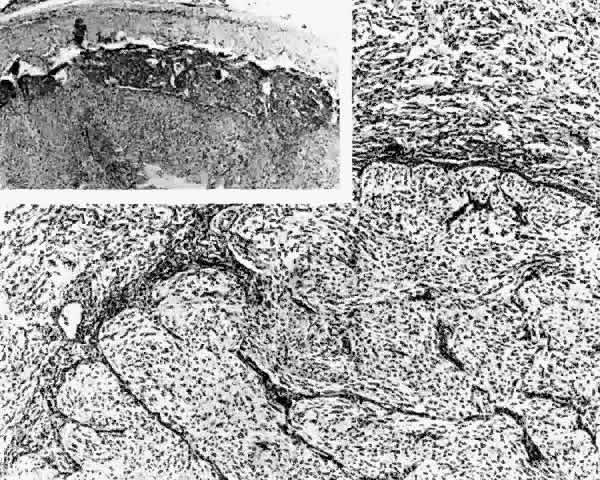
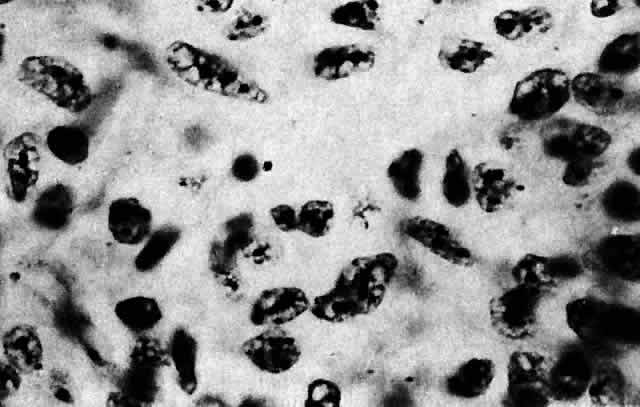
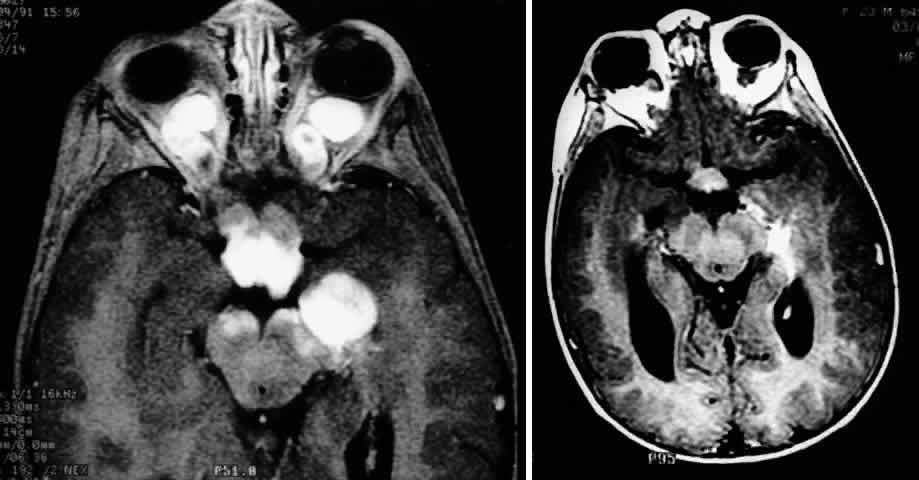
1. Scarpa A: Trattato delle Principali Maletties degli Occhi, Edizion Quinta, pp 507–509. Pavia, Pietro Bizzoni, 1816 2. Alvord EC, Lofton S: Gliomas of the optic nerve or chiasm: Outcome by patient's age, tumor
site, and treatment. J Neurosurg 68:85, 1988 3. Dutton JJ: Gliomas of the anterior visual pathway. Surv Ophthalmol 38:427, 1994 4. Eggers H, Jakobiec FA, Jones IS: Optic nerve gliomas. In Tasman W, Jaeger
EA (eds): Duane's Clinical Ophthalmology, Vol 2, pp 1–17. Philadelphia, JB
Lippincott, 1994 5. Davis FA: Primary tumors of the optic nerve (a phenomenon of von Recklinghausen's
disease): A clinical and pathologic study with a report of five
cases and a review of the literature. Arch Ophthalmol 23:735, 1940 6. King DL, Chang CH, Pool JL: Radiotherapy in management of meningiomas. Acta Radiol (Ther) 5:26, 1966 7. Reese AB: Tumors of the Eye, 3rd ed, pp 148–154. Hagerstown, MD, Harper & Row, 1976 8. Russell DC, Rubinstein LJ: Pathology of Tumors of the Nervous System, 4th
ed. Baltimore, Williams & Wilkins, 1977 9. Bucy PC, Russell JR, Whitsell FM: Surgical treatment of tumors of the optic nerve. Arch Ophthalmol 44:411, 1950 10. Mohan H, Sen DK: Astrocytoma of the optic nerve. Br J Ophthalmol 54:284, 1970 11. Eggers H, Jakobiec FA, Jones IS: Optic nerve gliomas. In Jones IS, Jakobiec
FA (eds): Diseases of the Orbit, pp 417–443. Hagerstown, MD, Harper & Row, 1979 12. Baudet D, Nicaise A, Javalet A et al: Gliome du nerf optique chez un adulte. Rev Otoneuroophtalmol 55:279, 1983 13. Wilson WB: Optic nerve gliomas: Treatment differences for the benign and
malignant varieties. In Tusa RJ, Newman SA (eds): Neuro-ophthalmological
Disorders: Diagnostic Work-up and Management, pp 163–172. New
York, Marcel Dekker, 1995 14. Porterfield JF: Orbital tumors in children: A report on 214 cases. Int Ophthalmol Clin 2:319, 1962 15. Cohen ME, Duffner PK: Optic Pathway Tumors. Neurol Clin 9:467, 1991 16. Fowler FD, Matson DD: Gliomas of optic pathways in childhood. J Neurosurg 14:515, 1957 17. Wisoff J: Management of optic pathway tumors of childhood. Pediatr Neurooncol 3:791, 1992 18. Miller NR, Iliff WJ, Green WR: Evaluation and management of gliomas of the anterior visual pathways. Brain 97:743, 1974 19. Davis PC, Hoffman JC Jr, Weidenheim KM: Large hypothalamic and optic chiasm gliomas in infants: Difficulties in
distinction. AJNR 5:579, 1984 20. Walker RW: Optic pathway gliomas (abstr). Proceedings of the North American
