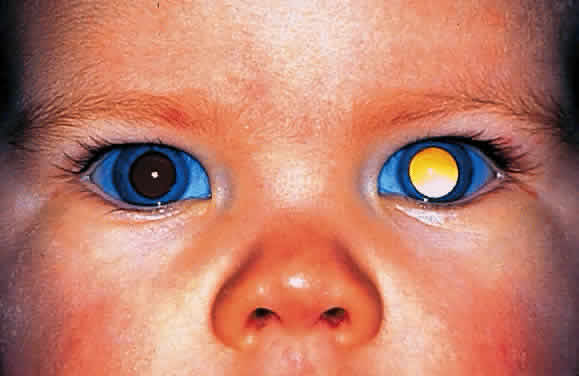
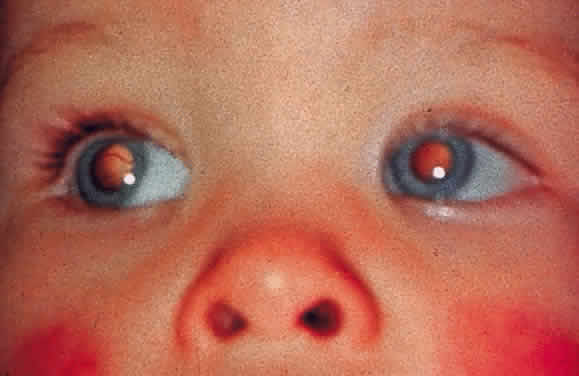
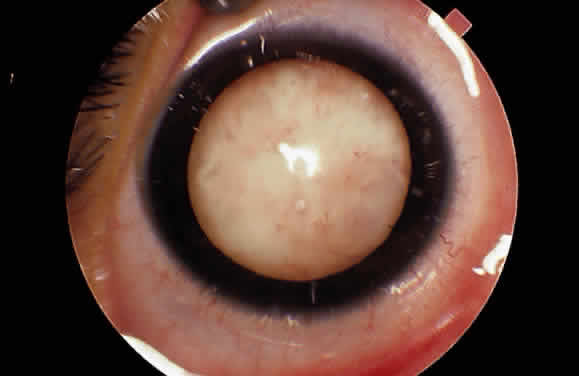
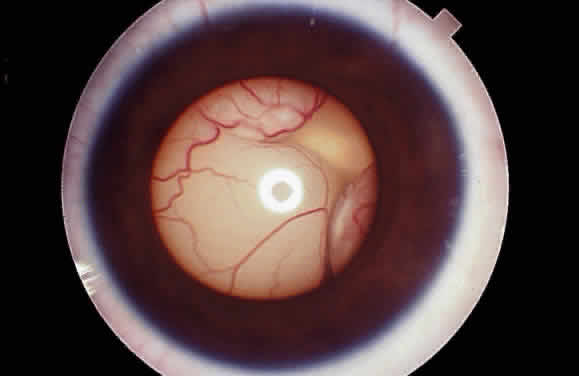
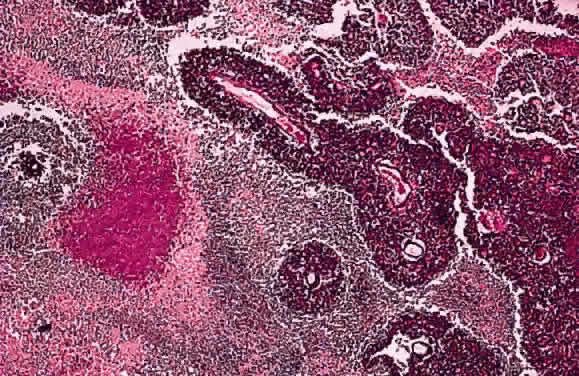
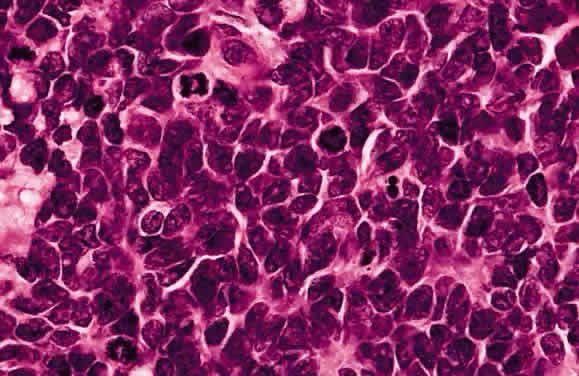
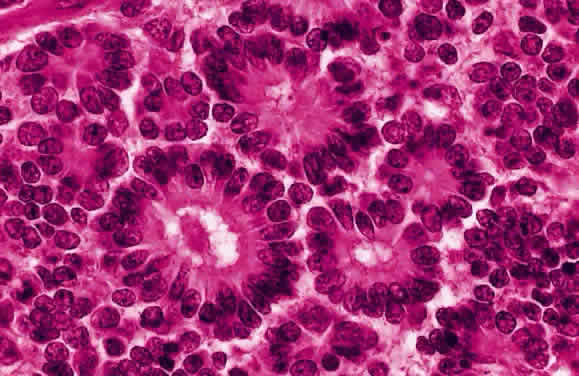
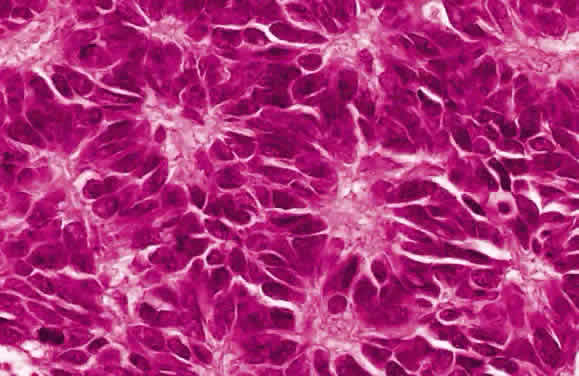
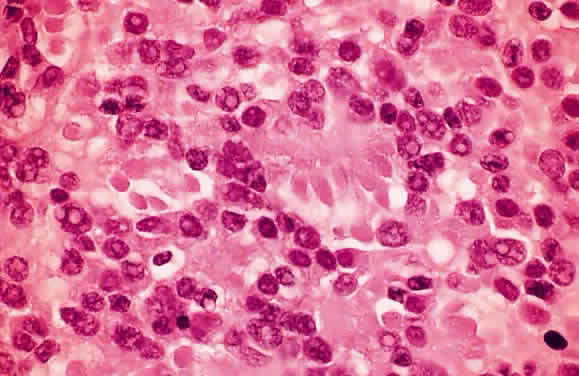
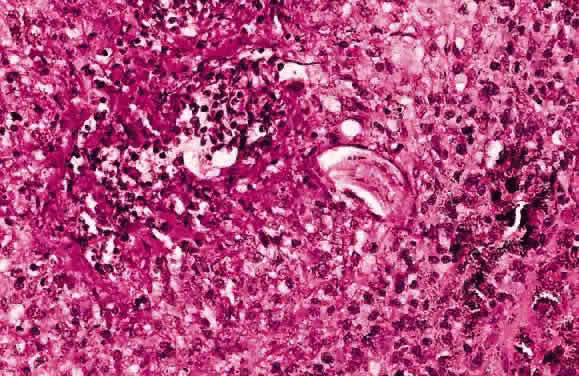
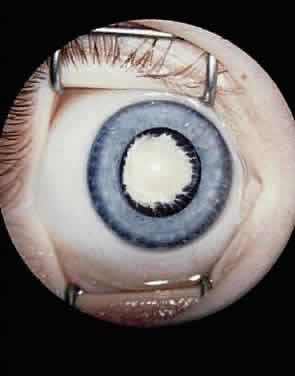
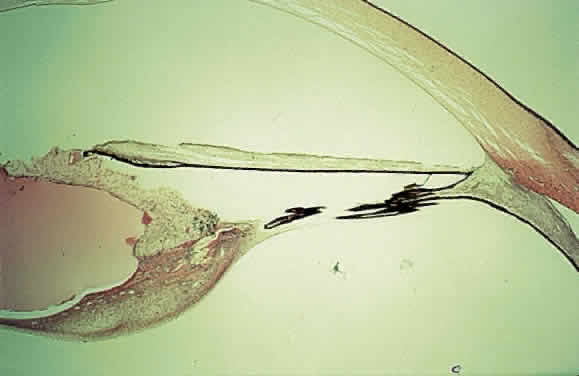
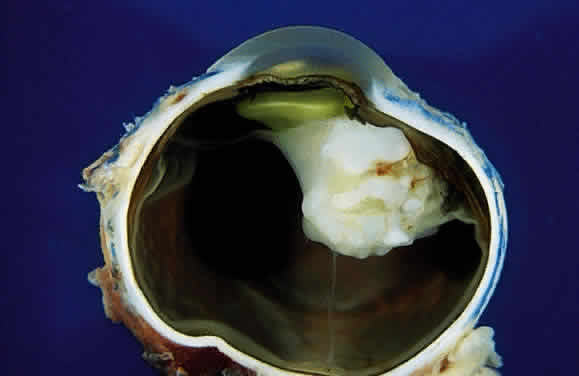
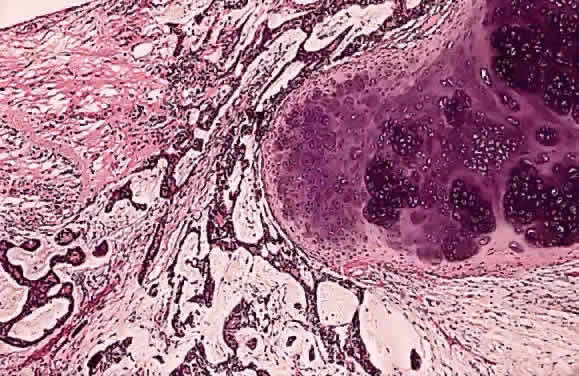
EPIDEMIOLOGY
Abramson DH: Retinoblastoma incidence in the United States [letter; comment]. Arch
Ophthalmol 108:1514, 1990
al-Idrissi I, al-Kaff AS, Senft SH: Cumulative incidence of retinoblastoma
in Riyadh, Saudi Arabia. Ophthalmic Paediatr Genet 13:9, 1992
BenEzra D, Chirambo MC: Incidence of retinoblastoma in Malawi. J Pediatr
Ophthalmol 13:340, 1976
Berkow RL, Fleshman JK: Retinoblastoma in Navajo Indian children. Am J
Dis Child 137:137, 1983
Devesa SS: The incidence of retinoblastoma. Am J Ophthalmol 80:263, 1975
Fitzgerald PH, Stewart J, Suckling RD: Retinoblastoma mutation rate in
New Zealand and support for the two-hit model. Hum Genet 64:128, 1983
Freedman J, Goldberg L: Incidence of retinoblastoma in the Bantu of South
Africa. Br J Ophthalmol 60:655, 1976
Kock E, Naeser P: Retinoblastoma in Sweden 1958-1971. A clinical and histopathological
study. Acta Ophthalmol (Copenh) 57:344, 1979
Tarkkanen A, Tuovinen E: Retinoblastoma in Finland 1912—1964. Acta
Ophthalmol 49:293, 1971
CLINICAL FEATURES
Abramson DH, Frank CM, Susman M et al: Presenting signs of retinoblastoma. J
Pediatr 132:505, 1998
Abramson DH, Gombos DS: The topography of bilateral retinoblastoma lesions. Retina 16:232, 1996
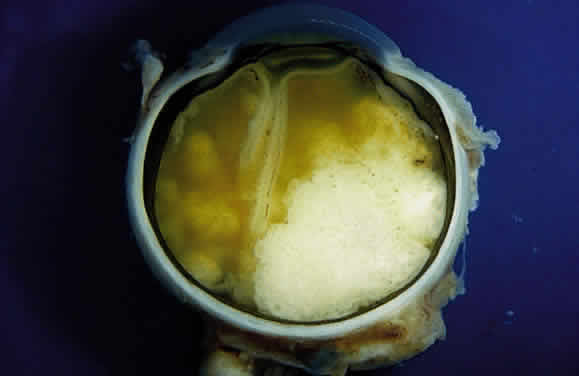
Bhatnagar R, Vine AK: Diffuse infiltrating retinoblastoma. Ophthalmology 98:1657, 1991
Byrnes GA, Shields CL, Shields JA, De Potter P, Eagle RC Jr: Retinoblastoma
presenting with spontaneous hyphema and dislocated lens. J Pediatr
Ophthalmol Strabismus 30:334, 1993
Fontanesi J, Pratt C, Meyer D et al: Asynchronous bilateral retinoblastoma: the
St. Jude Children's Research Hospital experience. Ophthalmic
Genet 16:109, 1995
Mietz H, Hutton WL, Font RL: Unilateral retinoblastoma in an adult: report
of a case and review of the literature [see comments]. Ophthalmology 104:43, 1997
Morgan G: Diffuse infiltrating retinoblastoma. Br J Ophthalmol 55: 600, 1971
Nicholson DH, Norton EW: Diffuse infiltrating retinoblastoma. Trans Am
Ophthalmol Soc 78:265, 1980
Plotsky D, Quinn G, Eagle R Jr, Shields J, Granowetter L: Congenital retinoblastoma: a
case report. J Pediatr Ophthalmol Strabismus 24:120, 1987
Rubenfeld M, Abramson DH, Ellsworth RM, Kitchin FD: Unilateral vs. bilateral
retinoblastoma. Correlations between age at diagnosis and stage
of ocular disease. Ophthalmology 93:1016, 1986
Schofield PB: Diffuse infiltrating retinoblastoma. Br J Ophthalmol 44:35, 1960
Shields CL, Shields JA, Shah P: Retinoblastoma in older children. Ophthalmology 98:395, 1991
Shields JA, Michelson JB, Leonard BC et al: Retinoblastoma in an 18-year-old. J
Pediatr Ophthalmol 13:275, 1976
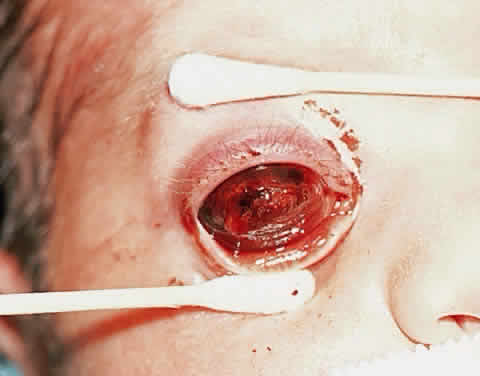
Shields JA, Shields CL, Eagle RC, Blair CJ: Spontaneous pseudohypopyon
secondary to diffuse infiltrating retinoblastoma. Arch Ophthalmol 106:1301, 1988
Shields JA, Shields CL, Eagle RC, De Potter P, Douglas CH: Calcified intraocular
abscess simulating retinoblastoma [letter]. Am J Ophthalmol 114:227, 1992
Shields JA, Shields CL, Eagle RC Jr, Barrett J, De Potter P: Endogenous
endophthalmitis simulating retinoblastoma. Retina 15:213, 1995
Shields JA, Shields CL, Suvarnamani C, Schroeder RP, DePotter P: Retinoblastoma
manifesting as orbital cellulitis. Am J Ophthalmol 112:442, 1991
Spaulding AG: Rubeosis iridis in retinoblastoma and pseudoglioma. Trans
Am Ophthalmol Soc 76:584, 1978
Stafford WR, Yanoff M, Parnell B: Retinoblastoma initially misdiagnosed
as primary ocular inflammation. Arch Ophthalmol 82:771, 1969
Takahashi T, Tamura S, Inoue M et al: Retinoblastoma in a 26-year-old man. Ophthalmology 90:179, 1979
Verhoeff FH: Retinoblastoma: Report of a case in a male age 48. Arch Ophthalmol 2:643, 1929
Walton DS, Grant WM: Retinoblastoma and iris neovascularization. Am J Ophthalmol 65:598, 1968
Yoshizumi MO, Thomas JV, Smith TR: Glaucoma-inducing mechanisms in eyes
with retinoblastoma. Arch Ophthalmol 96:105, 1978
Zimmerman LE: Retinoblastoma and retinocytoma. In Spencer WH (ed): Ophthalmic
Pathology: An Atlas and Textbook, 3rd ed, vol 2, pp 1292–1351. Philadelphia, WB
Saunders, 1985
Zimmerman LE: Retinoblastoma, including a report of illustrative cases. Med
Ann DC 38:366, 1969
PROGNOSTIC FACTORS
Carbajal UM: Metastasis in retinoblastoma. Am J Ophthalmol 48:47, 1959
DeBuen S: Retinoblastoma with spread by direct continuity to the contralateral
optic nerve: report of a case. Am J Ophthalmol 49:815, 1960
Dunkel IJ, Gerald WL, Rosenfield NS et al: Outcome of patients with a history
of bilateral retinoblastoma treated for a second malignancy: the
Memorial-Sloan Kettering experience. Med Pediatr Oncol 30:59, 1998
Howarth C, Meyer D, Hustu HO et al: Stage-related combined modality treatment
of retinoblastoma: results of a prospective study. Cancer 45:851, 1980
Khelfaoui F, Validire P, Auperin A et al: Histopathologic risk factors
in retinoblastoma: a retrospective study of 172 patients treated in a
