|
|
| Chapter 16 Pathology of the Optic Nerve THOMAS B. CROTTY, R. JEAN CAMPBELL, and ALFREDO A. SADUN Table Of Contents |
|
NORMAL ANATOMY AND HISTOLOGY CONGENITAL MALFORMATIONS OPTIC ATROPHY NEOPLASMS NONNEOPLASTIC MASS LESIONS OF THE OPTIC NERVE REFERENCES |
The optic stalk develops from the optic vesicle at about the seventh week
of gestation; from this is derived the optic vesicle, which matures
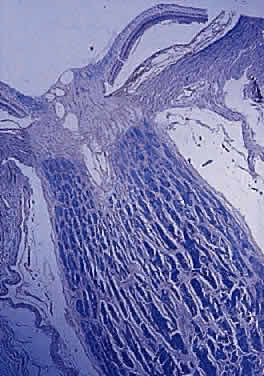
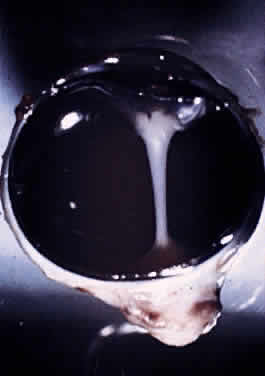
into the globe. The optic nerve is continuous with the optic tract (Fig. 1). The number of axons in the human optic nerve rapidly increases during
early embryogenesis to reach a peak of about 3 million during the second
trimester, after which loss of fibers occurs until about 1 million
fibers remain at birth.1 The diameter of the optic nerve increases throughout embryogenesis—from
about 0.15 mm at the ninth week of gestation until full term, when
it averages 2.7 mm.2 Myelination by oligodendroglial cells begins near the optic chiasm at
about the 30th week of gestation, extends distally as far as the laminar
cribrosa, and is complete at or soon after birth (Fig. 2).2
|
| NORMAL ANATOMY AND HISTOLOGY | |||||||
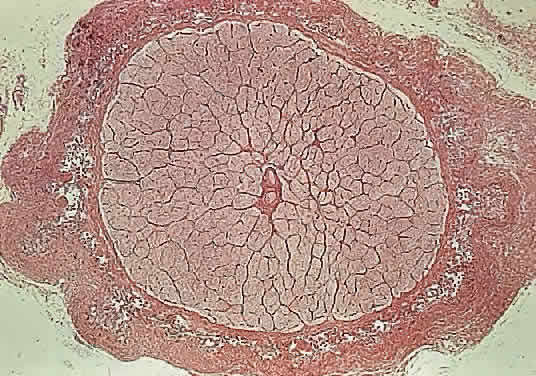
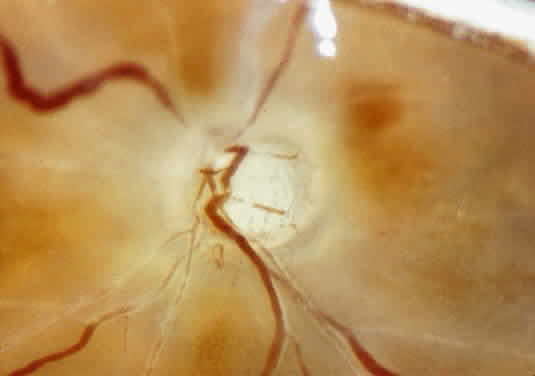
The adult human optic nerve has a cross-sectional area of about 9 mm2 (Fig. 3). It measures about 50 mm in length from the back of the eye to the optic
chiasm and is divided into intraocular, intraorbital, and intracanalicular
segments. The intraocular portion of the nerve, which is about 0.7 mm
in length, has a prelaminar and a laminar portion. The former
is divided into the inner retinal and middle choroidal layers. The outer
scleral layer constitutes the laminar portion. Axons from the retinal
ganglion cells exit the eye through the optic disc, which is 1.5 mm
in diameter. The physiologic cup is the smaller disc-shaped depression—the
edges of which parallel those of the optic disc. It lies
slightly temporal to the disc center and is lined by glial tissue (tissue
of Kuhnt). The nerve exits the globe slightly nasal to and above the
posterior pole. The choroidal layer of the prelaminar portion consists
of nonmyelinated fibers that are anterior to the laminar cribrosa. Astrocytes
are also present. The scleral lamina is composed of astrocytes, collagen, and
vessels.
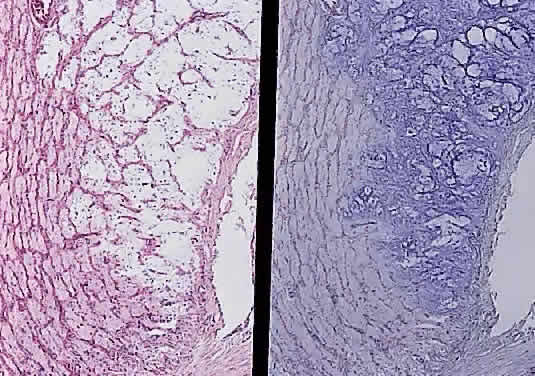
In the adult, the normal optic nerve contains about 1,000,000 nerve fibers.3,4 No difference in nerve-fiber counts between males and females has been observed but some studies have found differences between right and left eyes. Axon density is higher in the inferotemporal segment of the nerve because the fovea is slightly inferior to the horizontal midpoint of the retina and the major portion of the papillomacular bundle exits through this quadrant. Axonal diameter varies considerably (from 0.3 to 5 μm) within individual nerves and on average is smaller in the temporal and inner regions of the optic nerve than in the nasal and outer area, respectively.5 Although embryologically part of the central nervous system (CNS), the optic nerve is structurally more reminiscent of the peripheral nervous system because its nerve fibers are arranged in fascicles (Fig. 4).6 The number of fascicles in the optic nerve appears to vary only slightly between individuals but changes substantially along the length of the nerve. Immediately behind the globe and in the region of the optic canal, there are about 200 to 300 fascicles; in contrast, in the midorbital segment there are fewer than 120 fascicles. Teleologically, this variation maximizes compliance and tensile strength throughout the nerve.