Neuro-Ophthalmology Society Meeting, February 19–23, Tucson, AZ, 1995 21. Housepian EM: Management and results of 114 cases of optic glioma (abstr). Neurosurgery 1:67, 1977 22. Chutorian AM, Housepian EM, Hilal S: Optic gliomas of multicentric origin with favorable response to radiotherapy. Trans Am Neurol Assoc 101:229, 1976 23. Borit A, Richardson EP Jr: The biological and clinical behaviour of pilocytic astrocytomas of the
optic pathways. Brain 105:161, 1982 24. Dosoretz DE, Blitzer PH, Wang CC et al: Management of glioma of the optic nerve and/or chiasm. Cancer 45:1467, 1980 25. Rush JA, Younge BR, Campbell RJ et al: Optic glioma: Long-term follow-up of 85 histopathologically verified cases. Ophthalmology 89:1213, 1982 26. Brand WN, Hoover SV: Optic glioma in children: Review of 16 cases given mega voltage radiation
therapy. Childs Brain 5:459, 1979 27. Maitland CG, Abiko S, Hoyt WF et al: Chiasmal apoplexy: Report of four cases. J Neurosurg 56:118, 1982 28. Dodge HW Jr, Love JG, Craig WM et al: Gliomas of the optic nerves. Arch Neurol Psychiatry 79:607, 1958 29. Chutorian AM, Schwartz JF, Evans RA et al: Optic gliomas in children. Neurology 14:83, 1964 30. Rothman SJ, Stern J, Tenner M et al: Subclinical optic lesions in childhood neurofibromatosis. Neurology 37(suppl 1):148, 1987 31. MacCarty CS, Boyd AS Jr, Childs DS Jr: Tumors of the optic nerve and optic chiasm. J Neurosurg 33:439, 1970 32. Bynke H, Kagstrom E, Tjernstrom K: Aspects on the treatment of gliomas of the anterior visual pathway. Acta Ophthalmol 55:269, 1977 33. Lowes M, Bojsen-Moller M, Vorre P et al: An evaluation of gliomas of the anterior visual pathways: A 10-year survey. Acta Neurochir 43:201, 1978 34. Packer RJ, Siegel KR, Sutton LN et al: Leptomeningeal dissemination of primary central nervous system tumors of
childhood. Ann Neurol 18:217, 1985 35. Lourie GL, Osborne DR, Kirks DR: Involvement of posterior visual pathways by optic nerve gliomas. Pediatr Radiol 16:271, 1986 36. Grimson BS, Perry DD: Enlargement of the optic disk in childhood optic nerve tumors. Am J Ophthalmol 97:627, 1984 37. Buchanan TAS, Hoyt WF: Optic nerve glioma and neovascular glaucoma: Report of a case. Br J Ophthalmol 66:96, 1982 38. Hovland KR, Ellis PP: Hemorrhagic glaucoma with optic nerve glioma. Arch Ophthalmol 75:806, 1966 39. Donaldson DD: Atlas of External Diseases of the Eye, Anterior Chamber, Iris, and
Ciliary Body, p 238. St. Louis, CV Mosby, 1973 40. Hoyt WF, Imes RI: Optic gliomas of neurofibromatosis 1. In Ishibashi Y, Hori
Y (eds): Tuberous Sclerosis and Neurofibromatosis. Amsterdam, Elsevier, 1990 41. Byrne SF, Glaser JS: Orbital tissue differentiation with standardized echography. Ophthalmology 90:1071, 1983 42. Gans MS, Frazier Byrne S, Glaser JS: Standardized A-scan echography in optic nerve disease. Arch Ophthalmol 105:1232, 1987 43. Halpern J: Spasmus nutans. Arch Neurol 37:737, 1980 44. Koenig SB, Naidich TP, Zaparackas Z: Optic glioma masquerading