single institution. Cancer 77:1206, 1996
Kopelman JE, McLean IW, Rosenberg SH: Multivariate analysis of risk factors
for metastasis in retinoblastoma treated by enucleation. Ophthalmology 94:371, 1987
Lennox EL, Draper GJ, Sanders BM: Retinoblastoma: a study of natural history
and prognosis of 268 cases. Br Med J 3:731, 1975
MacKay CJ, Abramson DH, Ellsworth RM: Metastatic patterns of retinoblastoma. Arch
Ophthalmol 102:391, 1984
Magramm I, Abramson DH, Ellsworth RM: Optic nerve involvement in retinoblastoma. Ophthalmology 96:217, 1989
McLean IW, Rosenberg SH, Messmer EP et al: Prognostic factors in cases
of retinoblastoma: analysis of 974 patients from Germany and the United
States treated by enucleation. In Bornfeld N, Gragoudas ES, Lommatzsch
PK (eds): Tumors of the Eye. Proceedings of the International Symposium
on Tumors of the Eye, pp 69–72. Amsterdam, Kugler Publications, 1991
Messmer EP, Heinrich T, Hopping W et al: Risk factors for metastases in
patients with retinoblastoma. Ophthalmology 98:136, 1991
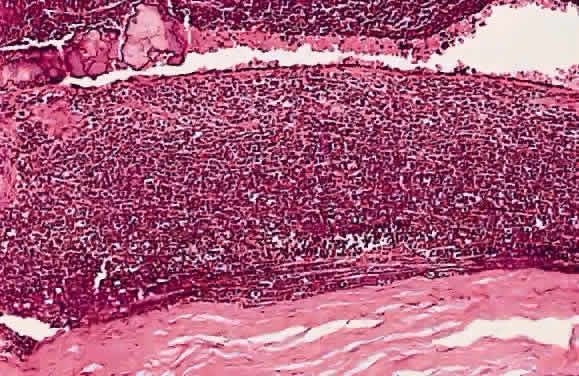
Redler LD, Ellsworth RM: Prognostic importance of choroidal invasion in
retinoblastoma. Arch Ophthalmol 90:294, 1973
Rootman J, Ellsworth RM, Hofbauer J, Kitchen D: Orbital extension of retinoblastoma: a
clinicopathological study. Can J Ophthalmol 13:72, 1978
Rootman J, Hofbauer J, Ellsworth RM, Kitchen D: Invasion of the optic nerve
by retinoblastoma: a clinicopathological study. Can J Ophthalmol 11:106, 1976
Rubin CM, Robison LL, Cameron JD et al: Intraocular retinoblastoma group
V: an analysis of prognostic factors. J Clin Oncol 3:680, 1985
Shields CL, Shields JA, Baez K, Cater JR, De Potter P: Optic nerve invasion
of retinoblastoma. Metastatic potential and clinical risk factors. Cancer 73:692, 1994
Shields CL, Shields JA, Baez KA, Cater J, De Potter PV: Choroidal invasion
of retinoblastoma: metastatic potential and clinical risk factors [see
comments]. Br J Ophthalmol 77:544, 1993
Taktikos A: Investigation of retinoblastoma with special reference to histology
and prognosis. Br J Ophthalmol 50:225, 1966
Wiggs J, Nordenskjold M, Yandell D et al: Prediction of the risk of hereditary
retinoblastoma, using DNA polymorphisms within the retinoblastoma
gene. N Engl J Med 318:151, 1988
Wolter JR: Retinoblastoma extension into the choroid. Pathological study
of the neoplastic process and thoughts about its prognostic significance. Ophthalmic
Paediatr Genet 8:151, 1987
RETINOMA/RETINOCYTOMA/SPONTANEOUS REGRESSION
Aaby AA, Price RL, Zakov ZN: Spontaneously regressing retinoblastomas, retinoma, or
retinoblastoma group 0. Am J Ophthalmol 96:315, 1983
Abramson DH: Retinoma, retinocytoma, and the retinoblastoma gene [editorial]. Arch
Ophthalmol 101:1517, 1983
Andersen SR, Jensen OA: Retinoblastoma with necrosis of central retinal
artery and vein and partial spontaneous regression. Acta Ophthalmol 52:183, 1974
Balmer A, Munier F, Gailloud C: Retinoma. Case studies. Ophthalmic Paediatr
Genet 12:131, 1991
Boniuk M, Zimmerman LE: Spontaneous regression of retinoblastoma. Int Ophthalmol
Clin 2:525, 1962
Eagle RC Jr, Shields JA, Donoso L, Milner RS: Malignant transformation
of spontaneously regressed retinoblastoma, retinoma/retinocytoma variant. Ophthalmology 96:1389, 1989
Gallie BL, Ellsworth RM, Abramson DH et al: Retinoma: spontaneous regression
of retinoblastoma or benign manifestation of the mutation? Br J
Cancer 45:513, 1982
Gallie BL, Phillips RA, Ellsworth RM, Abramson DH: Significance of retinoma
and phthisis bulbi for retinoblastoma. Ophthalmology 89:1393, 1982
Gangwar DN, Jain IS, Gupta A et al: Bilateral spontaneous regression of
retinoblastoma with dominant transmission. Ann Ophthalmol 14:479, 1982
Margo C, Hidayat A, Kopelman J, Zimmerman LE: Retinocytoma. A benign variant
of retinoblastoma. Arch Ophthalmol 101:1519, 1983
Smith JLS: Histology and spontaneous regression of retinoblastoma. Trans
Ophthalmol Soc UK 94:953, 1974
HISTOLOGY AND ULTRASTRUCTURE
Albert DM, Craft J, Sang DN: Ultrastructure of retinoblastomas: transmission
and scanning electron microscopy. In Jakobiec FA (ed): Ocular and
Adnexal Tumors, pp 157–171. Birmingham, AL, Aesculapius, 1978
Bunt AH, Tso MO: Feulgen-positive deposits in retinoblastoma. Incidence, composition, and
ultrastructure. Arch Ophthalmol 99:144, 1981
Burnier MN, McLean IW, Zimmerman LE, Rosenberg SH: Retinoblastoma. The
relationship of proliferating cells to blood vessels. Invest Ophthalmol
Vis Sci 31:2037, 1990
Datta BN: DNA coating of blood vessels in retinoblastomas. Am J Clin Pathol 62:94, 1974
Kyritsis AP, Tsokos M, Triche TJ, Chader GJ: Retinoblastoma: a primitive
tumor with multipotential characteristics. Invest Ophthalmol Vis Sci 27:1760, 1986
Loeffler KU, McMenamin PG: An ultrastructural study of DNA precipitation
in the anterior segment of eyes with retinoblastoma. Ophthalmology 94:1160, 1987
Mullaney J: Retinoblastomas with DNA precipitation. Arch Ophthalmol 82:454, 1969
Nork TM, Millecchia LL, de Venecia GB, Myers FL, Vogel KA: Immunocytochemical
features of retinoblastoma in an adult. Arch Ophthalmol 114:1402, 1996
Nork TM, Schwartz TL, Doshi HM, Millecchia LL: Retinoblastoma. Cell of
origin. Arch Ophthalmol 113:791, 1995
Pe'er J, Neufeld M, Baras M et al: Rubeosis iridis in retinoblastoma. Histologic
findings and the possible role of vascular endothelial growth
factor in its induction. Ophthalmology 104:1251, 1997
