. J Pediatr Ophthalmol Strabismus 15:82, 1978 106. Siatkowski RM, Sanchez JC, Andrade R, Alvarez A: The clinical, neuroradiographic, and endocrinologic profile of patients
with bilateral optic nerve hypoplasia. Ophthalmology 104:493, 1997 107. Willnow S, Kiess W, Butenandt O et al: Endocrine disorders in septo-optic dysplasia (De Morsier syndrome): Evaluation
and follow up of 18 patients. Eur J Pediatr 155:179, 1996 108. Brodsky MC, Glasier CM: Optic nerve hypoplasia: Clinical significance of associated central nervous
system abnormalities on magnetic resonance imaging. Arch Ophthalmol 111:66, 1993 109. Margalith D, Tze WJ, Jan JE: Congenital optic nerve hypoplasia with hypothalamic-pituitary dysplasia: A
review of 16 cases. Am J Dis Child 139:361, 1985 110. Kaufman LM, Miller MT, Mafee MF: Magnetic resonance imaging of pituitary stalk hypoplasia: A discrete midline
anomaly associated with endocrine abnormalities in septo-optic dysplasia. Arch Ophthalmol 107:1485, 1989 111. Robinson GC, Conry RF: Maternal age and congenital optic nerve hypoplasia: A possible clue to
etiology. Dev Med Child Neurol 28:294, 1986 112. Lubinsky MS: Hypothesis: Septo-optic dysplasia is a vascular disruption sequence. Am J Med Genet 69:235, 1997 113. Hoyt CS, Billson FA: Maternal anticonvulsants and optic nerve hypoplasia. Br J Ophthalmol 62:3, 1978 114. West J, Burke JP, Strachan I: Carbamazepine, epilepsy, and optic nerve hypoplasia [letter]. Br J Ophthalmol 74: 511, 1990 115. Stromland K, Pinazo-Duran MD: Optic nerve hypoplasia: Comparative effects in children and rats exposed
to alcohol during pregnancy. Teratology 50:100, 1994 116. McKinna AJ: Quinine induced hypoplasia of the optic nerve. Can J Ophthalmol 1:261, 1966 117. Nelson M, Lessell S, Sadun AA: Optic nerve hypoplasia and maternal diabetes mellitus. Arch Neurol 43:20, 1986 118. Kim RY, Hoyt WF, Lessell S, Narahara MH: Superior segmental optic hypoplasia: A sign of maternal diabetes. Arch Ophthalmol 107:1312, 1989 119. Hittner HM, Desmond MM, Montgomery JR: Optic nerve manifestations of human congenital cytomegalovirus infection. Am J Ophthalmol 81:661, 1976 120. Lee JT, Hall TR, Bateman JB: Optic nerve hypoplasia secondary to intracranial teratoma. Am J Ophthalmol 124:705, 1997 121. Ichiyama T, Hayashi T, Nishikawa M, Furukawa S: Optic nerve hypoplasia with hypopituitarism and an arachnoid cyst. Brain Dev 18:234, 1996 122. Scheie H, Adler F: Aplasia of the optic nerve. Arch Ophthalmol 83:569, 1941 123. Lambert SR, Hoyt CS, Narahara MH: Optic nerve hypoplasia. Surv Ophthalmol 32:1, 1987 124. Hoyt CS, Billson FA: Optic nerve hypoplasia: Changing perspectives. Aust NZ J Ophthalmol 14:325, 1986 125. Brodsky MC: Hypothesis: Septo-optic dysplasia is a vascular disruption sequence. Surv Ophthalmol 42:489, 1998 126. Weiter JJ, McLean IW, Zimmerman LE: Aplasia of the optic nerve and disk. Am J Ophthalmol 83:569, 1977 127. Little LE, Whitmore PV, Wells TW Jr: Aplasia of the optic nerve. J Ped Ophthalmol 13:84, 1976 128. Storm RL, PeBenito R: Bilateral optic nerve aplasia associated with hydranencephaly. Ann Ophthalmol 16:988, 1984 129. Yanoff M, Rorke LB, Allman MI: Bilateral optic system aplasia with relatively normal eyes. Arch Ophthalmol 96:97, 1978 130. Margo CE, Hamed LM, Fang E, Dawson WW: Optic nerve aplasia. Arch Ophthalmol 110:1610, 1992 131. Kindler P: Morning glory syndrome: Unusual congenital optic disk anomaly. Am J Ophthalmol 69:376, 1970 132. Steinkuller PG: The morning glory disk anomaly: Case report and literature review. J Pediatr Ophthalmol Strabismus 17:81, 1980 133. Haik BG, Greenstein SH, Smith ME et al: Retinal detachment in the morning glory anomaly. Ophthalmology 91:1638, 1984 134. Irvine AR, Crawford JB, Sullivan JH: The pathogenesis of retinal detachment with morning glory disc and optic