as spasmus nutans. J Pediatr Ophthalmol Strabismus 19:20, 1982 45. O'Neill JF, Lavery MA, Chu FC: Acquired nystagmus in early childhood: An early sign of intracranial tumor. Ophthalmology 90(suppl):76, 1983 46. Albright AL, Sclabassi RJ, Slamovitz TL et al: Spasmus nutans associated with optic gliomas in infants. J Pediatr 105:778, 1984 47. Newman S: Spasmus nutans—or is it? Surv Ophthalmol 34:453, 1990 48. Albert D, Puliafito C: Foundations of Opthalmic Pathology. New York, Appleton-Century-Crofts, 1979 49. Laue L, Comite F, Hench K et al: Precocious puberty associated with neurofibromatosis and optic glioma. Am J Dis Child 139:1097, 1985 50. Brauner R, Malandry F, Rappaport R et al: Growth and endocrine disorders in optic glioma. Eur J Pediatr 149:825, 1990 51. Ellis BD, Kosmorsky GS: The phakomatoses, p 153. In Tomsak RL (ed): Pediatric
Neuro-Ophthalmology. Newton, Butterworth-Heinemann, 1995 52. Smirniotopoulos JG, Murphy FM: The phakomatoses. ANJR 13:725, 1992 53. National Institutes of Health: Neurofibromatosis: National Institutes of
Health Consensus Development Conference Statement, p 6. Bethesda, MD, The
Institute, 1987 54. Dossetor FM, Landau K, Hoyt WF: Optic disc glioma in neurofibromatosis type 2. Am J Ophthalmol 108:602, 1989 55. Landau K, Yasargil GM: Ocular fundus in neurofibromatosis type 2. Br J Ophthalmol 77:646, 1993 56. Burki E: Ueber den primaren Sehnerventumor und seine Beziehunge zur Recklinghausenchen
neurofibromatose. Bibl Ophthalmol 30:1, 1944 57. Marshall D: Glioma of the optic nerve as a manifestation of von Recklinghausen's
disease. Am J Ophthalmol 37:15, 1954 58. Lloyd L: Gliomas of the optic nerve and chiasm in childhood. Trans Am Ophthalmol Soc 71:488, 1973 59. Stern J, DiGiacinto GV, Housepian EM: Neurofibromatosis and optic glioma: Clinical and morphological correlations. Neurosurgery 4:524, 1979 60. Stern J, Jakobiec FA, Housepian EM: The architecture of optic nerve gliomas with and without neurofibromatosis. Arch Ophthalmol 98:505, 1980 61. Klug GL: Gliomas of the optic nerve and chiasm in children. Neuro-ophthalmology 2:217, 1982 62. Lewis RA, Gerson LP, Axelson KA et al: Von Recklinghausen neurofibromatosis: II. Incidence of optic gliomata. Ophthalmology 91:929, 1984 63. Listernick R, Charrow J, Greenwald MJ et al: Optic gliomas in children with neurofibromatosis type 1. J Pediatr 114:788, 1989 64. DiMario FJ, Ramsby G, Greenstein R et al: Neurofibromatosis type I: Resonance imaging findings. J Child Neurol 8:32, 1993 65. Listernick R, Charrow J, Greenwald M et al: Natural history of optic pathway tumors in children with neurofibromatosis
type I: A longitudinal study. J Pediatr 125:63, 1994 66. Listernick R, Charrow J, Greenwald M: Emergence of optic pathway gliomas in children with neurofibromatosis type 1 after