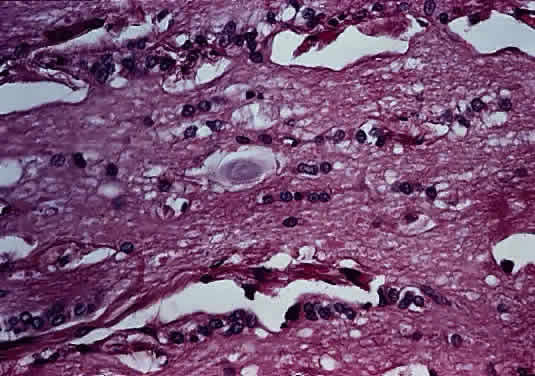
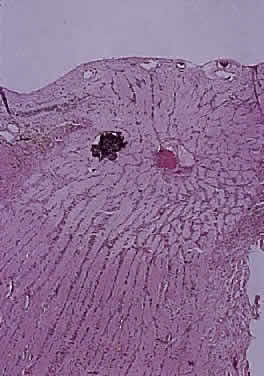
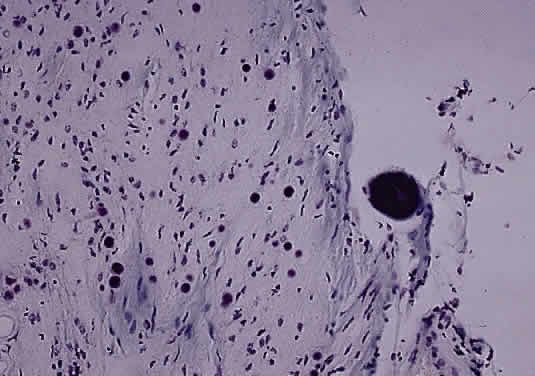
Anteriorly, the axons of the optic nerve traverse the lamina cribrosa, a sieve-like portion of connective tissue in the sclera; at this site, there appears to be some degree of constriction.7 The course of the axons through the lamina cribrosa varies in different areas and is longer and more tortuous peripherally than centrally.8 These phenomena are believed to have some role in the pathogenesis of glaucomatous optic neuropathy. Corpora amylacea are small (about 10 μm) homogeneous or laminated structures that are frequently found in the optic nerve, retina, brain, and spinal cord. These periodic acid-Schiff (PAS)-positive structures are considered by some authors to be located within the axoplasm of neurons. Ultrastructurally, they represent filamentous tangles of fibrils associated with microtubules (Figs. 5 and 6).9 Corpora amylacea increase in number with age and decrease in conditions associated with neuronal loss (e.g., glaucoma), which suggests that they represent intraneuronal aging products.10,11 Their consistent immunoreactivity for tau-2 protein, which is present in neurofibrillary tangles and plaques in various neurodegenerative disorders, supports this idea.12
BLOOD SUPPLY OF THE OPTIC NERVE Central vessels penetrate the optic nerve 8 to 15 mm behind the globe, having crossed the meninges to lie nasal to the nerve. The vein follows a course of variable length before entering the nerve. The artery penetrates the nerve at the same level or anterior to the vein (Fig. 7). Arrangement of arterial branches within the nerve varies between individuals and branches are from ophthalmic, posterior ciliary, central retinal artery (within orbit), and branches from the circle of Zinn-Haller. The intraocular portion of the nerve receives branches from the intracranial portion of the ophthalmic artery, anterior cerebral, internal carotid, and ophthalmic arteries.13,14
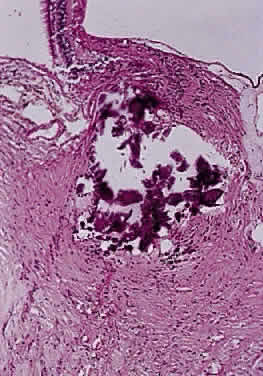
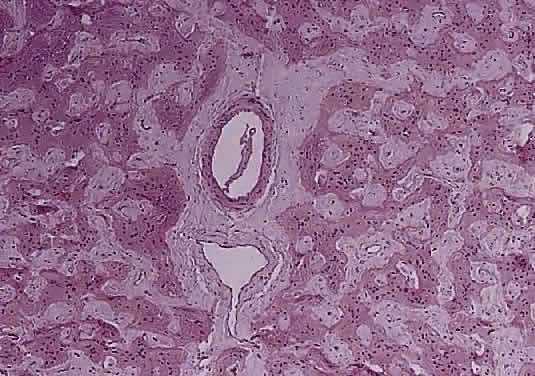
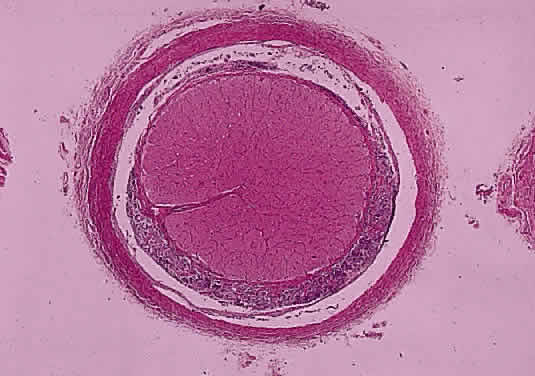
OPTIC NERVE SHEATH The optic nerve is invested by a sheath that is continuous posteriorly with the intracranial dura mater and the periorbita in the orbital apex; anteriorly, it fuses with the sclera. The arachnoid is composed of loose connective tissue continuous with that of the cranium. Meningocytes (arachnoid cells) may be found in nests, some of which contain calcified bodies (psammoma bodies; Fig. 8). The subarachnoid space is continuous with that of the cranium. Investing the nerve closely is the pia, which sends fibrovascular septa into the nerve, dividing it into fascicles. The pia blends with the lamina cribrosa and the connective tissue around the central vessels, so that the central retinal artery and vein have a common connective tissue sheath (Fig. 9). The optic nerve sheath, immediately behind the eye, is typically somewhat loose.
AGING CHANGES Most studies show that the number of axons within the optic nerve decreases with age at a rate of about 5000 axons per year, even in the absence of clinical eye disease.4,5,15,16 Whether this is a steady decrease throughout life or starts during adulthood is unclear. Accompanying this loss of axons is an increase in the proportion of the nerve occupied by connective tissue.10 Also noted with advancing age are increased corpora amylacea and lipofuscin deposits within astrocytes.11 |
| CONGENITAL MALFORMATIONS | |||||||||||||||||
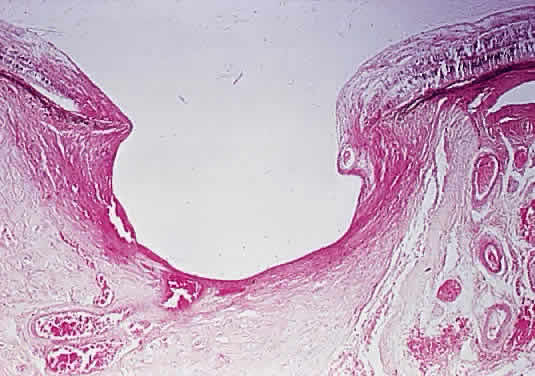
| The myopic disc is oriented obliquely to the horizontal meridian and is raised nasally. Temporally, it
appears flattened and has a white scleral crescent. Microscopically, the
nasal disc overlaps the neural retina, retinal pigment
epithelium (RPE), Bruch's membrane, and choroid. Temporally, the
RPE and Bruch's membrane do not extend to the disc margin. Pseudopapilledema or a structurally full optic disc head may be variously caused by hypermetropia, vascular anomalies, myelinated nerve fibers, drusen, epipapillary hamartomas, and tumors. Optic neuritis may also give the appearance of a full disc head. Drusen are calcific bodies that may be pre- or postlamina (Fig. 10). Those that are anterior to the lamina are usually bilateral and thought to result from mitochondrial calcification within the axons, which accompanies disturbance in axoplasmic flow. The presence of drusen may cause enlargement of the papilla and may be mistaken for papilledema (Fig. 11). Drusen may be idiopathic or occasionally inherited in an autosomal dominant pattern. The giant drusen of tuberous sclerosis are calcified astrocytic hamartomas (Fig. 12). An association with hypertension and vascular occlusion has also been noted.
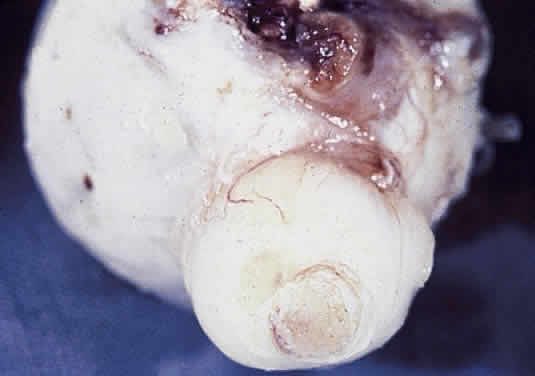
Aplasia of the optic nerve is rare and usually unilateral. The nerve, disc, retinal ganglion cells, and retinal blood vessels are absent and replaced by glial and fibrous tissue with capillaries and lobules of fatty tissue. The most favored etiology is that abnormal closure occurs during development of the inferior invagination. Multiple other ocular anomalies may be associated. The ipsilateral lateral geniculate ganglion shows depletion of ganglion cells of layers 2, 3, and 5; the contralateral geniculate body shows atrophy of ganglion cells in layers 1, 4, and 6. The optic tract and chiasm show axonal depopulation. Hypoplasia of the optic nerve is also rare but is more common than aplasia (Figs. 13 and 14). It represents one of the most important developmental abnormalities of the eye and is increasingly recognized as a significant cause of visual deficit in children. Congenital nerve hypoplasia is a nonprogressive condition associated with a reduction in the number of retinal ganglion cells and axons. It may be either unilateral or more commonly bilateral and may involve the entire nerve or be segmental. Although a hypoplastic optic nerve is usually idiopathic, an increased incidence has been noted in the offspring of diabetic mothers and with the maternal use of phenytoin, quinine and, alcohol. Rare reports of an inherited disorder may be found in the literature. Although it may be an isolated abnormality, the hypoplasia is frequently associated with other anomalies—both ocular and nonocular. Additional ocular abnormalities include microphthalmos and congenital nonattachment of the retina. The most common nonocular abnormalities associated with congenital optic nerve hypoplasia include midline CNS structural defects, including septo-optic dysplasia of the septum and ectropia of the posterior pituitary, producing diabetes insipidus.
