NEVI
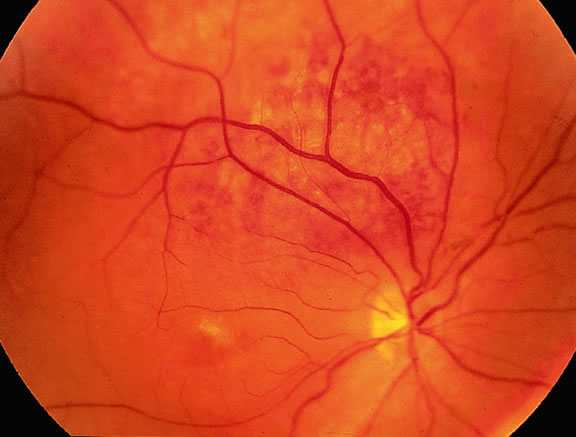
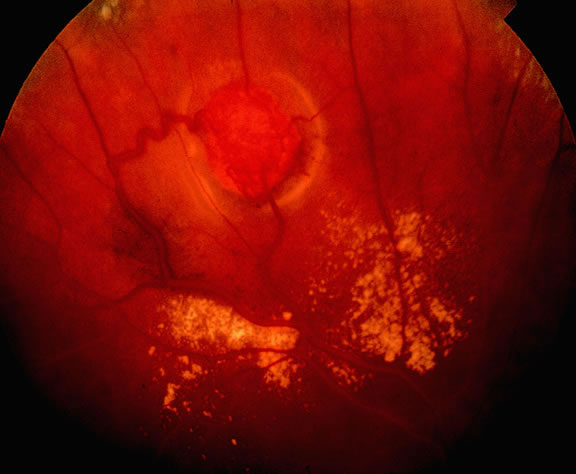
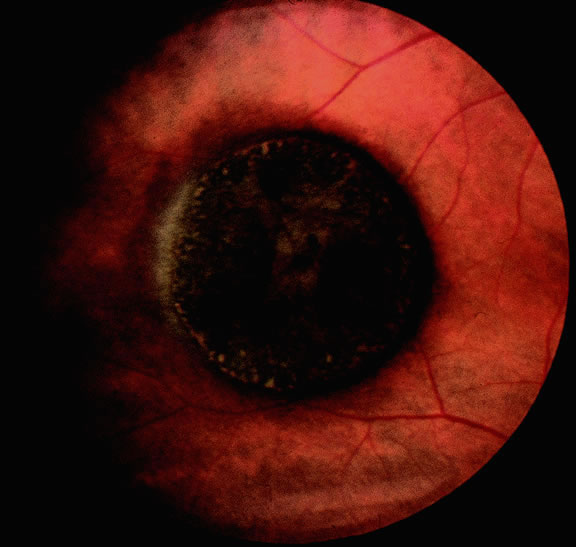
Uveal nevi are benign neoplasms derived from uveal melanocytes; 90% are located in the choroid posterior to the equator.2 Like nevi of the skin, uveal nevi are quite common; it has been estimated that they occur in more than 6% to 10% of the population.3,4 Nevi are often detected during routine fundus examinations (Fig. 1). Most choroidal nevi are flat or minimally elevated lesions that usually measure between 1.5 to 5 mm in diameter; most are less than 2 mm in thickness. Nevi may be pigmented or amelanotic, and they often have an irregular or jagged border. Drusen are quite common on the surface of nevi. Most nevi are stationary lesions that do not change on serial observation. Nevi have been observed to give rise to malignant melanomas, but this is a rare phenomenon. If one assumes the prevalence of uveal nevi to be 5% per year and the incidence of melanoma to be 7 per million per year, it can be estimated that the rate of transformation of nevi to melanoma is only 1 per 10,000 to 15,000 per year.5 Suspicious clinical features that suggest that a nevus actually may be a small melanoma that is likely to grow include the presence of orange pigment and subretinal fluid.6,7 It may be impossible to clinically differentiate large choroidal nevi from small malignant melanomas. The observation of growth is the only clinical criterion that is helpful.8