normal neuroimaging results. J Pediatr 121:584, 1992 67. Evans RA, Schwartz JF, Chutorian AM: Radiologic diagnosis in pediatric ophthalmology. Radiol Clin North Am 1:459, 1963 68. Spencer WH: Primary neoplasms of the optic nerve and its sheaths: Clinical features
and current concepts of pathogenetic mechanisms. Trans Am Ophthalmol Soc 70:490, 1972 69. Marquardt MD, Zimmerman LE: Histopathology of meningiomas and gliomas of the optic nerve. Hum Pathol 13:226, 1982 70. Holman CB: Roentgenologic manifestations of glioma of the optic nerve and chiasm. AJR 82:462, 1959 71. Miller NR: Tumors of neuroectodermal origin. In Miller NR (ed): Walsh and
Hoyt's Clinical Neuro-Ophthalmology, 4th ed, Vol 3, pp 1259–1275. Baltimore, Williams & Wilkins, 1988 72. Patronas NJ, Dwyer AJ, Papathanasiou M et al: Contributions of magnetic resonance imaging in the evaluation of optic
gliomas. Surg Neurol 28:367, 1987 73. Fletcher WA, Imes RK, Hoyt WF: Chiasmal gliomas: Appearance and long-term changes demonstrated by computerized
tomography. J Neurosurg 65:154, 1986 74. Hoyt WF, Fletcher WA, Imes RK: Chiasmal gliomas: Appearance and long-term changes demonstrated by computerized
tomography. Prog Exp Tumor Res 30:113, 1987 75. Tekkok IH, Tahta K, Saglam S et al: Optic nerve glioma presenting as a huge intrasellar mass: Case report. J Neurosurg Sci 38:137, 1994 76. Liu GT, Lessell S: Spontaneous visual improvement in chiasmal gliomas. Am J Ophthalmol 114:193, 1992 77. Hendrix LE, Kneeland JB, Houghton VM et al: MR imaging of optic nerve lesions: Value of gadopentate dimeglumine and
fat-suppression technique. AJNR 11:749, 1990 78. Haik BG, Saint-Louis L, Bierly J et al: Magnetic resonance imaging in the evaluation of optic nerve gliomas. Ophthalmology 94:709, 1987 79. Brown EW, Riccardi VM, Mawad M et al: MR imaging of optic pathways in patients with neurofibromatosis. AJNR 8:1031, 1987 80. Imes RD, Hoyt WF: Magnetic resonance imaging signs of optic nerve gliomas in neurofibromatosis 1. Am J Ophthalmol 111:729, 1991 81. Packer RJ, Bilaniuk LT, Cohen BH et al: Intracranial visual pathway gliomas in children with neurofibromatosis. Neurofibromatosis 1:212, 1988 82. Duffner PK, Cohen ME, Seidel FG et al: The significance of MRI abnormalities in children with neurofibromatosis. Neurology 39:373, 1989 83. Ng KL, McDermott N, Romanowski CAJ et al: Neurosarcoidosis masquerading as glioma of the optic chiasm in a child. Postgrad Med J 71:265, 1995 84. Jakobiec FA, Depot MJ, Kennerdell JS et al: Combined clinical and computed tomographic diagnosis of orbital glioma
and meningioma. Ophthalmology 91:137, 1984 85. Anderson DR, Spencer WH: Ultrastructural and histochemical observations of optic nerve gliomas. Arch Ophthalmol 83:324, 1970 86. Charles NC, Nelson L, Brookner AR et al: Pilocytic astrocytoma of the optic nerve with hemorrhage and extreme cystic