Popoff NA, Ellsworth RM: The fine structure of retinoblastoma: In vivo and in vitro observations. Lab Invest 25:389, 1971
Shields JA, Shields CL, Ehya H, Eagle RC Jr, De Potter P: Fine-needle aspiration
biopsy of suspected intraocular tumors. Ophthalmology 100:1677, 1993
Stowe GC, Zakov ZN, Albert DM et al: Vascular basophilia in ocular and
orbital tumors. Invest Ophthalmol Vis Sci 18:1068, 1979
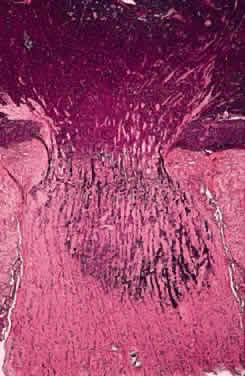
Ts'o MO, Fine BS, Zimmerman LE: The Flexner-Wintersteiner rosettes in retinoblastoma. Arch
Pathol 88:664, 1969
Ts'o MO, Fine BS, Zimmerman LE: The nature of retinoblastoma. II. Photoreceptor
differentiation: an electron microscopic study. Am J Ophthalmol 69:350, 1970
Ts'o MO, Zimmerman LE, Fine BS, Ellsworth RM: A cause of radioresistance
in retinoblastoma: photoreceptor differentiation. Trans Am Acad Ophthalmol
Otolaryngol 74:959, 1970
Ts'o MO, Zimmerman LE, Fine BS: The nature of retinoblastoma. I. Photoreceptor
differentiation: a clinical and histopathologic study. Am J Ophthalmol 69:339, 1970
Tsokos M, Kyritsis AP, Chader GJ, Triche TJ: Differentiation of human retinoblastoma in vitro into cell types with characteristics observed in embryonal or mature retina. Am
J Pathol 123:542, 1986
Verhoeff FH, Jackson E: Minutes of Proceedings, 62nd Annual Meeting. Trans
Am Ophthalmol Soc 24:38, 1926
Vrabec T, Arbizo V, Adamus G et al: Rod cell-specific antigens in retinoblastoma. Arch
Ophthalmol 107:1061, 1989
THE RETINOBLASTOMA GENE AND THE GENETICS OF RETINOBLASTOMA
Balaban G, Gilbert F, Nichols W et al: Abnormalities of chromosome 13 in
retinoblastoma from individuals with normal constitutional karyotypes. Cancer
Genet Cytogenet 6:213, 1982
Benedict WF, Murphree AL, Banerjee A et al: Patient with chromosome deletion: evidence
that the retinoblastoma gene is a recessive cancer gene. Science 219:973, 1983
Bookstein R, Lee WH: Molecular genetics of the retinoblastoma suppressor
gene. Crit Rev Oncog 2:211, 1991
Cavenee WK, Dryja TP, Phillips RA et al: Expression of recessive alleles
by chromosomal mechanisms in retinoblastoma. Nature (Lond) 305:779, 1983
Cavenee WK, Hansen MF, Norenskjold M et al: Genetic origin of mutations
predisposing to retinoblastoma. Science 228:501, 1985
Cavenee WK, Murphree AL, Shull MM et al: Prediction of familial predisposition
to retinoblastoma. N Engl J Med 314:1201, 1986
Dryja TP, Bruns GAP, Orkin SH et al: Isolation of DNA fragments from chromosome 13. Retina 3:121, 1983
Dryja TP, Cavenee W, White R et al: Homozygosity of chromosome 13 in retinoblastoma. N
Engl J Med 310:550, 1984
Dryja TP, Rapaport J, McGee TL, Nork TM, Schwartz TL: Molecular etiology
of low-penetrance retinoblastoma in two pedigrees. Am J Hum Genet 52:1122, 1993
Dryja TP, Rapaport JM, Weichselbaum R, Bruns GAP: Chromosome 13 restriction
fragment length polymorphisms. Hum Genet 65:320, 1984
Gallie BL, Dunn JM, Chan HS et al: The genetics of retinoblastoma: relevance
to the patient. Pediatr Clin North Am 38:299, 1991
Gallie BL, Phillips RA: Retinoblastoma: a model of oncogenesis. Ophthalmology 91:666, 1984
Godbout R, Dryja TP, Squire J et al: Somatic inactivation of genes on chromosome 13 is
a common event in retinoblastoma. Nature 304:451, 1983
Gordon H: Family studies in retinoblastoma. Birth Defects 10:185, 1974
Harbour JW: Overview of RB gene mutations in patients with retinoblastoma. Implications
for clinical genetic screening. Ophthalmology 105:1442, 1998
Knudson AG Jr, Hethcote HW, Brown BW: Mutation and childhood cancer: a
probabilistic model for the incidence of retinoblastoma. Proc Natl Acad
Sci USA 71:5116, 1975
Knudson AG Jr: Mutation and cancer: a statistical study of retinoblastoma. Proc
Natl Acad Sci USA 68:820, 1971
Knudson AG Jr: Retinoblastoma: a prototypic hereditary neoplasm. Semin
Oncol 5:57, 1978
Kratzke RA, Otterson GA, Hogg A et al: Partial inactivation of the RB product
in a family with incomplete penetrance of familial retinoblastoma
and benign retinal tumors. Oncogene 9:1321, 1994
McGee TL, Yandell DW, Dryja TP: Structure and partial genomic sequence
of the human retinoblastoma susceptibility gene. Gene 80:119, 1989
Murphree AL, Benedict WF: Retinoblastoma: clues to human oncogenesis. Science 223:1028, 1984
Murphree AL: Molecular genetics of retinoblastoma. Ophthalmol Clin North
Am 8:155, 1995
Nielson M, Goldschmidt E: Retinoblastoma among offspring of adult survivors
in Denmark. Acta Ophthalmol 46:736, 1968
Otterson GA, Chen W, Coxon AB, Khleif SN, Kaye FJ: Incomplete penetrance
of familial retinoblastoma linked to germ-line mutations that result
in partial loss of RB function. Proc Natl Acad Sci USA 94:12036, 1997
Shields CL, Shields JA, Donoso LA: Clinical genetics of retinoblastoma. Int
Ophthalmol Clin 33:67, 1993
Vogel F: Genetics of retinoblastoma. Hum Genet 52:1, 1979
Weichselbaum RR, Zakov ZN, Albert DM et al: New findings in the chromosome 13 long-arm
deletion syndrome and retinoblastoma. Ophthalmology 86:1191, 1979
Weinberg RA: The retinoblastoma gene and gene product. Cancer Surv 12:43, 1992
Yunis JJ, Ramsay N: Retinoblastoma and subband deletion of chromosome 13. Am
J Dis Child 132:161, 1978
SECONDARY NONOCULAR TUMORS
Abramson DH, Ellsworth RM, Zimmerman LE: Nonocular cancer in retinoblastoma
survivors. Trans Am Acad Ophthalmol Otolaryngol 81:454, 1976
Bader JL, Meadows AT, Zimmerman LE et al: Bilateral retinoblastoma with
ectopic intracranial retinoblastoma: trilateral retinoblastoma. Cancer
Genet Cytogenet 5:203, 1982
Holladay DA, Holladay A, Montebello JF, Redmond KP: Clinical presentation, treatment, and