|
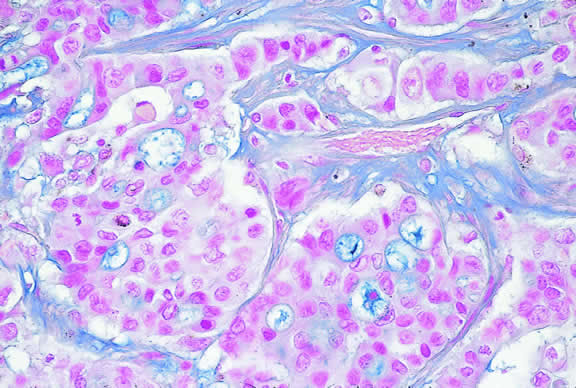
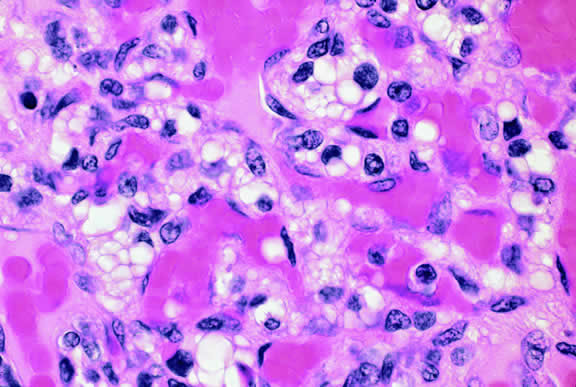
Nevi constitute the benign end of the biologic spectrum of melanocytic neoplasms. Most nevi are bland spindle cells tumors. Histopathologically, the choroidal stroma typically is replaced by a compact infiltrate of slender pigmented or nonpigmented spindle cells (Fig. 2). The nevus cells have bland oval or cigar-shaped nuclei that have finely dispersed chromatin and lack nucleoli or nuclear folds. Intranuclear cytoplasmic inclusions are common in some cases. Mitotic activity is absent. In some cases, the nevus cells are plump and dendritic in shape.9 Foamy balloon cells that appear to be undergoing lipoidal degeneration are found in 4% of nevi.4 Maximally pigmented, plump, polyhedral nevus cells comprise the magnocellular variant of nevus called melanocytoma (see later).
|
Most pigmented tumors of the iris are low-grade spindle cell nevi that remain stationary and do not enlarge on follow-up (Fig. 3). The iris freckle is a small, stationary form of iris nevus that is found in half of the adult population.1
|
MELANOCYTOMA
A melanocytoma is a characteristic type of uveal nevus that is composed of plump, polyhedral nevus cells filled with copious quantities of maximally pigmented cytoplasm.10,11 Melanocytomas also are called magnocellular nevi.12 In contrast to most nevi, melanocytoma may be relatively large and may be difficult to distinguish clinically from melanoma.
