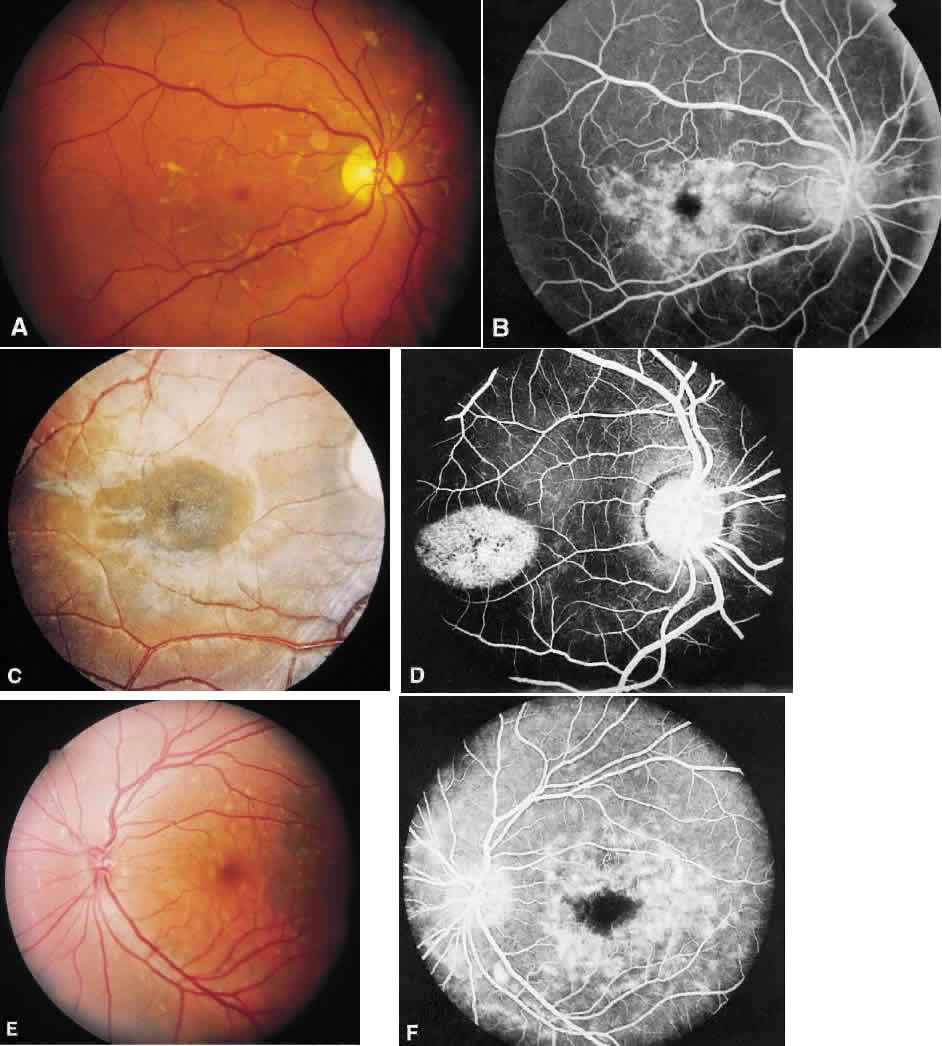
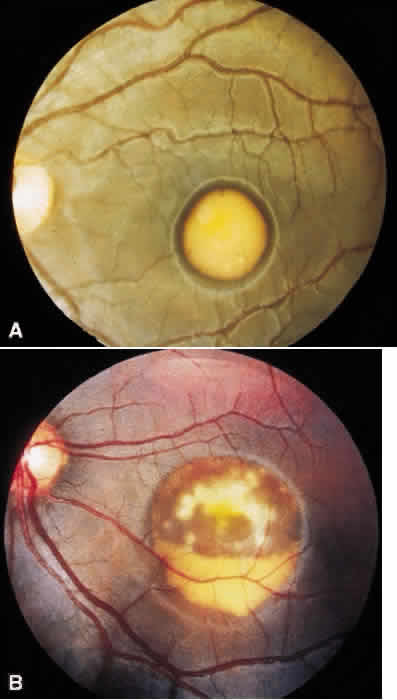
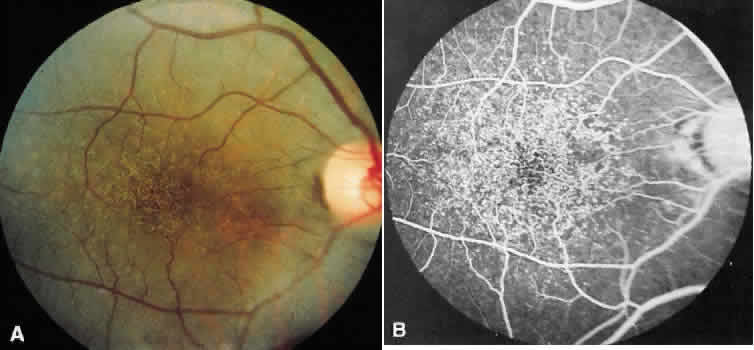
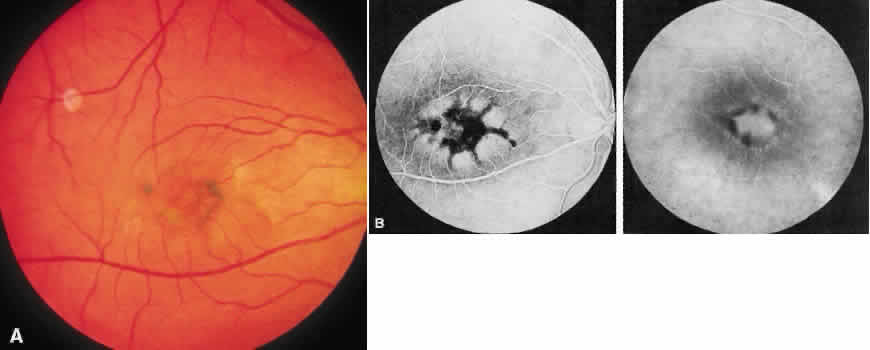
1. Stargardt K: Uber familiare, progressive Degeneration in der Makulagegend des Auges. Graefes Arch Ophthalmol 71:534, 1909. 2. Stargardt K: llber familiare progressive Degeneration in der Makulagegend des Auges. Z Augenheilkd 30:95, 1913. 3. Stargardt K: Zur Kasuistik der Familairen, progressive Degeneration der Makulagegend
des Auges. Z Augenheilkd 35:249, 1916. 4. Stargardt K: Uber familiare Degeneration in der Makulagegend des Auges, Nit und ohne
psychische Storungen. Arch Psychiatr Nervenkr 58:52, 1917. 5. Stargardt K: Fin Fall von familiarer progressives Makuladegeneration. Klin Monatsbl Augenheilkd 75:246, 1925. 6. Franceschetti A: Uber tapeto-retinal Degeneration im Kindesalter. In: Sautter
H, ed. Entwicklung und Rortschritt in der Augenheilkunde. Stuttgart, Enke, 1963:107. 7. Franceschetti A, Francois J: Fundus flavimaculatus. Arch Ophthalmol 25:505, 1965. 8. Hadden OB Gass JDM: Fundus flavimaculatus and Star-gardt's disease. Am J Ophthalmol 82:527, 1976. 9. Noble KG, Carr RE: Stargardt's disease and fundus flavimaculatus. Arch Ophthalmol 97:1281, 1979. 10. Aaberg TM: Stargardt's disease and fundus flavimaculatus: evaluation of morphologic
progression and intrafamilial co-existence. Trans Am Ophthalmol Soc 84:435, 1986. 11. Cibis CG, Morey M, Harris DJ: Dominantly inherited macular dystrophy with flecks (Stargardt's). Arch Ophthalmol 98:1785, 1980. 12. Wu G, Welter JJ, Hirose T: Autosomal dominant Star-gardt's disease. Ophthalmology 94(suppl):116, 1987. 13. Fishman GA: Fundus flavimaculatus: a clinical classification. Arch Ophthalmol 94:2061, 1976. 14. Klein BA, Krill AE: Fundus flavimaculatus: clinical, functional and histopathologic observations. Am J Ophthalmol 64:3, 1967. 15. Eagle RC Jr, Lucier AC, Bernardino VB Jr et al: Retinal pigment epithelial abnormalities in fundus flavimaculatus. Ophthalmology 87:189, 1980. 16. McDonnell PJ, Kivlin JD, Maumenee IH et al: Fundus flavimaculatus with maculopathy: a clinicopathologic study. Ophthalmology 93:116, 1986. 17. Fishman GA, Farber F, Patel BS, Derlacki DJ: Visual acuity in patients with Stargardt's macular dystrophy. Ophthalmology 94:809, 1987. 18. Bonin P, Passot M, Triolaire MT. Le signe du silence choroidien dans les
degenerescences tapeto-retiennes posterieures. In: De Lacy JJ, ed. International