outcome of trilateral retinoblastoma. Cancer 67:710, 1991
Jakobiec FA, Tso MOM, Zimmerman LE et al: Retinoblastoma and intracranial
malignancy. Cancer 39:2048, 1977
Moll AC, Imhof SM, Bouter LM et al: Second primary tumors in patients with
retinoblastoma. A review of the literature. Ophthalm Genet 18:27, 1997
Pesin SR, Shields JA: Seven cases of trilateral retinoblastoma. Am J Ophthalmol 107:121, 1989
Roarty JD, McLean IW, Zimmerman LE: Incidence of second neoplasms in patients
with bilateral retinoblastoma. Ophthalmology 95:1583, 1988
Wong FL, Boice JD, Abramson DH et al: Cancer incidence after retinoblastoma: radiation
dose and sarcoma risk. JAMA 278:1262, 1997
LESIONS THAT SIMULATE RETINOBLASTOMA
General References
Apple DJ, Hamming NA, Gieser DK: Differential diagnosis of leukocoria. In
Peyman GA et al (eds): Intraocular Tumors, pp 235–283. New York, Appleton/Century/Crofts, 1977
Francois J: Differential diagnosis of leukokoria in children. Ann Ophthalmol 10:1375, 1978
Francois J: Neonatal or juvenile leucocoria. Ophthalmologica 179:129, 1979
Howard GM, Ellsworth RM: Differential diagnosis of retinoblastoma: a statistical
survey of 500 children: 1. Relative frequency of the lesions
which simulate retinoblastoma. Am J Ophthalmol 60:610, 1965
Kogan L, Boniuk M: Causes for enucleation in children with special reference
to pseudoglioma and unsuspected retinoblastoma. Int Ophthalmol Clin 2:507, 1962
Margo CE, Zimmerman LE: Retinoblastoma: the accuracy of clinical diagnosis
in children treated by enucleation. J Pediatr Ophthalmol Strabismus 20:227, 1983
Robertson DM, Campbell RJ: Analysis of misdiagnosed retinoblastoma in a
series of 726 enucleated eyes. Mod Probl Ophthalmol 18:156, 1977
Shields JA, Parsons HM, Shields CL, Shah P: Lesions simulating retinoblastoma. J
Pediatr Ophthalmol Strabismus 28:338, 1991
Shields JA, Shields CL: Differential diagnosis of retinoblastoma. In: Intraocular
Tumors: A Text and Atlas, pp 341–362. Philadelphia, WB
Saunders, 1992
Shields JA, Shields CL, Parsons HM: Differential diagnosis of retinoblastoma. Retina 11:232, 1991
Smirniotopoulos JG, Bargallo N, Mafee MF: Differential diagnosis of leukokoria: radiologic-pathologic correlation. Radiographics 14:1059, 1994
Toxocariasis
Ashton N: Larval granulomatosis of the retina due to toxocara. Br J Ophthalmol 44:129, 1960
Belmont JB, Irvine A, Benson W, O'Connor GR: Vitrectomy in ocular toxocariasis. Arch
Ophthalmol 100:1912, 1982
Benitez del Castillo JM, Herreros G, Guillen JL et al: Bilateral ocular
toxocariasis demonstrated by aqueous humor enzyme-linked immunosorbent
assay. Am J Ophthalmol 119:514, 1995
Brown DH: Ocular Toxocara canis. J Pediatr Ophthalmol 7:182, 1970
Dernouchamps JP, Verougstraete C, Demolder E: Ocular toxocariasis: a presumed
case of peripheral granuloma. Int Ophthalmol 14:383, 1990
Ellis GS Jr, Pakalnis VA, Worley G et al: Toxocara canis infestation. Clinical and epidemiological associations with seropositivity
in kindergarten children. Ophthalmology 93:1032, 1986
Kayes SG: Human toxocariasis and the visceral larva migrans syndrome: correlative
immunopathology. Chem Immunol 66:99, 1997
Luxenberg MN: An experimental approach to the study of intraocular Toxocara canis. Trans Am Ophthalmol Soc 77:542, 1979
Maguire AM, Green WR, Michels RG, Erozan YS: Recovery of intraocular Toxocara canis by pars plana vitrectomy. Ophthalmology 97:675, 1990
Rockey JH, Donnelly JJ, Stromberg BE, Laties AM, Soulsby EJ: Immunopathology
of ascarid infection of the eye. Role of IgE antibodies and mast
cells. Arch Ophthalmol 99:1831, 1981
Rockey JH, Donnelly JJ, Stromberg BE, Soulsby EJ: Immunopathology of Toxocara canis and Ascaris suum infections of the eye: the role of the eosinophil. Invest Ophthalmol Vis
Sci 18:1172, 1979
Rockey JH, John T, Donnelly JJ et al: In vitro interaction of eosinophils from ascarid-infected eyes with Ascaris suum and Toxocara canis larvae. Invest Ophthalmol Vis Sci 24:1346, 1983
Safar EH, Abd-el Ghaffar FM, Saffar SA et al: Incidence of Toxoplasma and
Toxocara antibodies among outpatients in the Ophthalmic Research Institute, Egypt. J
Egypt Soc Parasitol 25:839, 1995
Schantz PM, Weis PE, Pollard ZF, White MC: Risk factors for toxocaral ocular
larva migrans: a case-control study. Am J Public Health 70:1269, 1980
Searl SS, Moazed K, Albert DM, Marcus LC: Ocular toxocariasis presenting
as leukocoria in a patient with low ELISA titer to Toxocara canis. Ophthalmology 88:1302, 1981
Shields JA, Shields CL, Eagle RC Jr, Barrett J, De Potter P: Endogenous
endophthalmitis simulating retinoblastoma. Retina 15:213, 1995
Shields JA: Ocular toxocariasis. A review. Surv Ophthalmol 28:361, 1984
Watzke RC: Ocular Toxocara canis infection: clinical and experimental features. Trans New Orleans Acad
Ophthalmol 31:263, 1983
Wilder HC: Nematode endophthalmitis. Trans Am Acad Ophthalmol Otolaryngol 54:99, 1950
Zinkham WH: Visceral larva migrans. A review and reassessment indicating
two forms of clinical expression: visceral and ocular. Am J Dis Child 132:627, 1978
Persistent Hyperplastic Primary Vitreous (Persistent Fetal Vasculature)
Boeve MH, van der Linde-Sipman JS, Stades FC, Vrensen GF: Early morphogenesis
of persistent hyperplastic tunica vasculosa lentis and primary
vitreous. A transmission electron microscopic study. Invest Ophthalmol
Vis Sci 31:1886, 1990
Boeve MH, Vrensen GF, Willekens BL et al: Early morphogenesis of persistent