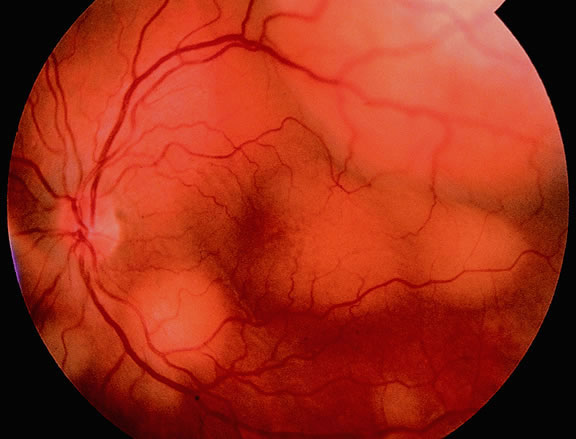
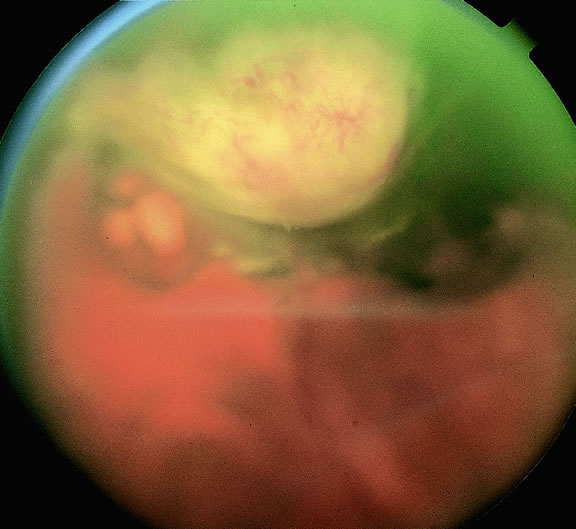
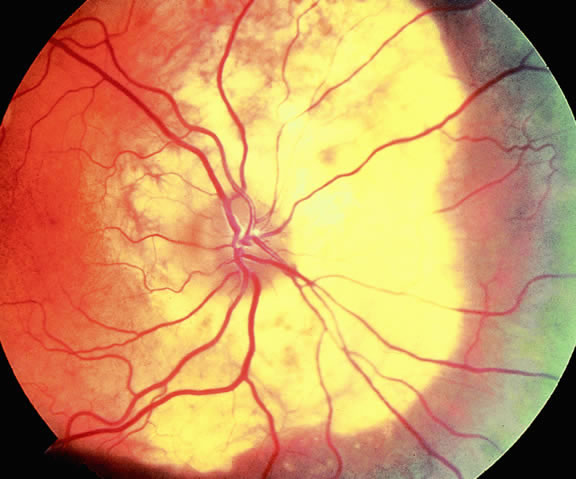
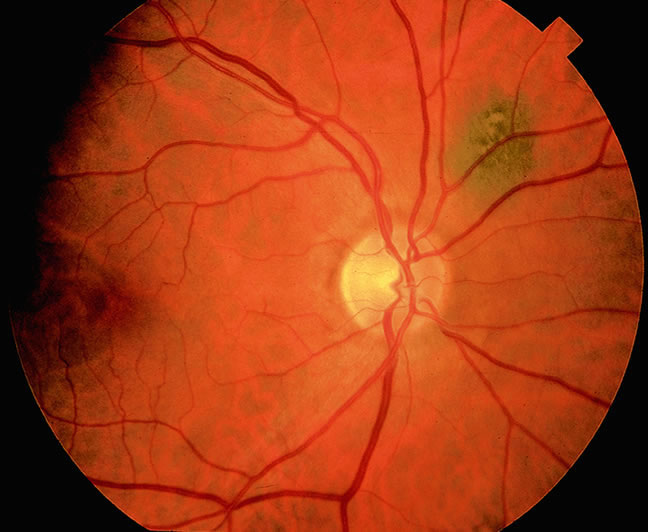
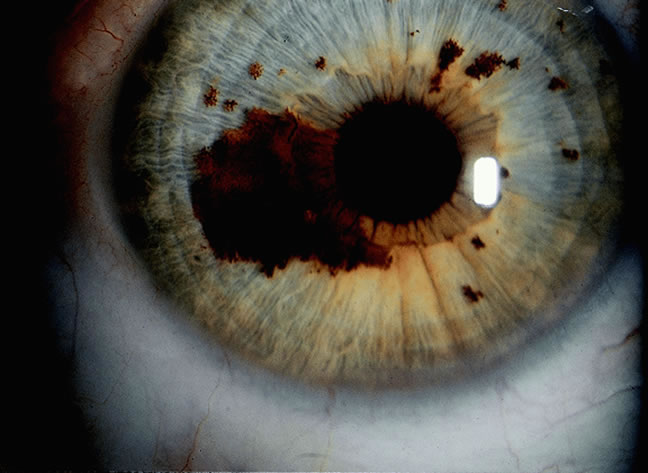
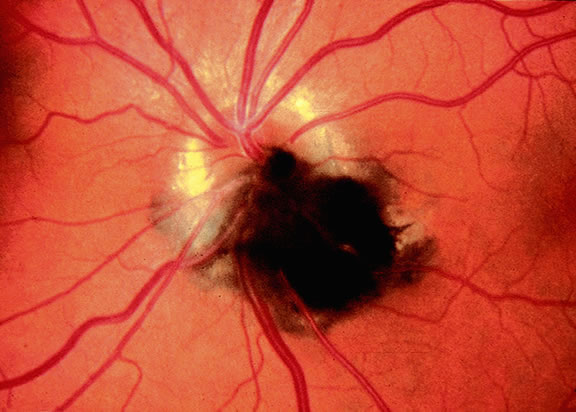
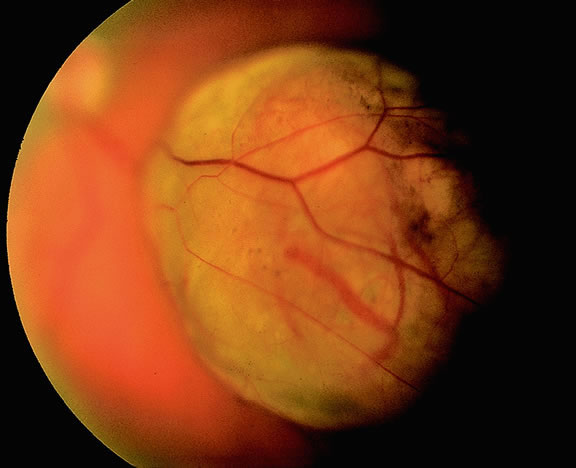
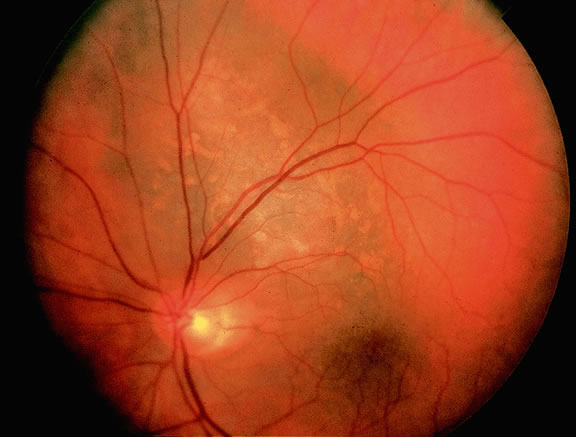
Although best know as a pigmented lesion of the optic disc (Fig. 4), melanocytoma can arise from the stroma of the iris, ciliary body, or choroid13, 14, 15 (Fig. 5). Clinically, melanocytomas are intensely pigmented and often occur in young patients. Unlike uveal melanoma, they do not have a predilection for Caucasians; 37% to 50% of optic disc melanocytomas have been reported in African Americans.10,16 Melanocytomas of the iris are uniformly dark brown in color and often have an irregular friable surface.
|
|
Most melanocytomas of the optic disc have a feathery margin caused by infiltration of the nerve fiber layer. In approximately one half of cases, a juxtapapillary component in the choroid identical to a choroidal nevus is present.16 Visual acuity is often good, but 30% of patients have an afferent pupillary defect.17 Visual field examination may show enlargement of the blind spot or nerve fiber bundle defects.17 About 15% of optic disc melanocytomas enlarge on follow-up.16 Rare cases of malignant transformation