Symposium on Fluorescein Angiography. The Hague, The Netherlands, Dr. W. Junk, 1976:461. 19. Fish G, Grey R, Sehmi KS et al: The dark choroid in posterior retinal dystrophies. Br J Ophthalmol 65:359, 1981. 20. Barkman Y: A clinical study of a central tapetoretinal degeneration. Acta Ophthalmol 39:663, 1961. 21. Wiznia RA, Perina B, Noble KG: Vitelliform macular dystrophy of late onset. Br J Ophthalmol 65:866, 1981. 22. Braley AE: Dystrophy of the macula. Am J Ophthalmol 61:1, 1966. 23. Falls HF: The polymorphous manifestations of Best's disease (vitelliform eruptive
disease of the retina). Trans Am Ophthalmol Soc 67:265, 1969. 24. Benson WE, Kolker AE, Enoch JM et al: Best's vitelliform macular dystrophy. Am J Ophthalmol 79:59, 1975. 25. Miller SA, Bresnick GH, Chandra SR: Choroidal neovascular membrane in Best's vitelliform dystrophy. Arch Ophthalmol 82:252, 1976. 26. Miller SA: Multifocal Best's vitelliform dystrophy. Arch Ophthalmol 95:984, 1977. 27. Maloney WF, Robertson DM, Duboff SM: Hereditary vitelliform macular degeneration: variable fundus findings within
a single pedigree. Arch Ophthalmol 95:979, 1977. 28. Fletcher RC, Jampol LM, Rimm W: An unusual presentation of Best's disease. Br J Ophthalmol 61:719, 1977. 29. Noble KG, Scher BM, Carr RE: Polymorphous presentations in vitelliform macular dystrophy: subretinal
neovascularization and central choroidal atrophy. Br J Ophthalmol 62:561, 1978. 30. Deutman AF: Electro-oculography in families with itelliform dystrophy of the fovea: detection
of the carrier state. Arch Ophthalmol 81:305, 1969. 31. Krill AE, Morse PA, Potts AM et al: Hereditary vitelliruptive macular degeneration of the macula. Am J Ophthalmol 61:1405, 1966. 32. Francois J DeRouch A, Fernandez-Sasso D: Electro-oculography in vitelliform degeneration of the macula. Arch Ophthalmol 77:726, 1967. 33. Thorburn W, Nordstrom S: EOG in a large family with hereditary macular degeneration. Acta Ophthalmol 56:455, 1978. 34. Cross HE, Bard L: Electro-oculography in Best's macular dystrophy. Am J Ophthalmol 77:46, 1974. 35. Frangieh GT, Green WR, Fine SL: A histopathologic study of Best's macular dystrophy. Arch Ophthalmol 100:1115, 1982. 36. Weingeist TA, Kobrin JL, Watzke RC: Histopathology of Best's macular dystrophy. Arch Ophthalmol 100:1108, 1982. 37. O'Gorman S. Flaherty WA, Fishtnan GA, Bersori EL: Histopathologic findings in Best's vitelliform macular dystrophy. Arch Ophthalmol 106:1261, 1988. 38. Birndorf LA, Dawson NW: A normal electro-oculogram in a patient with a typical vitelliform macular