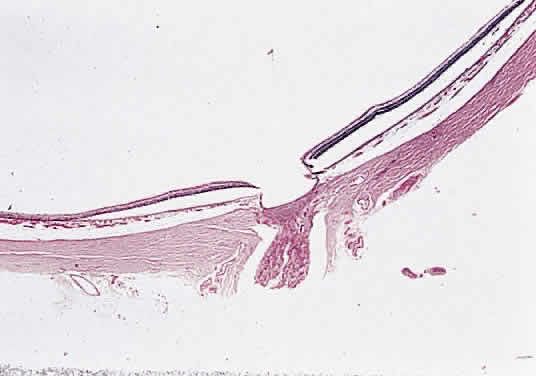
In optic nerve hypoplasia, the optic disc appears gray and is about half its normal size but retinal vessels are present because the optic stalk is invaginated by mesoderm. The optic foramen is also small. Clinically, there may be strabismus, nystagmus, and decreased visual acuity. Severe cerebral abnormalities may be present but the hypoplastic nerve may be the sole abnormality. Dysplasia or abnormal development of the optic nerve is usually associated with a coloboma or other major defects. An optic pit is often referred to as an “atypical coloboma” because it is positioned inferolaterally (Fig. 15). It consists of unilateral anomalous development of the optic nerve papilla, which is accompanied by a small depression that is an outpouching of neuroectoderm surrounded by a connective tissue capsule that passes through the lamina into the subarachnoid space. Occasionally, the affected disc is larger than the contralateral disc. Macular changes consisting of hemorrhages, cysts, holes, or retinal detachment are present in as many as 50% of affected patients. An outpouching of neuroectoderm surrounded by connective tissue capsule passes into the subarachnoid space through a defect in the lamina.
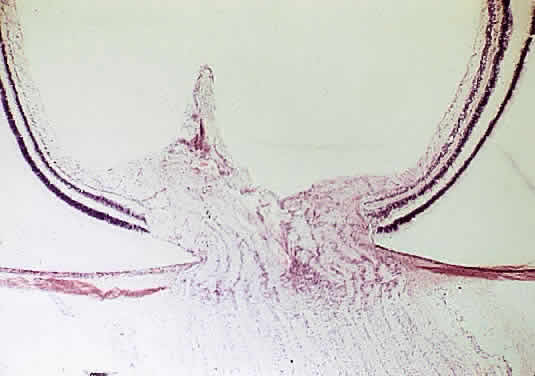
A coloboma, which is due to a failure of fusion, is confined to the disc or involves the entire embryonic fissure (Fig. 16). It is usually unilateral and varies in its appearance from a deep physiologic cup to a large hole with a retrobulbar cyst. Other ocular anomalies (e.g., microphthalmos with cyst) may be associated. The wall of the defect may contain adipose tissue and occasionally muscle fibers. The latter may lead to a contractile peripapillary staphyloma.
Vascular anomalies stem from a failure of the hyaloid vessel to regress completely or partially. Such anomalies include a Bergmeister's papilla (Fig. 17), persistent hyperplastic primary vitreous (Fig. 18), anomalous distribution of central retinal vessels, epipapillary vascular loops, abnormal chorioretinal anastomosis, and epipapillary membranes.
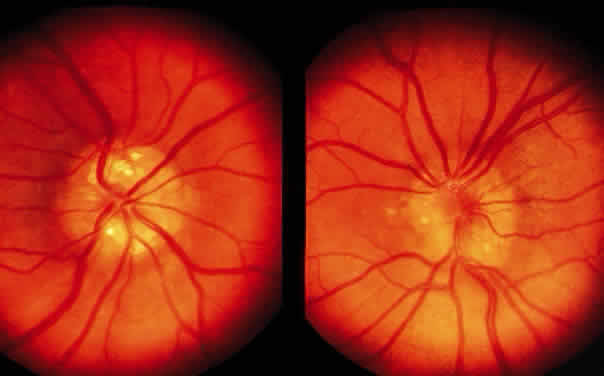
The term optic disc edema may be used for swelling that is not attributable to raised intracranial pressure, nor to inflammatory disease. Local factors at the level of the lamina cribrosa cause an increase in venous pressure and a disturbance in axoplasmic flow. Precipitating conditions include spontaneous central retinal vein occlusion, development of a carotid-cavernous fistula, and may occur as a complication of ophthalmic surgery. Neoplastic or inflammatory infiltrative pathology of the optic nerve or its sheath may also cause an increase in venous pressure and thus optic disc edema. A fall in intraocular pressure may also result in a swollen disc head. The term papilledema indicates swelling of the optic nerve head secondary to raised intracranial pressure (Fig. 19). The raised cerebrospinal fluid (CSF) pressure produces relative or absolute increase in venous pressure at the lamina cribrosa. The intracranial pathology that is responsible for the increased pressure includes space-occupying lesions, meningitis with or without encephalitis, subarachnoid hemorrhage, and malignant hypertension. Increased CSF pressure with normal or small ventricles may be seen with excessive vitamin A intake, administration of tetracyclines, and oral contraceptives. This condition, known as “pseudotumor cerebri,” may also be seen in pregnancy; in association with menstrual irregularities; and in young, frequently obese women.
Papillitis is the term used for an inflamed swollen optic nerve head. Regardless of the cause of a swollen disc head, the histopathologic alterations are similar. The earliest change is nerve-fiber swelling in the region of the intermediary tissue of Kuhnt. Bulging fibers initially displace the retina laterally, and the outer layers eventually buckle. The glial lamina is displaced anteriorly, and the vessels are congested. Bundles of nerve fibers are pale staining because of accumulation of watery interstitial fluid. The earliest and most accentuated changes are seen in the outermost fibers around the disc margins (Fig. 20). The swollen nerve head protrudes forward against the vitreous and laterally displaces the central elements of the peripapillary retina from the disc margins. Proteinaceous material often accumulates between the sensory retina and the pigment epithelium around the disc margins, resulting in an enlarged blind spot.
Pathophysiologic mechanisms responsible for the disc swelling include a relative or absolute increase in venous pressure at the pre- or postlamina (e.g., acute glaucoma, orbital and brain tumors, hypotony, hypertension, and central retinal vein thrombosis). Blockage of axoplasmic flow at the lamina cribrosa with accumulation of mitochondria and other organelles within the axon results in prelamina axonal swelling. Because the condensed axoplasm appears similar to a nucleus within a cell, the individual component of the cotton-wool spot is know as a cytoid body (Figs. 21 and 22). Visual acuity may be normal, or mild impairment of visual acuity accompanies marked edema. Transient attacks of blurred vision or blindness may occur. There is a gradual concentric enlargement of the blind spot.