hyperplastic tunica vasculosa lentis and primary vitreous (PHTVL/PHPV). Scanning
electron microscopic observations. Graefes Arch Clin Exp
Ophthalmol 231:29, 1993
Doro S, Werblin TP, Haas B, Iwamoto T, Jakobiec FA: Fetal adenoma of the
pigmented ciliary epithelium associated with persistent hyperplastic
primary vitreous. Ophthalmology 93:1343, 1986
Font RL, Yanoff M, Zimmerman LE: Intraocular adipose tissue and persistent
hyperplastic primary vitreous. Arch Ophthalmol 82:43, 1969
Goldberg MF, Mafee M: Computed tomography for diagnosis of persistent hyperplastic
primary vitreous (PHPV). Ophthalmology 90:442, 1983
Goldberg MF: Persistent fetal vasculature (PFV): an integrated interpretation
of signs and symptoms associated with persistent hyperplastic primary
vitreous (PHPV). Am J Ophthalmol 124:587, 1997
Haddad R, Font RL, Reeser F: Persistent hyperplastic primary vitreous. A
clinicopathologic study of 62 cases and review of the literature. Surv
Ophthalmol 23:123, 1978
Irvine AR, Albert DM, Sang DN: Retinal neoplasia and dysplasia. II. Retinoblastoma
occurring with persistence and hyperplasia of the primary
vitreous. Invest Ophthalmol Vis Sci 16:403, 1977
Kaste SC, Jenkins JJ 3rd, Meyer D, Fontanesi J, Pratt CB: Persistent hyperplastic
primary vitreous of the eye: imaging findings with pathologic
correlation. AJR Am J Roentgenol 162:437, 1994
Mafee MF, Goldberg MF: Persistent hyperplastic primary vitreous (PHPV): role
of computed tomography and magnetic resonance. Radiol Clin North
Am 25:683, 1987
Mittra RA, Huynh LT, Ruttum MS et al: Visual outcomes following lensectomy
and vitrectomy for combined anterior and posterior persistent hyperplastic
primary vitreous. Arch Ophthalmol 116:1190, 1998
Pollard ZF: Persistent hyperplastic primary vitreous: diagnosis, treatment
and results. Trans Am Ophthalmol Soc 95:487, 1997
Reese AB, Payne F: Persistence and hyperplasia of the primary vitreous. Am
J Ophthalmol 29:1, 1964
Reese AB: Persistence and hyperplasia of the primary vitreous. Am J Ophthalmol 40:317, 1955
Spaulding AG, Naumann G: Persistent hyperplastic primary vitreous in an
adult. A brief review of the literature and a histopathologic study. Arch
Ophthalmol 77:666, 1967
Spitznas M, Koch F, Pohl S: Ultrastructural pathology of anterior persistent
hyperplastic primary vitreous [see comments]. Graefes
Arch Clin Exp Ophthalmol 228:487, 1990
Coats' Disease
Chang MM, McLean IW, Merritt JC: Coats' disease: a study of 62 histologically
confirmed cases. J Pediatr Ophthalmol Strabismus 21:163, 1984
Coats G: Forms of retinal disease with massive exudation. R London Ophthalmol
Hosp Rep 17:440, 1907/1908
Coats G: Uber retinitis exsudativa (Retinitis hemorrhagica externa). Albrecht
Von Graefes Arch Klin Exp Ophthalmol 81:275, 1912
Egbert PR, Chan CC, Winter FC: Flat preparations of the retinal vessels
in Coats' disease. J Pediatr Ophthalmol 13:336, 1976
Egerer I, Tasman W, Tomer TT: Coats disease. Arch Ophthalmol 92:109, 1974
Farkas TG, Potts AM, Boone C: Some pathologic and biochemical aspects of
Coats' disease. Am J Ophthalmol 75:289, 1973
Goel SD, Augsburger JJ: Hemorrhagic retinal macrocysts in advanced Coats
disease. Retina 11:437, 1991
Haik BG: Advanced Coats' disease. Trans Am Ophthalmol Soc 89:371, 1991
Henkind P, Morgan G: Peripheral retinal angioma with exudative retinopathy
in adults (Coats' lesion). Br J Ophthalmol 50:2, 1967
Jaffe MS, Shields JA, Canny CL, Eagle RC Jr, Fry RL: Retinoblastoma simulating
Coats' disease: a clinicopathologic report. Ann Ophthalmol 9:863, 1977
Kremer I, Nissenkorn I, Ben-Sira I: Cytologic and biochemical examination
of the subretinal fluid in diagnosis of Coats' disease. Acta Ophthalmol (Copenh) 67:342, 1989
Leber T: Uber eine durch Vorkommen multipler Miliaraneurysmen charakterisierte
Form von Retinaldegeneration. Arch Ophthalmol 81:1, 1912
Patel HK, Augsburger JJ, Eagle RC Jr: Unusual presentation of advanced
Coats' disease. J Pediatr Ophthalmol Strabismus 32:120, 1995
Reese AB: Telangiectasis of the retina and Coats' disease. Am J Ophthalmol 42:1, 1956
Ridley ME, Shields JA, Brown GC, Tasman W: Coats' disease. Evaluation of
management. Ophthalmology 89:1381, 1982
Shields JA, Eagle RC Jr, Fammartino J, Shields CL, De Potter P: Coats' disease
as a cause of anterior chamber cholesterolosis [letter]. Arch
Ophthalmol 113:975, 1995
Silodor SW, Augsburger JJ, Shields JA, Tasman W: Natural history and management
of advanced Coats' disease. Ophthalmic Surg 19:89, 1988
Steidl SM, Hirose T, Sang D, Hartnett ME: Difficulties in excluding the
diagnosis of retinoblastoma in cases of advanced Coats' disease: a clinicopathologic
report. Ophthalmologica 210:336, 1996
Stewart J, Halliwell T, Gupta RK: Cytodiagnosis of Coats' disease from
an ocular aspirate. A case report. Acta Cytol 37:717, 1993
Retinopathy of Prematurity
Ashton N, Graymore C, Pedler C: Studies on developing retinal vessels: V. Mechanism
of vasoobliteration. A preliminary report. Br J Ophthalmol 41:449, 1957
Brockhurst RJ, Chishti M: Cicatricial retrolental fibroplasia: its occurrence
without oxygen administration and in full-term infants. Albrecht
Von Graefes Arch Klin Exp Ophthalmol 195:113, 1975
Committee for the Classification of Retinopathy of Prematurity: An International
Classification of Retinopathy of Prematurity. Arch Ophthalmol 102:1130, 1984
de Juan E Jr, Machemer R, Flynn JT, Green WR: Surgical pathoanatomy in
stage 5 retinopathy of prematurity. Birth Defects Orig Artic Ser 24:281, 1988