lesion. Invest Ophthalmol 12:830, 1973. 39. Fishman GA, Trimble S, Raab MF, Fishman M: Pseudovitelliform macular degeneration. Arch Ophthalmol 95:73, 1977. 40. Kingham JD, Lochen GP: Vitelliform macular degeneration. Am J Ophthalmol 84:526, 1977. 41. Snyder DA, Fishman GA, Witteman G, Fishman M: Vitelliform lesions associated with retinal pigment epithelial detachments. Ann Ophthalmol 10:1711, 1978. 42. Gass JDM: Pathogenesis of disciform detachment of the neuroepithelium: I. General
concepts and classification. Am J Ophthalmol 63:573, 1967. 43. Gass JDM: Pathogenesis of disciform detachment of the neuroepithelium: III. Senile
disciform macular degeneration. Am J Ophthalmol 63:617, 1967. 44. Sarks SH: Ageing and degeneration in the macular region: a clinicopathological study. Br J Ophthalmol 60:324, 1976. 45. Sarks SH: Drusen and their rlationship to senile macular degeneration. Aust J Ophthalmol 8:117, 1980. 46. Gass JDM, Jallow S, Davis B: Adult vitelliform macular detachment occurring in patients with basal laminar
drusen. Am J Ophthalmol 99:445, 1985. 47. Sjogren H: Dystrophia reticularis lantinae pigmentosue retinae. Acta Ophthalmol 28:279, 1950. 48. Mesker RP, Oosterhuis JA, Delleman JW: A retinal lesion resembling Sjogren's
dystrophy reticulates laminae pigmentosae retina. In: Winkelman
JE, Crone RA, eds. Perspectives in ophthalmology, vol 2. Amsterdam, Excerpta
Medica, 1970:45 49. Deutman AF, van Blomniestein IDA, Hehkes HE et al: Butterfly-shaped pigment dystrophy of the fovea. Arch Ophthalmol 83:558, 1970. 50. Hsieh RC, Fine BS, Lyons JS: Patterned dystrophies of the retinal pigment epithelium. Arch Ophthalmol 95:429, 1977. 51. Marmor MF, Byers B: Pattern dystrophy of the pigment epithelium. Am J Ophthalmol 84:32, 1977. 52. deJong PTV, Dellman W: Pigment epithelial pattern dystrophy. Arch Ophthalmol 100:1416, 1982. 53. Cortin P, Archer D, Maumenee IH et al: A patterned macular dystrophy with yellow plaques and atrophic changes. Br J Ophthalmol 64:127, 1980. 54. Burgess D: Subretinal neovascularization in pattern dystrophy of the retinal pigment
epithelium. Retina 1:151, 1981. 55. Gutman I, Walsh JB, Henkind P: Vitelliform macular dystrophy and butterfly-shaped epithelial dystrophy: a
continuum? Br J Ophthalmol 66:170, 1982. 56. Giuffre G, Lodato G: Vitelliform dystrophy and pattern dystrophy of the retinal pigment epithelium: concomitant
presence in a family. Br J Ophthalmol 70:526, 1986. 57. Klein R, Lewis RA, Meyers SM et al: Subretinal neovascularization associated with fundus flavimaculatus. Arch Ophthalmol 96:2054, 1978. 58. Leveille AS, Morse PH, Burch JV: Fundus flavimaculatus and subretinal neovascularization. Ann Ophthalmol 14:331, 1982. 59. Bastiacnsen LAK, Hoefnages KLJ: Patterned anomalies of the retinal pigment eithelium: dystrophy or syndrome? Doe Ophthalmol 55:17, 1983. 