Prelaminar disease (e.g., hypotony secondary to trauma or uveitis, acute angle closure and vascular disease) that is local or systemic may also be accompanied by a swollen disc head. Venous stasis, retinopathy, and hypertensive retinopathy are other frequent causes. Microscopically, the earliest changes consist of a transudate in and between the axons, with accumulation of mitochondria. As a result, the nonmyelinated axons are separated and there is an anterior bowing of the lamina cribrosa. The optic cup appears shallow and the sensory retina is displaced laterally, so that the blind spot is enlarged. The vessels are congested, particularly the veins. Any hemorrhages that occur are predominantly in the nerve-fiber layer around the disc margin. Edema fluid may accumulate in the subarachnoid space of the optic nerve. Persistence of an edematous disc is accompanied by the appearance of cotton-wool spots and eventually atrophy of the axons. Loss of axons and myelin leads to proliferation of astrocytes or gliosis. Optic neuropathy is a general term that is applied to optic nerve dysfunction. It may be caused by a variety of conditions such as ischemia (e.g., giant cell arteritis, compression by tumor); metabolic diseases (e.g., diabetes mellitus); drug toxicity (e.g., chloramphenicol); and vitamin deficiencies (e.g., B12 deficiency). Inflammation of the optic nerve may be secondary to bacterial and mycotic infections of adjacent anatomic structures (eye, sinus), or it may occur as a part of a systemic infection, particularly in an immunosuppressed patient (Fig. 23). Clinically, there is unilateral acute blurred vision, with pain or discomfort on movement or palpation of the globe. On examination, visual acuity with a central scotoma is decreased to colored objects but the disc appears normal. Classification depends on ophthalmoscopy topography (Table 1).
TABLE 16-1. Classification of Optic Neuropathy Ophthalmoscopy Retrobulbar Topographic Periaxial
Transverse Inflammation that extends from the meninges into the pia affects the peripheral nerve bundles. A continuous exudate, such as is seen in a granulomatous inflammation due to fungi or acid-fast bacilli, leads to atrophy of the peripheral nerve bundles and thus a periaxial atrophy. In axial neuropathy, the macular fibers are predominantly affected. These fibers are particularly susceptible to involvement in multiple sclerosis and in tobacco or alcohol amblyopia. Both periaxial and axial forms may extend and affect the optic nerve transversely. Papillitis refers to inflammation of the disc; it is usually accompanied by a vitreous haze due to the presence of inflammatory cells. Commonly, it is seen with suppurative endophthalmitis and panophthalmitis. The disc shows edema, perivenous cuffing by chronic inflammatory cells, and astrocytic proliferation. Thin bands of connective tissue may spread from the disc surface into the vitreous and cover the adjacent retina. Loss of myelin may occur with all the pathologic processes described above and also with trauma and neoplasm; this type of demyelination is secondary. Primary demyelination refers to demyelinating disease of unknown etiology (Figs. 24 and 25). Loss of myelin occurs early but initially the axons are undamaged (disseminated sclerosis, acute disseminating myelitis), so that visual function may return. The damaged myelin is removed by macrophages followed by astrocytic proliferation, which produces a glial scar or plaque (Fig. 26).
Dysmyelination is the term applied to the formation of abnormal myelin in the fetus (e.g., leukodystrophy). |
| OPTIC ATROPHY | |||||||
| The etiology of optic neuropathy is outlined in Table 2. The portion of the optic nerve that is nearer to the lateral geniculate
body is noted as “central” and the portion nearer to the
retina as “peripheral.” Conventionally, optic atrophy is
classified as ascending or descending. In the ascending form, there is
loss of retinal ganglion cells due to glaucoma or infarction, resulting
in degeneration of the axons. Descending atrophy results from pathologic
changes within the cranial cavity or orbit.
TABLE 16-2. Etiology of Optic Neuropathy Inflammation of Eye, Orbit Sinuses, Brain and Meninge Atherosclerosis, arteriosclerosis Elevated intraocular pressure Systemic hypotension Hematologic disorders Compression of optic nerve blood vessels Degenerative Disease Demyelinating diseases Idiopathic Multiple sclerosis Secondary to Trauma Dysmyelinating disease (leukodystrophy) Krabbe's disease Toxic Tobacco, alcohol, lead, drugs Systemic disease Endocrine (Graves' disease, diabetes mellitus)
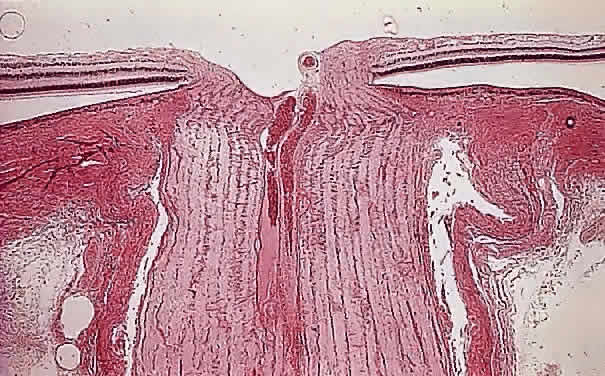
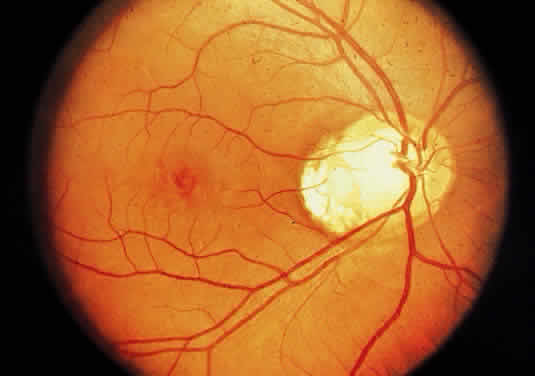
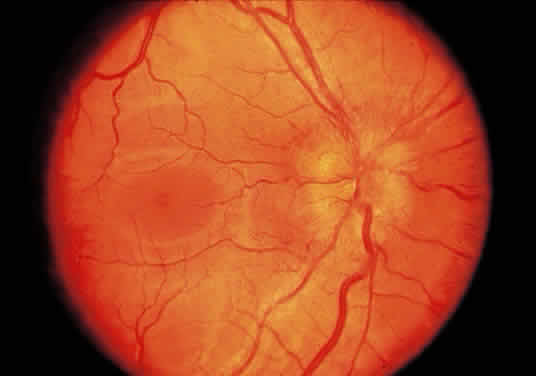
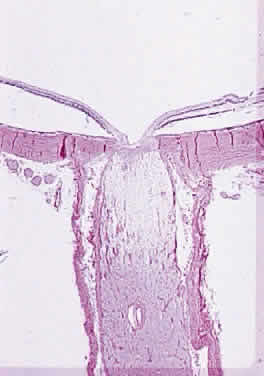
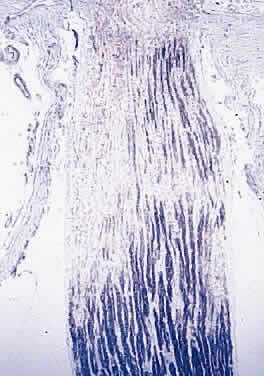
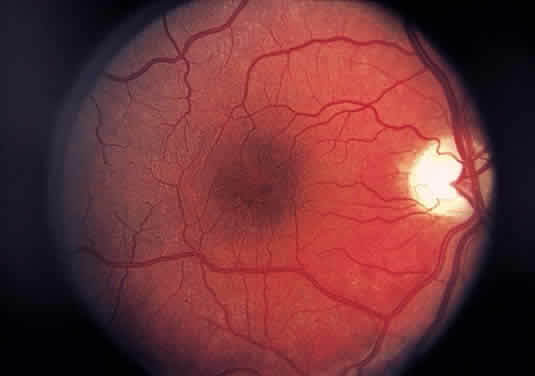
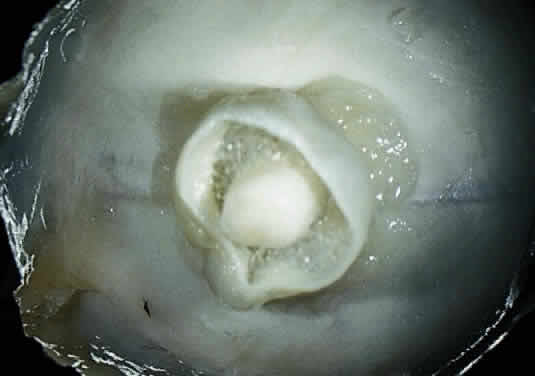
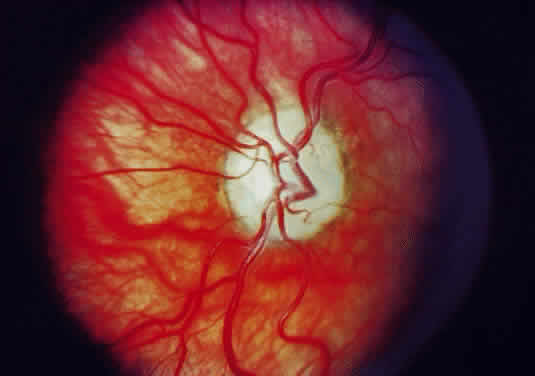
Injury to the retina or any peripheral portion of the optic nerve results in rapid ascending atrophy of the central portion. Initially, the axons at the peripheral portion swell. Retrograde degeneration of the axon then occurs, with loss of retinal ganglion cells. Clinically, there is pallor of the optic disc (Fig. 27), and early decreased fluorescence is seen with angiography. The atrophic optic nerve is surrounded by a loose sheath (Fig. 28). Microscopically, there is shrinkage of the nerve parenchyma secondary to axonal and myelin loss and gliosis (Fig. 29). Corpora amylacea accumulate in the subpial and perivascular areas (Fig. 30). With shrinkage of the nerve, the subarachnoid space is widened and the pial strands and arteriolar walls are thickened. Atrophy that is secondary to glaucoma is initially seen in the prelaminar region. Loss of astrocytes and axons results in baring of the lamina and deep excavation of the optic cup (Figs. 31 and 32).