Faris B, Tolentino F, Freeman HM et al: Retrolental fibroplasia in the
cicatricial stage. Arch Ophthalmol 85:661, 1971
Flower RW, Patz A: Oxygen studies in retrolental fibroplasia: IX. The effects
of elevated arterial oxygen tension on retinal vascular dynamics
in the kitten. Arch Ophthalmol 85:197, 1972
Flynn JT, O'Grady GE, Herrera J et al: Retrolental fibroplasia: 1. Clinical
observations. Arch Ophthalmol 95:217, 1977
Flynn JT: Acute proliferative retrolental fibroplasia: evolution of the
lesion. Albrecht Von Graefes Arch Klin Exp Ophthalmol 195:101, 1975
Foos RY: Acute retrolental fibroplasia. Albrecht Von Graefes Arch Klin
Exp Ophthalmol 195:87, 1975
Foos RY: Chronic retinopathy of prematurity. Ophthalmology 92:563, 1985
Green WR: Retrolental fibroplasia (RLF). In Spencer WH (ed): Ophthalmic
Pathology: An Atlas and Textbook, vol 2, 3rd ed., pp 647–651. Philadelphia, WB
Saunders, 1985
Hittner HM, Rhodes LM, McPherson AR: Anterior segment abnormalities in
cicatricial retinopathy of prematurity. Ophthalmology 86:803, 1979
Hittner HM, Rudolph AJ, Kretzer FL: Suppression of severe retinopathy of
prematurity with vitamin E supplementation: ultrastructural mechanism
of clinical efficacy. Ophthalmology 91:1512, 1984
Karlsberg RC, Green WR, Patz A: Congenital retrolental fibroplasia. Arch
Ophthalmol 89:122, 1973
Kinsey VE, Arnold HJ, Kalina RE et al: PaO2 levels and retrolental fibroplasia: a report of the cooperative study. Pediatrics 60:655, 1977
Kushner BJ, Essner D, Cohen IJ et al: Retrolental fibroplasia: pathologic
correlation. Arch Ophthalmol 95:29, 1977
Naiman J, Green WR, Patz A: Retrolental fibroplasia in hypoxic newborn. Am
J Ophthalmol 88:55, 1979
Patz A: Current status of role of oxygen in retrolental fibroplasia. Invest
Ophthalmol Vis Sci 15:337, 1976
Patz A: Retrolental fibroplasia survey. Surv Ophthalmol 14:1, 1969
Reese AB, Stepanik J: Cicatricial stage of retrolental fibroplasia. Am
J Ophthalmol 38:308, 1954
Stefani FH, Ehalt H: Nonoxygen-induced retinitis proliferans and retinal
detachment in full-term infants. Br J Ophthalmol 58:490, 1974
Tasman W: Retinal detachment in retrolental fibroplasia. Albrecht Von Graefes
Arch Klin Exp Ophthalmol 195:129, 1975
Tasman W: The natural history of active retinopathy of prematurity. Ophthalmology 91:1499, 1984
Tasman W: Vitreoretinal changes in cicatricial retrolental fibroplasia. Trans
Am Ophthalmol Soc 68:548, 1970
Incontinentia Pigmenti
Brown CA: Incontinentia pigmenti: the development of pseudoglioma. Br J
Ophthalmol 72:452, 1988
Catalano RA: Incontinentia pigmenti. Am J Ophthalmol 110:696, 1990
Fowell SM, Greenwald MJ, Prendiville JS, Jampol LM: Ocular findings of
incontinentia pigmenti in a male infant with Klinefelter syndrome. J Pediatr
Ophthalmol Strabismus 29:180, 1992
Francis JS, Sybert VP: Incontinentia pigmenti. Semin Cutan Med Surg 16:54, 1997
Goldberg MF, Custis PH: Retinal and other manifestations of incontinentia
pigmenti (Bloch-Sulzberger syndrome). Ophthalmology 100:1645, 1993
Goldberg MF: The blinding mechanisms of incontinentia pigmenti. Ophthalmic
Genet 15:69, 1994
Heathcote JG, Schoales BA, Willis NR: Incontinentia pigmenti (Bloch-Sulzberger
syndrome): a case report and review of the ocular pathological
features. Can J Ophthalmol 26:229, 1991
Mensheha-Manhart O, Rodrigues MM, Shields JA, Shannon GM, Mirabelli RP: Retinal
pigment epithelium in incontinentia pigmenti. Am J Ophthalmol 79:571, 1975
Rahi J, Hungerford J: Early diagnosis of the retinopathy of incontinentia
pigmenti: successful treatment by cryotherapy. Br J Ophthalmol 74:377, 1990
Rosenfeld SI, Smith ME: Ocular findings in incontinentia pigmenti. Ophthalmology 92:543, 1985
Shah GK, Summers CG, Walsh AW, Neely KA: Optic nerve neovascularization
in incontinentia pigmenti. Am J Ophthalmol 124:410, 1997
Soltau JB, Lueder GT: Bilateral macular lesions in incontinentia pigmenti. Bloch-Sulzberger
syndrome. Retina 16:38, 1996
Spallone A: Incontinentia pigmenti (Bloch-Sulzberger syndrome): seven case
reports from one family. Br J Ophthalmol 71:629, 1987
Wald KJ, Mehta MC, Katsumi O, Sabates NR, Hirose T: Retinal detachments
in incontinentia pigmenti. Arch Ophthalmol 111:614, 1993
Watzke RC, Stevens TS, Carney RG Jr: Retinal vascular changes of incontinentia
pigmenti. Arch Ophthalmol 94:743, 1976
Norrie's Disease and Associated Disorders
Apple DJ, Fishman GA, Goldberg MF: Ocular histopathology of Norrie's
disease. Am J Ophthalmol 78:196, 1974
Caballero M, Veske A, Rodriguez JJ et al: Two novel mutations in the Norrie
disease gene associated with the classical ocular phenotype. Ophthalmic
Genet 17:187, 1996
Chynn EW, Walton DS, Hahn LB, Dryja TP: Norrie disease. Diagnosis of a
simplex case by DNA analysis. Arch Ophthalmol 114:1136, 1996
Enyedi LB, de Juan E Jr, Gaitan A: Ultrastructural study of Norrie's
disease. Am J Ophthalmol 111:439, 1991
Holmes LB: Norrie's disease—an X-linked syndrome of retinal
malformation, mental retardation and deafness. N Engl J Med 284:367, 1971
Johnson K, Mintz-Hittner HA, Conley YP, Ferrell RE: X-linked exudative
vitreoretinopathy caused by an arginine to leucine substitution (R121L) in
the Norrie disease protein. Clin Genet 50:113, 1996
Mintz-Hittner HA, Ferrell RE, Sims KB et al: Peripheral retinopathy in
offspring of carriers of Norrie disease gene mutations. Possible transplacental
effect of abnormal norrin. Ophthalmology 103:2128, 1996
Parsons MA, Curtis D, Blank CE, Hughes HN, McCartney AC: The ocular pathology