have been reported.18, 19, 20 Central retinal vein occlusion has been reported in a patient with a necrotic optic disc melanocytoma and does not always imply malignant transformation.21 Primary malignant melanomas of the optic nerve head have been reported.22 Retinal pigment epithelial (RPE) adenoma may mimic optic disc melanocytoma.23
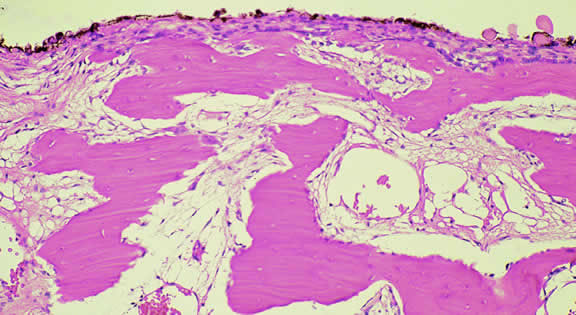
Histopathologically, melanocytomas are composed of cells that have abundant quantities of maximally pigmented cytoplasm that often obscures nuclear details, making microscopic assessment impossible11 (see Fig. 5). Bleached sections, which are a requisite for examination, show that the cells have bland nuclei and a low nuclear-to-cytoplasmic ratio (Fig. 6). Nucleoli usually are inconspicuous, but there are exceptions to the rule. Electron microscopic studies also have revealed a second population of spindle cells.15,24 Melanocytoma cells resemble the benign melanocytes found in the uvea of patients with ocular melanocytosis.
|
Melanocytomas are predisposed to undergo spontaneous necrosis.14 One is much more likely to find extensive tumor necrosis in a melanocytoma than a melanoma of comparable size, and totally necrotic melanocytomas occasionally are encountered. Numerous plump melanophages typically are found when necrosis is present. It may be difficult to differentiate the melanophages from magnocellular nevus cells in cytologic preparations.