pit. Trans Am Ophthalmol Soc 84:280, 1986 135. Coll GE, Chang S, Flynn TE, Brown GC: Communication between the subretinal space and the vitreous cavity in the
morning glory syndrome. Graefes Arch Clin Exp Ophthalmol 233:441, 1995 136. Bartz-Schmidt KU, Heimann K: Pathogenesis of retinal detachment associated with morning glory disc. Int Ophthalmol 19:35, 1995 137. von Fricken MA, Dhungel R: Retinal detachment in the morning glory syndrome: Pathogenesis and management. Retina 4:97, 1984 138. Chang S, Haik BG, Ellsworth RM et al: Treatment of total retinal detachment in morning glory syndrome. Am J Ophthalmol 97:596, 1984 139. Brown GC, Brown MM: Repair of retinal detachment associated with congenital excavated defects
of the optic disc. Ophthalmic Surg 26:11, 1995 140. Morioka M, Marubayashi T, Masumitsu T et al: Basal encephaloceles with morning glory syndrome, and progressive hormonal
and visual disturbances: Case report and review of the literature. Brain Dev 17:196, 1995 141. Itakura T, Miyamoto K, Uematsu Y et al: Bilateral morning glory syndrome associated with sphenoid encephalocele: Case
report. J Neurosurg 77:949, 1992 142. Hope-Ross M, Johnston SS: The morning glory syndrome associated with sphenoethmoidal encephalocele. Ophthalmic Paediatr Genet 11:147, 1990 143. Jackson WE, Freed S: Ocular and systemic abnormalities associated with morning glory syndrome. Ophthalmic Paediatr Genet 5:111, 1985 144. Cennamo G, Liguori G, Pezone A, Iaccarino G: Morning glory syndrome associated with marked persistent hyperplastic primary
vitreous and lens colobomas. Br J Ophthalmol 73:684, 1989 145. Brown GC, Gonder J, Levin A: Persistence of the primary vitreous in association with the morning glory
disc anomaly. J Pediatr Ophthalmol Strabismus 21:5, 1984 146. Goldhammer Y, Smith JL: Optic nerve anomalies in basal encephalocele. Arch Ophthalmol 93:115,1975 147. Savell J, Cook JR: Optic nerve colobomas of autosomal-dominant heredity. Arch Ophthalmol 94:395, 1976 148. Daufenbach DR, Ruttum MS, Pulido JS, Keech RV: Chorioretinal colobomas in a pediatric population. Ophthalmology 105:1455, 1998 149. Apple DJ: New aspects of colobomas and optic nerve anomalies. Int Ophthalmol Clin 24:109, 1984 150. Brodsky MC: Morning glory disc anomaly or optic disc coloboma [letter]? Arch Ophthalmol 112:153, 1994 151. Traboulsi EI: Morning glory disc anomaly or optic disc coloboma [letter]? Arch Ophthalmol 112:153, 1994 152. Pagon RA: Ocular coloboma. Surv Ophthalmol 25:223, 1981 153. Pagon RA, Graham JM Jr, Zonana J, Yong SL: Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE
association. J Pediatr 99:223, 1981 154. Maumenee IH, Mitchell TN: Colobomatous malformations of the eye. Trans Am Ophthalmol Soc 88:123, 1990 155. Cogan DG: Congenital anomalies of the retina. Birth Defects 7:41, 1971 156. Jesberg D, Schepens C: Retinal detachment associated with coloboma of the choroid. Arch Ophthalmol 65:163, 1961 157. McDonald HR, Lewis H, Brown G, Sipperley JO: Vitreous surgery for retinal detachment associated with choroidal coloboma. Arch Ophthalmol 109:1399, 1991 158. Riise D: The nasal fundus ectasia. Acta Ophthalmol Suppl 3:108, 1975 159. Riise D: Visual field defects in optic disc malformation with ectasia of the fundus. Acta Ophthalmol 44:906, 1966 160. Young SE, Walsh FB, Knox DL: The tilted disk syndrome. Am J Ophthalmol 82:16, 1976 161. Malinowski SM, Pulido JS, Flickinger RR: The protective effect of the tilted disc syndrome in diabetic retinopathy. Arch Ophthalmol 114:230, 1996 162. Wise JB, MacLean AL, Gass JD: Contractile peripapillary staphyloma. Arch Ophthalmol 75:626, 1966 163. Sugar HS, Beckman H: Peripapillary staphyloma with respiratory pulsation. Am J Ophthalmol 68:895, 1969 164. Gottlieb JL, Prieto DM, Vander JF et al: Peripapillary staphyloma. Am J Ophthalmol 124:249, 1997 165. Caldwell JB, Sears ML, Gilman M: Bilateral peripapillary staphyloma with normal vision. Am J Ophthalmol 71(suppl):423, 1971 166. Kral K, Svarc D: Contractile peripapillary staphyloma. Am J Ophthalmol 71:1090, 1971 167. Cennamo G, Sammartino A, Fioretti F: Morning glory syndrome with contractile peripapillary staphyloma. Br J Ophthalmol 67:346, 1983 168. Font RL, Zimmerman LE: Intrascleral smooth muscle in coloboma of the optic disk: Electron microscopic