of Norrie disease in a fetus of 11 weeks' gestational age. Graefes
Arch Clin Exp Ophthalmol 230:248, 1992
Perez-Vilar J, Hill RL: Norrie disease protein (norrin) forms disulfide-linked
oligomers associated with the extracellular matrix. J Biol Chem 272:33410, 1997
Schroeder B, Hesse L, Bruck W, Gal A: Histopathological and immunohistochemical
findings associated with a null mutation in the Norrie disease
gene. Ophthalmic Genet 18:71, 1997
Shastry BS, Pendergast SD, Hartzer MK et al: Identification of missense
mutations in the Norrie disease gene associated with advanced retinopathy
of prematurity. Arch Ophthalmol 115:651, 1997
Shastry BS: Identification of a recurrent missense mutation in the Norrie
disease gene associated with a simplex case of exudative vitreoretinopathy. Biochem
Biophys Res Commun 246:35, 1998
Townes PL, Roca PD: Norrie's disease (hereditary oculo-acoustic-cerebral
degeneration). Report of a United States family. Am J Ophthalmol 76:797, 1973
Warburg M: Norrie's disease—differential diagnosis and treatment. Acta
Ophthalmol (Copenh) 53:217, 1975
Wolff G, Mayerova A, Wienker TF et al: Clinical reinvestigation and linkage
analysis in the family with Episkopi blindness (Norrie disease). J
Med Genet 29:816, 1992
Retinal Dysplasia
Albert DM, Lahav M, Colby ED, Shadduck JA, Sang DN: Retinal neoplasia and
dysplasia. I. Induction by feline leukemia virus. Invest Ophthalmol
Vis Sci 16:325, 1977
Godel V, Nemet P, Lazar M: Retinal dysplasia. Doc Ophthalmol 51:277, 1981
Hoepner J, Yanoff M: Ocular anomalies in trisomy 13–15. Am J Ophthalmol 74:729, 1972
Lahav M, Albert DM, Wyand S: Clinical and histopathologic classification
of retinal dysplasia. Am J Ophthalmol 75:648, 1973
Lloyd IC, Colley A, Tullo AB, Bonshek R: Dominantly inherited unilateral
retinal dysplasia. Br J Ophthalmol 77:378, 1993
Silverstein AM, Osburn BI, Prendergast RA: The pathogenesis of retinal
dysplasia. Am J Ophthalmol 72:13, 1971
Medulloepithelioma
Brownstein S, Barsoum-Homsy M, Conway VH, Sales C, Condon G: Nonteratoid
medulloepithelioma of the ciliary body. Ophthalmology 91:1118, 1984
Broughton WL, Zimmerman LE: A clinicopathologic study of 56 cases of intraocular
medulloblastoma. Am J Ophthalmol 85:407, 1978
Canning CR, McCartney ACE, Hungerford JL: Medulloepithelioma (diktyoma). Br
J Ophthalmol 72:764, 1988
Carrillo R, Streeten BW: Malignant teratoid medulloepithelioma in an adult. Arch
Ophthalmol 97:695, 1979
Floyd BB, Minckler DS, Valentin L: Intraocular medulloepithelioma in a 79-year-old
man. Ophthalmology 89:1088, 1982
Green WR, Iliff WJ, Trotter RR: Malignant teratoid medulloepithelioma of
the optic nerve. Arch Ophthalmol 91:451, 1974
Hamburg A: Medulloepithelioma arising from the posterior pole. Ophthalmologica 181:152, 1980
Hausmann N, Stefani FH: Medulloepithelioma of the ciliary body. Acta Ophthalmol (Copenh) 69:398, 1991
Hennis HL, Saunders RA, Shields JA: Malignant teratoid medulloepithelioma
of the ciliary body. J Clin Neuroophthalmol 10:291, 1990
Husain SE, Husain N, Boniuk M, Font RL: Malignant nonteratoid medulloepithelioma
of the ciliary body in an adult. Ophthalmology 105:596, 1998
Jakobiec FA, Howard GM, Ellsworth RM, Rosen M: Electron microscopic diagnosis
of medulloepithelioma. Am J Ophthalmol 79:321, 1975
Kivela T, Tarkkanen A: Recurrent medulloepithelioma of the ciliary body. Immunohistochemical
characteristics. Ophthalmology 95:1565, 1988
Mullaney J: Primary malignant medulloepithelioma of the retinal stalk. Am
J Ophthalmol 77:499, 1974
O'Keefe M, Fulcher T, Kelly P, Lee W, Dudgeon J: Medulloepithelioma of
the optic nerve head. Arch Ophthalmol 115:1325, 1997
Pe'er J, Hidayat AA: Malignant teratoid medulloepithelioma manifesting
as a black epibulbar mass with expulsive hemorrhage. Arch Ophthalmol 102:1523, 1984
Shields JA, Eagle RC Jr, Shields CL et al: Fluorescein angiography and
ultrasonography of malignant intraocular medulloepithelioma. J Pediatr
Ophthalmol Strabismus 33:193, 1996
Shields JA, Eagle RC Jr, Shields CL, Potter PD: Congenital neoplasms of
the nonpigmented ciliary epithelium (medulloepithelioma). Ophthalmology 103:1998, 1996
Shields JA, Shields CL, Schwartz RL: Malignant teratoid medulloepithelioma
of the ciliary body simulating persistent hyperplastic primary vitreous. Am
J Ophthalmol 107:296, 1989
Vadmal M, Kahn E, Finger P, Teichberg S: Nonteratoid medulloepithelioma
of the retina with electron microscopic and immunohistochemical characterization. Pediatr
Pathol Lab Med 16:663, 1996
Zimmerman LE: Verhoeff's “terato-neuroma.” A critical
reappraisal in light of new observations and current concepts of embryonic
tumors. Am J Ophthalmol 72:1039, 1971
Retinal Astrocytoma and Astrocytic Hamartoma
Arnold AC, Hepler RS, Yee RW et al: Solitary retinal astrocytoma. Surv
Ophthalmol 30:173, 1985
Bornfeld N, Messmer EP, Theodossiadis G, Meyer-Schwickerath G, Wessing
A: Giant cell astrocytoma of the retina. Clinicopathologic report of a
case not associated with Bourneville's disease. Retina 7:183, 1987
de Juan E Jr, Green WR, Gupta PK, Baranano EC: Vitreous seeding by retinal
astrocytic hamartoma in a patient with tuberous sclerosis. Retina 4:100, 1984
Destro M, D'Amico DJ, Gragoudas ES et al: Retinal manifestations of neurofibromatosis. Diagnosis
and management. Arch Ophthalmol 109:662, 1991
Jakobiec FA, Brodie SE, Haik B, Iwamoto T: Giant cell astrocytoma of the
retina. A tumor of possible Mueller cell origin. Ophthalmology 90:1565, 1983