The entity known as “cavernous optic atrophy of Schnabel” is characterized microscopically by large cystic spaces that are posterior to the lamina cribrosa. In these areas, the nerve fibers are destroyed and the cavernous spaces are filled with acid mucopolysaccharide-positive material (Fig. 33). Neither gliosis nor mesenchymal reaction are evident. This intracystic material was initially thought to be vitreous that penetrated into the parenchyma through the internal limiting membrane of the optic nerve head. The condition, however, is an acute infarction of the nerve secondary to acute glaucoma, vascular disease, or “deficient oxygenation states” (hypotension).
|
| NEOPLASMS | ||||||||||||||||
| Neoplastic proliferations of the optic nerve are rare, comprising about 1% of
all ocular and ocular adnexal neoplasms and 10% of all orbital
neoplasms at one referral center.17 The neoplasm may be confined to the optic disc, the retrobulbar portion
of the nerve, or may involve both sites by direct extension from one
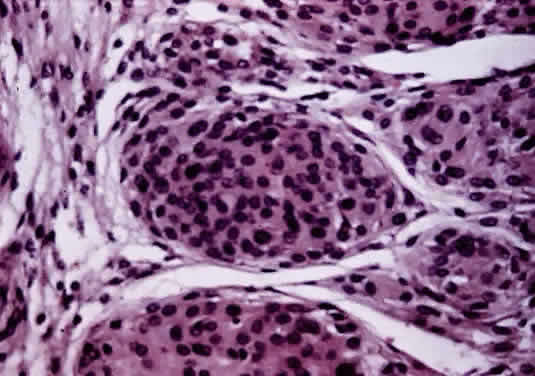
site to the other. Primary tumors of the disc head include the astrocytic hamartoma, which may be seen with tuberous sclerosis or neurofibromatosis, rare pigment epithelial proliferations, hemangioma, peripapillary choroidal melanoma, and melanocytoma. The melanocytoma is a benign, deeply pigmented tumor that arises from dendritic melanocytes incorporated within the optic disc (Fig. 34). It may also arise in any part of the uveal tract. Commonly, it is situated eccentrically on the disc and projects for 2 mm or less into the vitreous. It extends into the lower temporal retina and posteriorly beyond the lamina. Bleached sections show closely apposed plump cells, with abundant cytoplasm. The nuclei have appreciable chromatin; nucleoli, when present, are small and regular. The intense pigmentation blocks fluorescein angiography. As an entity, it is more common in blacks and Mediterranean races. It also occurs in whites; as the tumor may grow and undergo necrosis, there may be difficulty in distinguishing it clinically from melanoma. The melanocytoma, however, does not metastasize. Distinguishing this tumor from a peripapillary uveal melanoma that grows over the disc and may invade the vitreous and the optic nerve is important. The borders of a melanocytoma, as seen by ophthalmoscopy, tend to feather out, in contrast to the borders of a melanoma.
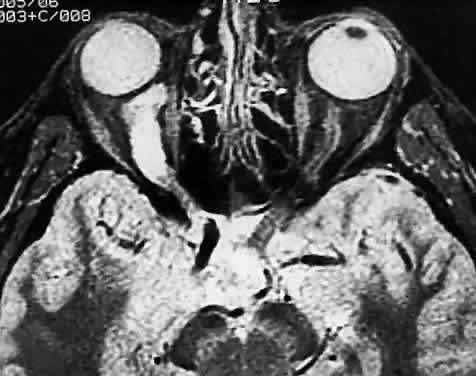
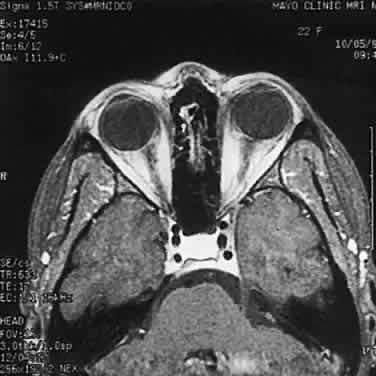
Most examples of malignant melanoma involving the optic nerve represent peripapillary choroidal melanomas with secondary involvement of the optic nerve. Rare examples of primary optic nerve malignant melanomas have been reported, however.18 GLIAL NEOPLASMS With rare exceptions, glial neoplasms of the optic nerve are of the juvenile or pilocytic type, which almost always occur during childhood and are associated with a good outcome. Less common is the diffuse high-grade glioma, a highly malignant neoplasm that is largely confined to the adult population. JUVENILE PILOCYTIC ASTROCYTOMA Juvenile pilocytic astrocytoma is the most common optic nerve neoplasm. It may arise in any part of the visual pathway from the junction of the optic nerve with the globe to the optic tract. About 75% of tumors involve the optic chiasm, with or without involvement of one or both nerves; a single nerve is affected in 25%.19,20 There is a close association with neurofibromatosis type 1 (von Recklinghausen disease or NF-1).21 Indeed, optic nerve glioma is one of the most common ocular manifestations of this autosomal dominant genetic disorder. Clinical or radiographic evidence of optic nerve glioma is present in about 15% of patients with NF-1.22,23 The prevalence of NF-1 in patients with optic nerve glioma is about 25%.24,25 Most patients with optic nerve glioma remain asymptomatic. The most common tumor-related signs and symptoms at diagnosis are decreased visual acuity (including blindness), optic atrophy, and proptosis.19,24,26 The role of visually evoked potentials in the detection and monitoring of optic nerve glioma is controversial.27,28 Computed tomographic (CT) and magnetic resonance imaging (MRI) have had a significant impact on the management of patients.29 Many tumors are diagnosed based on imaging features alone, without tissue confirmation (Fig. 35). They typically show a demarcated fusiform enlargement of the nerve, which may be kinked and indent the globe. On T2-weighted MRI, a circumferential component surrounds a compact core of low signal intensity.23 MRI is particularly useful in assessing the extent of the lesion.