degeneration. Am J Ophthalmol 92:691, 1981 87. Levin LA, Jakobiec FA: Peripheral nerve sheath tumors of the orbit. In
Albert DM, Jakobiec FA (eds): Principles and Practice of Ophthalmology, Vol 3, pp 1997–1998. Philadelphia, WB Saunders, 1994 88. Goldman JE, Corbin E: Rosenthal fibers contain ubiquitinated alpha B-crystallin. Am J Pathol 139:933, 1991 89. Hossmann K-A, Wechsler W: Zur Feinstruktur menschlicher Spongioblastome. Dtsch Z Nervenheilkd 187:327, 1965 90. Brzowski AE, Bazan C, Mumma JV et al: Spontaneous regression of optic glioma in a patient with neurofibromatosis. Neurology 42:679, 1992 91. Hudson AC: Primary tumors of the optic nerve. R Ophthalmol Hosp Rep 18:317, 1912 92. Hoyt WF, Baghdassarian SA: Optic glioma of childhood: Natural history and rationale for conservative
management. Br J Ophthalmol 53:793, 1969 93. Burnstine MA, Levin LA, Louis DN et al: Nucleolar organizer regions in optic gliomas. Brain 116:1465, 1993 94. de Keizer RJW, de Wolff-Rouendaal, Bots GTAM et al: Optic glioma with intraocular tumor and seeding in a child with neurofibromatosis. Am J Ophthalmol 108:717, 1989 95. Bruggers CS, Friedman HS, Phillips PC et al: Leptomeningeal dissemination of optic pathway gliomas in three children. Am J Ophthalmol 111:719, 1991 96. Civitello LA, Packer RJ, Rorke LB et al: Leptomeningeal dissemination of low-grade gliomas in childhood. Neurology 38:562, 1988 97. Kocks W, Kalff R, Reinhardt V et al: Spinal metastasis of pilocytic astrocytoma of the chiasma opticum. Child Nerv Syst 5:118, 1989 98. Trigg ME, Swanson JD, Letellier MA: Metastasis of an optic glioma though a ventriculoperitoneal shunt. Cancer 52:599, 1983 99. Marejeva TG, Rostotskaya VI, Sokolova ON et al: Tumours of the optic nerve and chiasma in children. Acta Neurochir (Suppl) 28:409, 1979 100. Wong IG, Lubow M: Management of optic glioma of childhood: A review of 42 cases. In
Smith JL (ed): Neuro-ophthalmology Symposium of the Bascom
Palmer Eye Institute and the University of Miami, Vol 6, pp 51–60. St. Louis, CV
Mosby, 1972 101. Heiskenan O, Raitta C, Torsti R: The management and prognosis of gliomas of the optic pathways in children. Acta Neurochir 43:193, 1978 102. Oxenhandler DC, Sayers MP: The dilemma of childhood optic gliomas. J Neurosurg 48:34, 1978 103. Glaser JS, Hoyt WF, Corbett J: Visual morbidity with chiasmal glioma: Long-term studies of visual fields
in untreated and irradiated cases. Arch Ophthalmol 85:3, 1971 104. Wilson WB, Feinsod M, Hoyt WF et al: Malignant evolution of childhood chiasmal pilocytic astrocytoma. Neurology 26:322, 1976 105. Danoff BF, Kramer S, Thompson N: The radiotherapeutic management of optic nerve gliomas in children. Int J Radiat Oncol Biol Phys 6:45, 1980 106. Imes RK, Hoyt WF: Childhood chiasmal gliomas: Update on the fate of the patients in the 1969 San