60. Noble KG: Pathology of the hereditary macular dystrophies. Semin Ophthalmol 2:110, 1987. 61. Vine AK, Schatz H: Adult-onset foveomacular pigment epithelial dystrophy. Am J Ophthalmol 89:680, 1980. 62. Sabates R, Pruett RC, Hirose T: Pseudovitelliform macular degeneration. Retina 2:197, 1982. 63. Miller SA: Fluorescence in Best's vitelliform dystrophy, lipofuscin and fundus
flavimaculatus. Br J Ophthalmol 62:256, 1978. 64. Sandvig K: Familial central, areolar, choroidal atrophy of autosomal dominant inheritance. Acta Ophthalmol 33:71, 1955. 65. Noble KG: Central areolar choroidal dystrophy. Am J Ophthalmol 84:310, 1977. 66. Noble KG, Carr RE, Siegel IM: Fluorescein and angiography of the hereditary choroidal dystrophies. Br J Ophthalmol 61:43, 1977. 67. Ashton N: Central areolar choroidal sclerosis: a histopathological study. Br J Ophthalmol 37:140, 1953. 68. Klein BA: Some aspects of classification in differential diagnosis of senile macular
degeneration. Am J Ophthalmol 58:927, 1964. 69. Babel MJ: Le role de la choriocapillare dans les afflictions degeneratives du pole
posterieur. Bull Mem Soc Fr Ophtalmol 71:389, 1958. 70. Ferry A, Llovera L, Shafer DM: Central areolar choroidal dystrophy. Arch Ophthalmol 88:39, 1972. 71. Lefler WH, Wadsworth JAC, Sidbury JB: Hereditary macular degeneration and aminoaciduria. Am J Ophthalmol 71:224, 1971. 72. Frank HR, Landers MB, Williams RJ, Sidbury JB: A new dominant progressive foveal dystrophy. Am J Ophthalmol 78:903, 1974. 73. Fetkenhour CL, Gurney N, Dobbie JG, Choromokos E: Central areolar pigment epithelial dystrophy. Am J Ophthalmol 81:745, 1976. 74. Leveille AS, Morse PH, Kiernan JP: Autosomal dominant central pigment epithelial and choroidal degeneration. Ophthalmology 89:1407, 1982. 75. Gass JDM: Stereoscopic atlas of macular diseases, 3rd ed. St. Louis, CV
Mosby, 1987. <en 76. Small KW: North Carolina macular dystrophy: revisited. Ophthalmology 96:1747, 1989. 77. Gass JDM: A clinicopathologic study of a peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc 72:139, 1974. 78. Patrinely JR, Lewis RA, Font RL: Foveomacular vitelliform dystrophy, adult type: a clinicopathologic study
ineluding electron microscopic observations. Ophthalmology 92:1712, 1985. 79. Jaffe GJ, Schatz H: Histopathologic features of adult onset foveomacular pigment epithelial
dystrophy. Arch Ophthalmol 106:958, 1988. 80. Sorsby A, Joll-Mason ME, Gardener N: A fundus dystrophy with unusual features. Br J Ophthalmol 33:67, 1949. 81. Duke-Elder S, Perkin ES: Diseases of the uveal tract. In: Duke-Elder S, ed. System