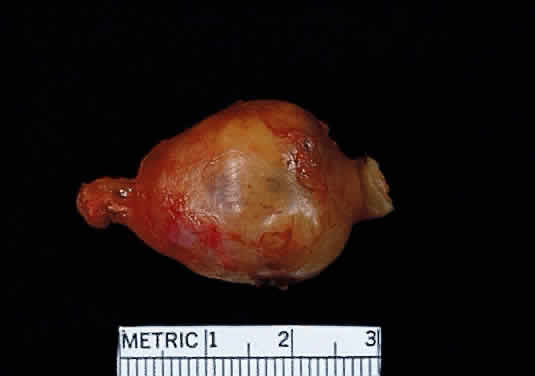
Grossly, optic gliomas that are confined to the orbit appear as fusiform, solid, and somewhat firm expansions of the optic nerve (Fig. 36). They usually blend imperceptibly with the grossly uninvolved nerve at either end. The enveloping dura may be stretched but is not breached. Microscopically, the tumor is composed of bipolar hair-like (pilocytic) astrocytes that infiltrate between preexisting axons and expand rather than compress the nerve fascicles. The neoplastic cells are only mildly atypical, and mitotic figures are typically absent. Not uncommonly, the tumor “spills over” into the subarachnoid space, forming a collar (Fig. 37).
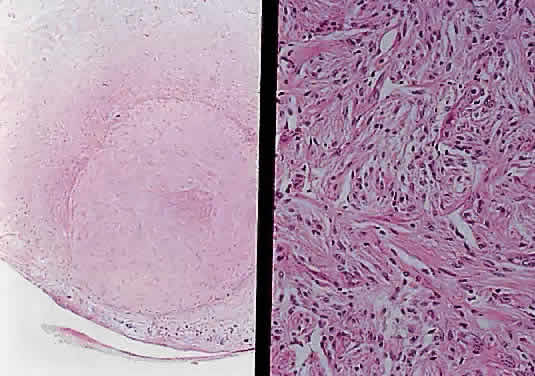
Two histologic features that are present in nearly every example of pilocytic astrocytoma are Rosenthal fibers and eosinophilic granular bodies. Rosenthal fibers are enlarged, brightly eosinophilic cell processes that are sausage or corkscrew-shaped. On Masson trichrome staining, they are bright red (Fig. 38). Ultrastructurally, Rosenthal fibers are defined by amorphous aggregates of electron-dense material within the nerve processes. The number of Rosenthal fibers in different tumors and even within an individual tumor varies considerably; rarely, they are absent entirely. Granular bodies, as the name suggests, consist of rounded eosinophilic proteinaceous structures (Fig. 39). Neither Rosenthal fibers nor granular bodies are unique to pilocytic astrocytomas and are commonly seen in other low-grade astrocytic neoplasms (e.g., ganglioglioma). Their presence, however, essentially excludes a high-grade glioma.
The neoplastic cells in optic nerve glioma frequently invade the surrounding meninges and may elicit a marked reactive proliferation of the arachnoid cells. As a result, a mistaken diagnosis of meningioma may be made if only a small, superficial biopsy is taken.30 There are rare reports of optic nerve tumors showing mixed features of glioma and meningioma. The management of optic nerve glioma has long been a controversial subject.20,24,26,31–33 Its rarity ensures that most studies will be retrospective and include only a few cases. The indolent nature of most tumors, the long natural history, the failure to differentiate between overall and tumor-related morbidity and mortality (especially important in patients with NF-1), and the multiplicity of therapeutic approaches that are available further complicate the picture. These tumors are slow-growing, and tumor-related deaths are unusual; indeed, examples of spontaneous involution of optic nerve glioma have been reported.34 Do optic nerve gliomas in patients with NF-1 differ from those in patients without NF? Some studies seem to indicate that optic nerve gliomas in patients with NF-1 are associated with invasion of the leptomeninges by neoplastic astrocytes accompanied by an exuberant proliferation of reactive astrocytes, fibroblasts, and arachnoid cells.35 In contrast, optic nerve expansion in patients without NF-1 is the result of thickening of the nerve parenchyma. Other studies have not confirmed this difference.24 Deliganis and colleagues19 reported that the presence or absence of NF-1 does not influence age at presentation or clinical signs or symptoms. Patients without NF-1 were more likely to have involvement of the hypothalamus or posterior optic pathways, although bilateral tumors were equally prevalent. In contrast, NF-1 was associated with delayed tumor progression. Most studies have concluded that NF-1 is not protective.19,24,36–38 FIBRILLARY ASTROCYTOMA Fibrillary (diffuse) astrocytomas comprise the largest group of all primary brain tumors, but examples of such tumors arising in the optic nerve are rare; about 30 cases are reported in the literature.20,39–41 Unlike the more common pilocytic astrocytoma, they generally affect older adults (mean age, about 50 years) and are not associated with neurofibromatosis.20 At some stage during their natural history, all malignant optic pathway gliomas involve the optic chiasm; most also involve intracerebral structures. Rare examples of such a tumor arising within an optic nerve have been reported (Fig. 40).42
Symptoms are typically rapid in onset and always include subjective vision loss. If accompanied by an acute onset of pain, a diagnosis of temporal arteritis or optic neuritis may be considered, particularly if there is a response to steroids. Absence of light perception and visual-field defects are present in 80% to 90% of patients. Because most tumors do not involve the intraorbital segment of the optic nerve, proptosis is present in only a minority of cases. The early radiographic features may be nonspecific, with only slight enlargement and enhancement of the nerve.42 The chiasm is typically expanded. Diffuse optic nerve gliomas produce enlargement of the nerve, which is typically described as reddish and smooth. Higher-grade neoplasms may have a soft and friable consistency. Histologically, there is diffuse expansion of the nerve by atypical cells (Fig. 41). Mitotic figures and foci of necrosis may be evident. The absence of Rosenthal fibers and granular bodies helps to exclude pilocytic astrocytoma and demyelination. Malignant gliomas of the optic nerve usually arise from the astrocyte. A single report of an optic nerve oligodendroglioma has been published.43
Malignant gliomas of the optic nerve are aggressive neoplasms. Most reported cases are examples of a frontal lobe glioma that has spread to involve the optic nerve. Rapid progression to involve the contralateral eye, posterior optic pathways, and adjacent brain tissue is the rule. Complete surgical excision of these tumors is not possible because of their infiltrative nature; surgery is confined to obtaining an adequate biopsy specimen for diagnosis. Adjuvant irradiation and chemotherapy have produced little benefit, although there is one report of a response to radiation therapy.44 Survival is less than a year in most cases, and there have been no reported instances of survival beyond 2 years. MENINGIOMA The meningioma is derived from arachnoidal cells that are distributed throughout the meninges, including the optic nerve sheath. Most (90%) orbital meningiomas are secondary, resulting from extension of intracranial tumors through the optic foramen. Primary optic nerve sheath meningiomas comprise about 1% of all meningiomas and 3% to 9% of all orbital tumors in adults.45,46 The meningiomas have a greater predilection for the orbit in children, in whom they comprise 5% to 10% of all meningiomas.47 The average age of patients with optic nerve meningioma is about 40 years, although the age range is broad (2 to 78 years). For intracranial tumors, most affected patients are female, with a male-to-female ratio of about 1:2. A meningioma of the optic nerve sheath has a lesser tendency than the glioma to be bilateral, with only about 5% of cases falling into this category. Of these, most are canalicular meningiomas, and it has been suggested that most bilateral meningiomas represent optic nerve involvement by a primarily retroorbital meningioma.48 There is an increased incidence of meningiomas, including optic nerve meningioma, in patients with neurofibromatosis. Other possible risk factors include prior radiation therapy and a positive family history. The patient usually presents nearly with decreased vision, with or without decreased color vision and visual field. Atrophy and swelling of the optic disc, decreased ocular motility, and the presence of optociliary shunts are well-recognized features (Fig. 42).