Francisco Study. Br J Ophthalmol 70:179, 1986 107. Wishart JH: Case of extirpation of the eyeball. Edinb Med Surg J 40:274, 1833 108. North American Study Group for Optic Glioma: Tumor spread in unilateral
optic glioma: Study report No. 2. Neurofibromatosis 2:195, 1989 109. Glaser JS: Gliomas of the anterior visual pathways in childhood: Rationale
for conservative management. In Brockhurst RJ, Borouchoff SA, Hutchinson
BT et al (eds): Controversy in Ophthalmology, pp 897–909. Philadelphia, WB
Saunders, 1977 110. Wright JE, McDonald WI, Call NB: Management of optic nerve gliomas. Br J Ophthalmol 64:545, 1980 111. Tenney RT, Laws ER, Young BR et al: The neurosurgical management of optic gliomas: Results in 104 patients. J Neurosurg 57:452, 1982 112. Wilson WB, Finkel RS, McCleary L et al: Large cystic optic glioma. Neurology 40:1898, 1990 113. Kovalic JJ, Grigsby PW, Shephard MJ et al: Radiation therapy for gliomas of the optic nerve and chiasm. Int J Radiat Oncol Biol Phys 18:927, 1990 114. Bataini JP, Delanian S, Ponvert D: Chiasmal gliomas: Results of irradiation management in 57 patients and
review of literature. J Radiat Oncol Biol Phys 21:615, 1991 115. Flickinger JC, Torres C, Deutsch M: Management of low-grade gliomas of the optic nerve and chiasm. Cancer 61:635, 1988 116. Horwich A, Bloom HJG: Optic gliomas: Radiation therapy and prognosis. Int J Radiat Oncol Biol Phys 11:1067, 1985 117. Furuya Y, Uemura K, Ryu H et al: Optic glioma decreasing in size after irradiation. J Child Neurol 1:173, 1986 118. Packer RJ, Savino PJ, Bilaniuk LT et al: Chiasmatic gliomas of childhood: A reappraisal of natural history and effectiveness
of cranial irradiation. Childs Brain 10:393, 1983 119. Kingsley DPE, Kendall BE: CT of the adverse effects of therapeutic radiation of the central nervous
system. AJNR 2:453, 1981 120. Epstein M, Packer R, Rorke L: Vascular malformation with radiation vasculopathy after treatment of chiasmatic
hypothalamic glioma. Cancer 70:887, 1992 121. Kestle J, Hoffman J, Mock A: Moyamoya phenomenon after radiation for optic glioma. J Neurosurg 79:32, 1993 122. Janss AJ, Grundy R, Cnaan A et al: Optic pathway and hypothalamic/chiasmatic gliomas in children younger than
age 5 years with a 6-year follow-up. Cancer 75:1051, 1995 123. Packer RA, Lange B, Ater J: Carboplatin and vincristine for progressive low grade gliomas of childhood. J Clin Oncol 11:850, 1993 124. Lassman LP, Pearce GW, Gang J: Effect of vincristine sulphate on the intracranial gliomata of childhood. Br J Surg 53:774, 1966 125. Packer RJ, Sutton LN, Bilaniuk LT et al: Treatment of chiasmatic/hypothalamic gliomas of childhood with chemotherapy: An