Berger B, Peyman GA, Juarez C et al: Massive retinal gliosis simulating
choroidal melanoma. Can J Ophthalmol 14:285, 1979
Margo CE, Barletta JP, Staman JA: Giant cell astrocytoma of the retina
in tuberous sclerosis. Retina 13:155, 1993
Mullaney PB, Jacquemin C, Abboud E, Karcioglu ZA: Tuberous sclerosis in
infancy. J Pediatr Ophthalmol Strabismus 34:372, 1997
Nork TM, Ghobrial MW, Peyman GA, Tso MO: Massive retinal gliosis. A reactive
proliferation of Muller cells. Arch Ophthalmol 104:1383, 1986
Robertson DM: Ophthalmic manifestations of tuberous sclerosis. Ann NY Acad
Sci 615:17, 1991
Sharma A, Ram J, Gupta A: Solitary retinal astrocytoma. Acta Ophthalmol (Copenh) 69:113, 1991
Shields JA, Shields CL, Ehya H et al: Atypical retinal astrocytic hamartoma
diagnosed by fine-needle biopsy [see comments]. Ophthalmology 103:949, 1996
Ulbright TM, Fulling KH, Helveston EM: Astrocytic tumors of the retina. Differentiation
of sporadic tumors from phakomatosis-associated tumors. Arch
Pathol Lab Med 108:160, 1984
Green WR: Bilateral Coats' disease. Massive gliosis of the retina. Arch
Ophthalmol 77:378, 1967
Yanoff M, Zimmerman LE, Davis RL: Massive gliosis of the retina. Int Ophthalmol
Clin 11:211, 1971
Other Rare Neoplasms
Freitag SK, Eagle RC Jr, Shields JA, Duker JS, Font RL: Melanogenic neuroectodermal
tumor of the retina (primary malignant melanoma of the retina). Arch
Ophthalmol 115:1581, 1997
Ghadially FN, Chisholm IA, Lalonde JM: Ultrastructure of an intraocular
lacrimal gland choristoma. J Submicrosc Cytol 18:189, 1986
Kivela T, Kauniskangas L, Miettinen P, Tarkkanen A: Glioneuroma associated
with colobomatous dysplasia of the anterior uvea and retina. A case
simulating medulloepithelioma. Ophthalmology 96:1799, 1989
Shields JA, Eagle RC Jr, Shields CL et al: Natural course and histopathologic
findings of lacrimal gland choristoma of the iris and ciliary body. Am
J Ophthalmol 119:219, 1995
Spencer WH, Jesberg DO: Glioneuroma (choristomatous malformation of the
optic cup margin). Arch Ophthalmol 89:387, 1973
Wilson ME, McClatchey SK, Zimmerman LE: Rhabdomyosarcoma of the ciliary
body. Ophthalmology 97:1484, 1990
Miscellaneous Lesions
Brown GC, Shields JA, Oglesby RG: Anterior polar cataracts associated with
bilateral retinoblastoma. Am J Ophthalmol 87:276, 1979
Chang MW, Frieden IJ, Good W: The risk intraocular juvenile xanthogranuloma: survey
of current practices and assessment of risk. J Am Acad Dermatol 34:445, 1996
Friendly DS, Parks MM: Concurrence of hereditary congenital cataract and
hereditary retinoblastoma. Arch Ophthalmol 84:525, 1970
Leonardy NJ, Rupani M, Dent G, Klintworth GK: Analysis of 135 eyes for
ocular involvement in leukemia. Am J Ophthalmol 109:436, 1990
Shields JA, Eagle RC Jr, Shields CL et al: Iris juvenile xanthogranuloma
studied by immunohistochemistry and flow cytometry. Ophthalmic Surg
Lasers 28:140, 1997
Zimmerman LE: Ocular lesions of juvenile xanthogranuloma, nevoxanthoendothelioma. Trans
Am Acad Ophthalmol Otolaryngol 69:412, 1965
Treatment of Retinoblastoma
Baumal CR, Shields CL, Shields JA, Tasman WS: Surgical repair of rhegmatogenous
retinal detachment after treatment for retinoblastoma. Ophthalmology 105:2134, 1998
Doz F, Khelfaoui F, Mosseri V et al: The role of chemotherapy in orbital
involvement of retinoblastoma: the experience of a single institution
with 33 patients. Cancer 74:722, 1994
Doz F, Neuenschwander S, Plantaz D et al: Etoposide and carboplatin in
extraocular retinoblastoma: a study by the Societe Francaise d'Oncologie
Pediatrique. J Clin Oncol 13:902, 1995
Freire J, Miyamoto C, Brady LW et al: Retinoblastoma after chemoreduction
and irradiation: preliminary results. Front Radiat Ther Oncol 30:88, 1997
Gunduz K, Shields CL, Shields JA et al: The outcome of chemoreduction treatment
in patients with Reese-Ellsworth group V retinoblastoma. Arch
Ophthalmol 116:1613, 1998
Hernandez JC, Brady LW, Shields JA et al: External beam radiation for retinoblastoma: results, patterns of failure, and a proposal for treatment
guidelines. Int J Radiat Oncol Biol Phys 35:125, 1996
Pratt CB, Fontanesi J, Chenaille P et al: Chemotherapy for extraocular
retinoblastoma. Pediatr Hematol Oncol 11:301, 1994
Shields CL, De Potter P, Himelstein BP et al: Chemoreduction in the initial
management of intraocular retinoblastoma. Arch Ophthalmol 114:1330, 1996
Shields CL, Shields JA, De Potter P et al: Plaque radiotherapy in the management
of retinoblastoma. Use as a primary and secondary treatment [see
comments]. Ophthalmology 100:216, 1993
Shields CL, Shields JA, DePotter P, Himelstein BP, Meadows AT: The effect
of chemoreduction on retinoblastoma-induced retinal detachment. J Pediatr
Ophthalmol Strabismus 34:165, 1997
Shields CL, Shields JA, Needle M et al: Combined chemoreduction and adjuvant
treatment for intraocular retinoblastoma [see comments]. Ophthalmology 104:2101, 1997
Shields JA, Shields CL, De Potter P, Needle M: Bilateral macular retinoblastoma
managed by chemoreduction and chemothermotherapy. Arch Ophthalmol 114:1426, 1996
Shields JA, Shields CL, De Potter P: Cryotherapy for retinoblastoma. Int
Ophthalmol Clin 33:101, 1993
Shields JA, Shields CL, De Potter P: Enucleation technique for children
with retinoblastoma. J Pediatr Ophthalmol Strabismus 29:213, 1992
Shields JA, Shields CL, De Potter P: Photocoagulation of retinoblastoma. Int
Ophthalmol Clin 33:95, 1993
Shields JA, Shields CL: Current management of retinoblastoma. Mayo Clin
Proc 69:50, 1994
|