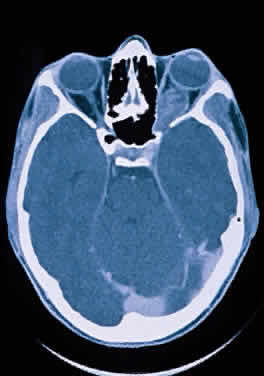
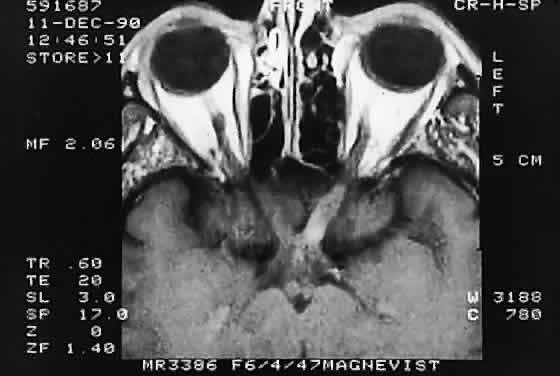
Modern imaging techniques have greatly facilitated the evaluation of optic nerve sheath meningioma and glioma.29 Plain orbital radiographs occasionally show abnormalities of the optic foramen and calcification. Imaging studies (CT and MRI) show diffuse tubular or less commonly, globular enlargement of the nerve sheath complex (Fig. 43).49,50 As with its intracranial counterpart, the optic nerve meningioma typically enhances brightly. “Tram-tracking” results when the thickened sheath outlines the uninvolved central nerve from which it is sharply demarcated. Although characteristic of optic nerve meningioma, this is not specific and can be seen in inflammatory conditions and rarely even in optic nerve glioma. Precise localization of the extent of the tumor is determined with MRI studies, particularly with fat-saturation techniques. Dilation of the anterior perioptic space (perioptic cyst) between the anterior edge of the tumor and the globe is seen in instances in which the tumor does not encroach on the globe and is a distinct and common feature.51,52
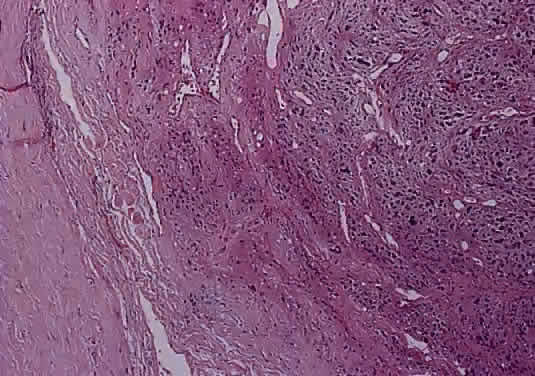
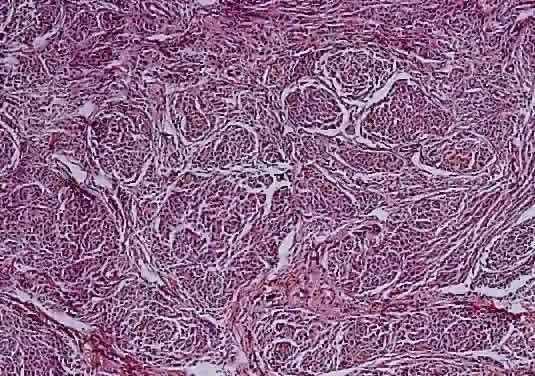
Histologically the common types of meningioma include the meningotheliomatous, transitional, fibroblastic, and psammomatous meningiomas. In most instances, the cells bear some resemblance to arachnoid cells (Fig. 44). Variations in their cytologic features and architectural arrangements produce a broad spectrum of microscopic appearances. The most common type within the orbit is the meningotheliomatous type that is composed of lobules and sheets of neoplastic cells with typically indistinct cell borders (syncytial arrangement). The ultrastructural basis for this latter phenomenon is the complex interdigitation of cytoplasmic extensions. Individual nuclei are oval with light-staining chromatin and small nucleoli. Intranuclear cytoplasmic pseudoinclusions are usually readily evident. The cytoplasm is uniform and lightly eosinophilic. Whorls of cells, in which the cells arrange themselves in concentric layers akin to the layers of an onion, are found in nearly all meningiomas and are one of the most helpful diagnostic features (Fig. 45) Psammoma bodies—concentric laminated mineralized concretions typically centered on whorls—are present in varying number in most meningiomas; when particularly abundant, the tumor is referred to as a psammomatous meningioma.
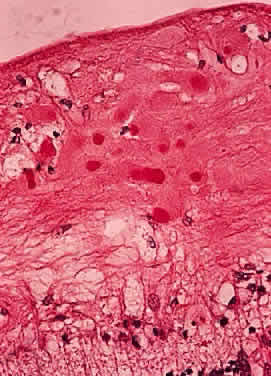
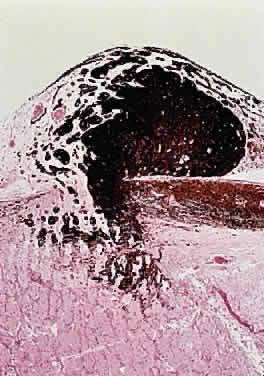
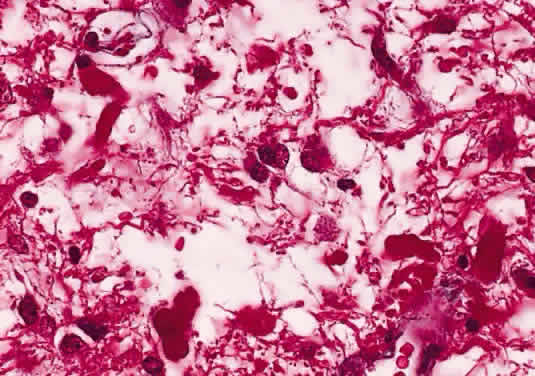
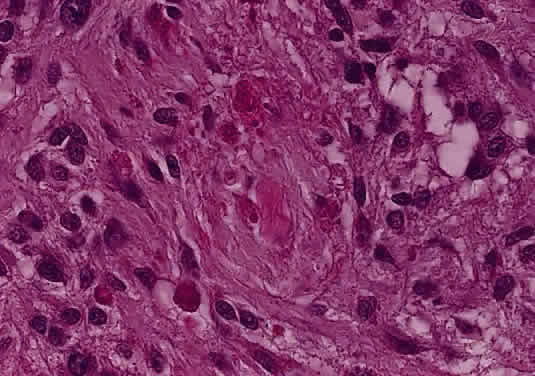
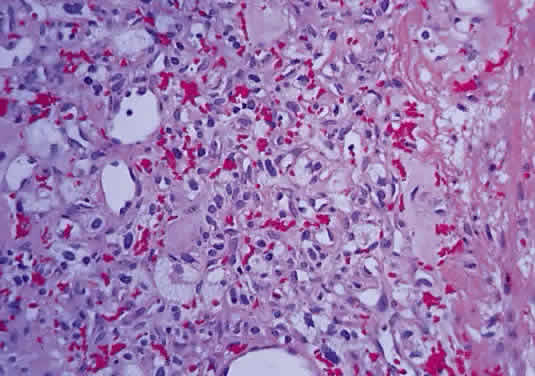
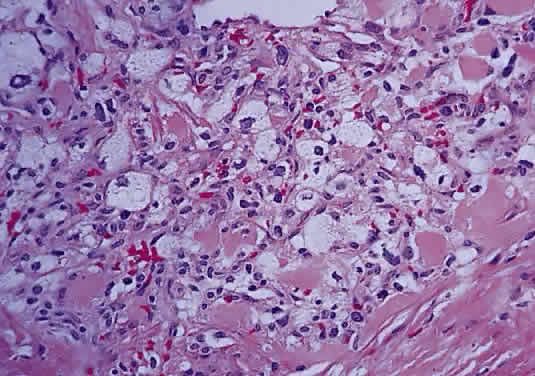
In fibroblastic meningiomas, the cellularity is less and much of the tumor is composed of spindle-shaped cells within a collagen-rich matrix. Transitional tumors, as the name suggests, have features of both meningotheliomatous and fibroblastic differentiation. Typically, the spindle cells are located around the periphery of whorls. In the past, the term “angioblastic” meningioma was used to describe tumors with numerous and prominent vascular spaces. Investigations, however, indicate that most of these tumors represent examples of hemangiopericytoma, a mesenchymalderived neoplasm with a distinctly more aggressive natural history than the ordinary meningioma. As with optic nerve glioma, the approach to the management of optic nerve sheath meningioma is a complicated and a controversial subject.46,53–55 Left untreated, the tumors continue to grow and eventually may extend intracranially and even to the contralateral nerve. Factors which must be considered when planning therapy include patient age, tumor location, size and extent, visual status, and degree of proptosis. The role of adjuvant radiotherapy remains unclear but early results are cause for encouragement.45,55,56 HEMANGIOBLASTOMA Hemangioblastoma of the optic nerve is rare; fewer than 20 cases are reported in the literature.57,58 Males and females have been equally affected; the patient age range is from 15 to 44 years. Most reported cases have occurred in association with von Hippel-Lindau syndrome (VHL), an inherited autosomal dominant condition associated with an abnormal tumor suppressor gene on the short arm of chromosome 3. Patients with VHL have a predilection to develop hemangioblastomas of the retina, optic nerve, cerebellum, and spinal cord; cysts of the pancreas, kidneys, and liver; and renal cell carcinoma. Overall, about 25% of patients with hemangioblastoma manifest the stigmata of VHL but this rises to 70% when the optic nerve is involved. All reported hemangioblastomas have been prechiasmal and all but one were unilateral. Tumors may either be exclusively intraorbital or intracranial, or they may extend through the optic foramen to involve both the intraorbital and intracranial portions of the optic nerve. The presenting symptom in nearly all cases was loss of vision, sometimes accompanied by headaches or a sensation of eye pressure. Proptosis is a common finding in intraorbital tumors. Imaging studies typically