update. Ann Neurol 23:79, 1988 126. Petronio J, Edwards SB, Prados M et al: Management of chiasmal and hypothalamic gliomas of infancy and childhood
with chemotherapy. J Neurosurg 74:701, 1991 127. Kretschmar CS, Linggood RM: Chemotherapeutic treatment of extensive optic pathway tumors in infants. J Neurooncol 10:263, 1991 128. Chamberlain MC: Recurrent chiasmatic-hypothalamic glioma treated with oral etoposide. Arch Neurol 52:509, 1995 129. Moghrabi A, Friedman HS, Burger PC et al: Carboplatin treatment of progressive optic pathway gliomas to delay radiotherapy. J Neurosurg 79:223, 1993 130. Feldon SE: Tumors of the anterior visual pathways. In Albert DM, Jakobiec
FA (eds): Principles and Practice of Ophthalmology, Vol 4, p 2578. Philadelphia, WB
Saunders, 1994 131. Housepien EM: Surgical treatment of unilateral optic nerve gliomas. J Neurosurg 31:604, 1969 132. Housepien EM: Transcranial orbital surgery. In Laws ER Jr (ed): The Diagnosis
and Management of Orbital Tumors. Mount Kisco, NY, Futura, 1988 133. Wolter JR: The special blood supply of the retina in optic nerve gliomas. J Pediatr Ophthalmol 13:198, 1976 134. Hoyt WF, Meshel LG, Lessell S et al: Malignant optic glioma of adulthood. Brain 96:121, 1973 135. Safneck JR, Napier LB, Halliday WC: Malignant astrocytoma of the optic nerve in a child. Can J Neurol Sci 19:498, 1992 136. Andoh A, Ohkuma H, Ebina K et al: Optic gliomas in neonates: Report of two cases. No Shinkei Geka 16:429, 1988 137. Manor RS, Israeli J, Sandbank U: Malignant optic glioma in a 70 year old patient. Arch Ophthalmol 94:1142, 1976 138. Harper CG, Stewart-Wynne EG: Malignant optic gliomas in adults. Arch Neurol 35:731, 1978 139. Albers GW, Hoyt WF, Forno LS et al: Treatment response in malignant optic glioma of adulthood. Neurology 38:1071, 1988 140. Spoor TC, Kennerdell JS, Martinez AJ et al: Malignant gliomas of the optic nerve pathways. Am J Ophthalmol 89:284, 1980 141. Hamilton AM, Garner A, Tripathi RC et al: Malignant optic glioma: Report of a case with electron microscope study. Br J Ophthalmol 57:253, 1973 142. Saebo J: Primary tumour of the optic nerve (glioblastoma multiforme). Br J Ophthalmol 33:701, 1949 143. Mattson RH, Peterson EW: Glioblastoma multiforme of the optic nerve. JAMA 196:799, 1966 144. Taphoorn MJB, de Vries-Knoppert WAEJ, Ponssen H et al: Malignant optic glioma in adults. J Neurosurg 70:277, 1989 145. Rudd A, Rees JE, Kennedy P et al: Malignant optic nerve gliomas in adults. J Clin Neuro-ophthalmol 5:238, 1985 146. Barbaro NM, Rosenblum ML, Maitlant CG et al: Malignant optic glioma presenting radiologically as a “cystic” suprasellar
mass: Case report and review of the literature. Neurosurgery 11:787, 1982 |