verification. Am J Ophthalmol 72:452, 1971 169. Vuori ML: Morning glory disc anomaly with pulsating peripapillary staphyloma: A case
history. Acta Ophthalmol 65:602, 1987 170. Buettner H: Congenital hypertrophy of the retinal pigment epithelium. Am J Ophthalmol 79:177, 1975 171. Shields JA, Tso MO: Congenital grouped pigmentation of the retina: Histopathologic description
and report of a case. Arch Ophthalmol 93:1153, 1975 172. Purcell JJ Jr, Shields JA: Hypertrophy with hyperpigmentation of the retinal pigment epithelium. Arch Ophthalmol 93:1122, 1975 173. Traboulsi EI, Murphy SF, de la Cruz ZC et al: A clinicopathologic study of the eyes in familial adenomatous polyposis
with extracolonic manifestations (Gardner's syndrome). Am J Ophthalmol 110:550, 1990 174. Traboulsi EI, Maumenee IH, Krush AJ et al: Congenital hypertrophy of the retinal pigment epithelium predicts colorectal
polyposis in Gardner's syndrome. Arch Ophthalmol 108:525, 1990 175. Shields J, Shields C: Intraocular Tumors: A Text and Atlas, pp 438–444. Philadelphia, WB
Saunders, 1992 176. Auerbach V: Inborn errors of metabolism originating in the eye. In Harley
R (ed): Pediatric Ophthalmology, pp 548–563. Philadelphia, WB
Saunders, 1975 177. Trevor-Roper P: Marriage of two complete albinos with normally pigmented offspring. Br J Ophthalmol 36:107, 1952 178. Spritz RA: Molecular genetics of oculocutaneous albinism. Hum Mol Genet 3:1469, 1994 179. Summers CG: Vision in albinism. Trans Am Ophthalmol Soc 94:1095, 1996 180. Oetting WS, Summers CG, King RA: Albinism and the associated ocular defects. Metab Pediatr Syst Ophthalmol 17:5, 1994 181. O'Donnell FE Jr, King RA, Green WR, Witkop CJ Jr: Autosomal recessively inherited ocular albinism: A new form of ocular albinism
affecting females as severely as males. Arch Ophthalmol 96:1621, 1978 182. Schiaffino MV, Baschirotto C, Pellegrini G et al: The ocular albinism type 1 gene product is a membrane glycoprotein localized
to melanosomes. Proc Natl Acad Sci USA 93:9055, 1996 183. Schnur RE, Gao M, Wick PA et al: OA1 mutations and deletions in X-linked ocular albinism. Am J Hum Genet 62:800, 1998 184. O'Donnell FE Jr, Green WR, Fleischman JA, Hambrick GW: X-linked ocular albinism in blacks: Ocular albinism cum pigmento. Arch Ophthalmol 96:1189, 1978 185. Fulton AB, Albert DM, Craft JL: Human albinism: Light and electron microscopy study. Arch Ophthalmol 96:305, 1978 186. Mietz H, Green WR, Wolff SM, Abundo GP: Foveal hypoplasia in complete oculocutaneous albinism: A histopathologic
study. Retina 12:254, 1992 187. Apkarian P, Shallo-Hoffmann J: VEP projections in congenital nystagmus: VEP asymmetry in albinism. A comparison
study. Invest Ophthalmol Vis Sci 32:2653, 1991 188. Bouzas EA, Caruso RC, Drews-Bankiewicz MA, Kaiser-Kupfer MI: Evoked potential analysis of visual pathways in human albinism. Ophthalmology 101:309, 1994 189. Fitzgerald K, Cibis GW: The value of flash visual evoked potentials in albinism. J Pediatr Ophthalmol Strabismus 31:18, 1994 190. Carden SM, Boissy RE, Schoettker PJ, Good WV: Albinism: Modern molecular diagnosis. Br J Ophthalmol 82:189, 1998 191. Collins B, Silver J: Recent experiences in the management of visual impairment in albinism. Ophthalmic Paediatr Genet 11:225, 1990 192. Gregg N: Congenital cataract following German measles in the mother. Trans Ophthalmol Soc Aust 3:35, 1941 193. Wolff SM: The ocular manifestations of congenital rubella. Trans Am Ophthalmol Soc 70:577, 1972 194. Hertzberg R: Twenty-five-year follow-up of ocular defects in congenital rubella. Am J Ophthalmol 66:269, 1968 195. Bonomo PP: Involution without disciform scarring of subretinal neovascularization