demonstrate a vascular, homogeneously enhancing mass that enlarges or replaces the optic nerve. Imaging is also useful in distinguishing hemangioblastoma from such entities as aneurysm, meningioma, glioma, and metastasis. Intraoperatively, hemangioblastoma appears as a reddish, circumscribed tumor that typically is well demarcated from the nerve, although often the central portion is involved. Microscopically, the most striking feature is the abundance of vascular channels, most of which have the appearance of capillaries. Reticulin-staining highlights the vascular network. Between them are stromal cells containing abundant vacuolated cytoplasm, which is due to the presence of lipid. Although bland, the nuclei of the stromal cells are variable in appearance and may exhibit focal marked hyperchromasia (Figs. 46 and 47). Electron microscopy shows that the large stromal cells contain intracytoplasmic particles of lipid and glycogen. Scattered foci of extramedullary hematopoiesis may be present as a consequence of erythropoietin production by the tumor cells. Mast cells also are often readily identifiable. Small cysts are frequent, and the cyst wall consists of gliosis, in which may be found numerous Rosenthal fibers.
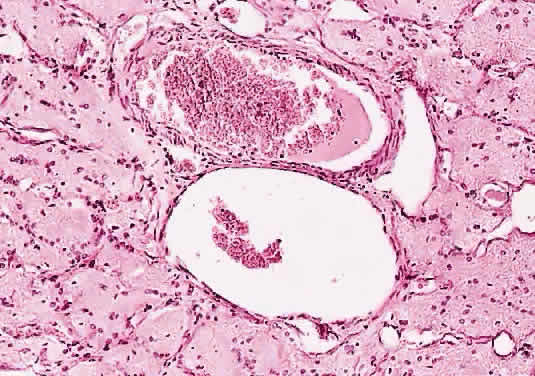
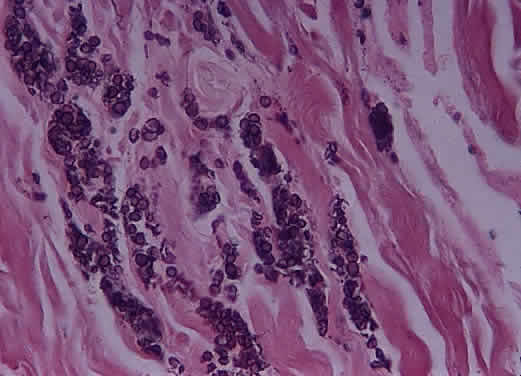
Immunohistochemical studies have failed to clarify the nature of the stromal cells. Scattered cells may react with glial fibrillary acidic protein but the significance is uncertain. They are consistently negative for epithelial markers, which is useful for differentiating these tumors from metastatic renal cell carcinoma; they also fail to react with vascular endothelial markers. The natural history of hemangioblastoma is characterized by progressive growth and vision loss. Metastasis does not occur. The optimal treatment is surgical resection, which is facilitated by the frequent sharp demarcation between tumor and nerve. Complete resection may be complicated, however, by the marked tumor vascularity, involvement of the central portion of the nerve, and extension into or through the optic canal. Neither irradiation nor chemotherapy have been effective treatment options in tumors at other locations. Recurrence of optic nerve hemangioblastoma has not been reported. SECONDARY TUMORS OF THE OPTIC NERVE Secondary tumors of the optic nerve are more common than primary neoplasms and develop through one of several routes: extension from an ocular tumor; extension from adjacent structures (orbit, nasopharynx, paranasal sinuses); involvement by meningeal neoplasms; and blood-borne metastasis.59 Primary intraocular tumors—particularly uveal melanoma and retinoblastoma—are the most common source of secondary tumors of the optic nerve. Involvement of the optic nerve by retinoblastoma occurs in about 25% of cases and may extend into the brain. The extent of optic nerve involvement correlates with survival: tumors that have invaded beyond the lamina cribrosa are associated with a worse prognosis. Optic nerve involvement is one of the most commonly employed prognostic indicators in retinoblastoma. Factors that predispose to optic nerve involvement by retinoblastoma include tumor seeding of the vitreous and the presence of glaucoma. Invasion of the optic nerve by a peripapillary choroidal malignant melanoma is less common that with retinoblastoma, occurring in about 5% of cases. In only a few of these cases (about 1% of all melanomas) does the tumor extend beyond the lamina cribrosa. Whether optic nerve involvement by uveal melanoma is a significant prognostic factor remains a source of controversy . Histologically confirmed metastatic tumors of the optic nerve are uncommon and usually are associated with widespread metastatic disease (Figs. 48 and 49). In only about 1% of cases is the optic nerve predominantly involved.60 The two most common primary sources are breast and lung. Isolated metastasis to the optic nerve is rare; fewer than 20 cases are reported.61–63 Visual disturbance often accompanies meningeal carcinomatosis, typically presenting as rapid visual loss occurring late in the course of the disease. Leukemia and lymphoma may also involve the optic nerve. The frequency with which this is occurring may be increasing because patients survive longer as a result of more effective therapy. Optic nerve involvement by medulloblastoma has also been described.64
|
| NONNEOPLASTIC MASS LESIONS OF THE OPTIC NERVE | ||
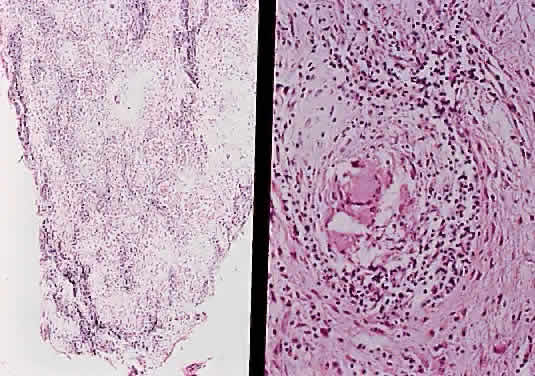
MENINGOCELES Saccular dilation of the optic nerve sheath occasionally occurs in association with neoplasms (glioma, meningioma) of the optic nerve. It may also be seen in neurofibromatosis without any neoplastic involvement, or it my be idiopathic.65,66 SARCOIDOSIS The optic nerve is affected in some manner in 1% to 5% of patients with sarcoidosis and is the second most frequently involved cranial nerve—the facial nerve being the most common.67,68 Rarely, sarcoidosis may present as tumefactive enlargement of the optic nerve without evidence of systemic disease. Most cases are misdiagnosed initially as meningioma or orbital pseudotumor (Figs. 50 and 51).67,69
|