of ophthalmology, vol 9. St. Louis, CV Mosby, 1966:715. 82. Carr RE, Noble KG, Nasaduke I: Hereditary hemorrhagic macular dystrophy. Am J Ophthalmol 85:318, 1978. 83. Hoskin A, Sehmi K, Bird AC: Sorsby's pseudoinflammatory macular dystrophy. Br J Ophthalmol 65:859, 1981. 84. Forsius HR, Eriksson AW, Suvanto EA, Alanko HI: Pseudoinflammatory fundus dystrophy with autosomal recessive inheritance. Am J Ophthalmol 94:634, 1982. 85. Ashton N, Sorsby A: Fundus dystrophy with unusual features: a histological study. Br J Ophthalmol 35:751, 1951. 86. Deutman AF, Pinckers AJLG, Aan De Kerk AL: Dominantly inherited cystoid macular edema. Am J Ophthalmol 82:540, 1976. 87. Pinckers AJLG, Deutman AF, Notting JGA: Retinal functions in dominant cystoid macular dystrophy. Acta Ophthalmol 54:579, 1976. 88. Notting JGA, Pinckers AJLG: Dominant cystoid macular dystrophy. Am J Ophthalmol 83:234, 1977. 89. Fishmar GA, Goldberg ME, Trautmann JC: Dominantly inherited cystoid macular edema. Ann Ophthalmol 11:21, 1979. 90. Lewis RA, Lee GB, Martonyi CL et al: Familial foveal retinoschisis. Arch Ophthalmol 95:1190, 1977. 91. Singerman LJ, Berkow JW, Patz A: Dominant slowly progressive macular dystrophy. Am JOphthalmol 83:680, 1977. 92. O'Donnell FE, Welch RB: Fenestrated sheen macular dystrophy. Arch Ophthalmol 97:1292, 1979. 93. Deutman AF: Benign concentric annular macular dystrophy. Am J Ophthalmol 78:384, 1974. 94. Coppetto J, Ayazi S: Annular macular dystrophy. Am J Ophthalmol 93:279, 1982. 95. van Den Biesen PR, Deutman AF, Pinckers AJLG: Evolution of benign concentric annular macular dystrophy. Am J Ophthalmol 100:73, 1985. 96. Noble KG, Sherman J: Central pigmentary sheen dystrophy. Am J Ophthalmol 108:255, 1989. 97. Duke-Elder S: System of ophthalmology, vol X. Diseases of the retina. St. Louis, CV
Mosby, 1967. 98. Franceschetti A, Francois J, Babel J: Chorioretina heredodegenerations. Springfield, IL, Charles
C. Thomas, 1974. 99. Weber BH, Vogt G, Pruett RC et al: Mutations in the tissue inhibitor of metalloproteinases-3 (TIMP 3) in patients
with Sorsby's fundus dystrophy. Nat Genet 8:352–356, 1994. 100. Allikmets R, Sing N, Sun H et al: A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated
in recessive Stargardt macular dystrophy. Nat Genet 15:236–246, 1997. 101. Petrukhin K, Kost MJ, Bakail B et al: Identification of the gene responsible for Best macular dystrophy. Nat Genet 19:241–247, 1998. 102. Stone EM, Lotery AJ, Munier FL et al: A single EFEMP 1 mutation associated with both Mallatia Leventinese and
Doyne honeycomb retinal dystrophy. Nat Genet 22:199–202, 1999. |