in presumed rubella retinopathy: A case report. Acta Ophthalmol 60:141, 1982 196. Slusher MM, Tyler ME: Rubella retinopathy and subretinal neovascularization. Ann Ophthalmol 14:292, 1982 197. Frank KE, Purnell EW: Subretinal neovascularization following rubella retinopathy. Am J Ophthalmol 86:462, 1978 198. Deutman AF, Grizzard WS: Rubella retinopathy and subretinal neovascularization. Am J Ophthalmol 85:82, 1978 199. Orth DH, Fishman GA, Segall M et al: Rubella maculopathy. Br J Ophthalmol 64:201, 1980 200. Boger WPd: Late ocular complications in congenital rubella syndrome. Ophthalmology 87:1244, 1980 201. Obenour LC: The electroretinogram in rubella retinopathy. Int Ophthalmol Clin 12:105, 1972 202. Deutman A: Rod-cone dystrophy: Primary, hereditary, pigmentary retinopathy, retinitis
pigmentosa. In Krill A (ed): Hereditary Retinal and Choroidal
Diseases, 552–556. Hagerstown, MD, Harper & Row, 1977 203. Perrault I, Rozet JM, Calvas P et al: Retinal-specific guanylate cyclase gene mutations in Leber's congenital
amaurosis. Nature Genet 14:461, 1996 204. Dagi LR, Leys MJ, Hansen RM, Fulton AB: Hyperopia in complicated Leber's congenital amaurosis. Arch Ophthalmol 108:709, 1990 205. Schroeder R, Mets MB, Maumenee IH: Leber's congenital amaurosis: Retrospective review of 43 cases and
a new fundus finding in two cases. Arch Ophthalmol 105:356, 1987 206. Traboulsi EI, Maumenee IH: Photoaversion in Leber's congenital amaurosis. Ophthalmic Genet 16:27, 1995 207. Heher IL, Traboulsi EI, Maumenee IH: The natural history of Leber's congenital amaurosis: Age-related findings
in 35 patients. Ophthalmology 99:241, 1992 208. Camuzat A, Rozet JM, Dollfus H et al: Evidence of genetic heterogeneity of Leber's congenital amaurosis (LCA) and
mapping of LCA1 to chromosome 17p13. Hum Genet 97:798, 1996 209. Camuzat A, Rozet JM, Dollfus H et al: A gene for Leber's congenital amaurosis maps to chromosome 17p. Hum Mol Genet 4:1447, 1995 210. Kodama T, Hayasaka S, Setogawa T: Myelinated retinal nerve fibers: Prevalence, location and effect on visual
acuity. Ophthalmologica 200:77, 1990 211. Lee MS, Gonzalez C: Unilateral peripapillary myelinated retinal nerve fibers associated with
strabismus, amblyopia, and myopia. Am J Ophthalmol 125:554, 1998 212. Schmidt D, Meyer JH, Brandi-Dohrn J: Wide-spread myelinated nerve fibers of the optic disc: Do they influence
the development of myopia? Int J Ophthalmol 20:263, 1996 213. Summers CG, Romig L, Lavoie JD: Unexpected good results after therapy for anisometric amblyopia associated
with unilateral peripapillary myelinated nerve fibers. J Pediatr Ophthalmol Strabismus 28:134, 1991 214. Kasmann B, Hoh H, Ruprecht KW: Results of occlusion therapy in anisomyopic amblyopia with myelinated nerve
fibers. Ger J Ophthalmol 5:241, 1996 215. Leys AM, Leys MJ, Hooymans JM et al: Myelinated nerve fibers and retinal vascular abnormalities. Retina 16:89, 1996 216. Silvestri G, Sehmi K, Hamilton P: Retinal vascular abnormalities: A rare complication of myelinated nerve
fibers? Retina 16:214, 1996 217. Lucas BC, Eckardt C: Venous vascular anomalies within the myelinated retinal nerve fiber area [in
German]. Klin Monatsbl Augenheilkd 199:454, 1991 218. Goldberg RE, Leonard BC, Kleineidam M, Augsburger JJ: Venous anomalies within the myelinated retinal nerve fiber area [letter; in
German]. Klin Monatsbl Augenheilkd 201:411, 1992 |