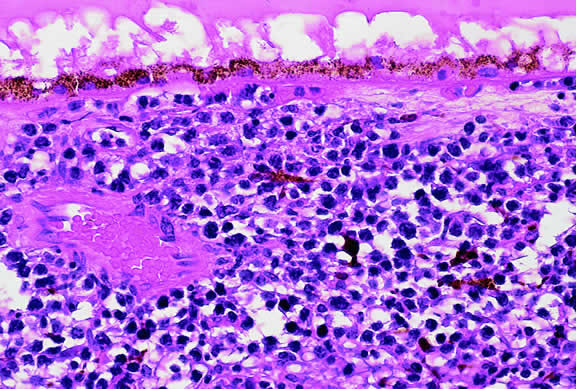
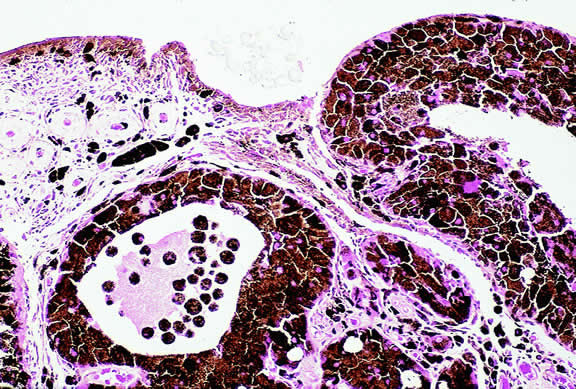
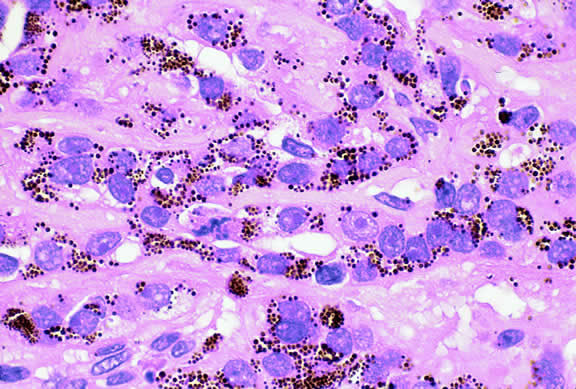
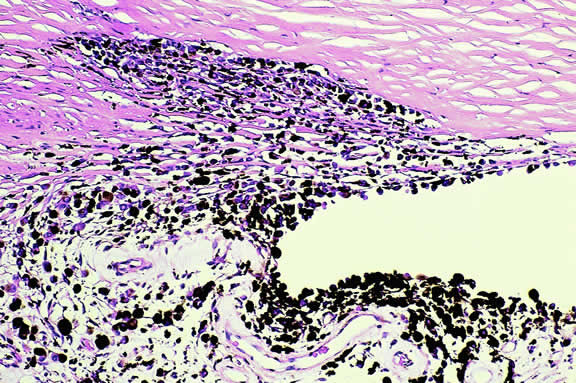
A rare type of secondary glaucoma called melanocytomalytic glaucoma may develop in patients who have partially necrotic melanocytomas of the iris and ciliary body.25, 26, 27 In such cases, the open angle is obstructed by macrophages that have ingested melanin pigment released by the necrotic tumor (Fig. 7).
|
UVEAL MELANOMA
On a worldwide basis, uveal melanoma is the most common primary intraocular tumor in adults. In the United States and Europe, uveal melanoma is also the most common primary intraocular tumor. In most of the rest of the world, the most common intraocular tumor is the pediatric retinal neoplasm retinoblastoma.28
Uveal melanoma is rare, even among persons of European ancestry. Less than 2000 cases occur yearly in the United States where the incidence is estimated to be 7 cases per million per year, less than 10% the incidence of lung cancer.29
Although pediatric and even rare congenital cases30,31 have been reported, uveal melanoma generally occurs in older persons. The mean age of patients eligible for treatment in the Collaborative Ocular Melanoma Study (COMS) I-125 brachytherapy study was 59 years.32 Less than 0.8% of cases occur in patients younger than age 20 years.33 In general, patients who are older tend to have larger tumors and are more likely to die from disseminated melanoma after enucleation. Recent data from COMS indicates that uveal melanoma affects both sexes equally.32
Predisposing Factors
Race is an important predisposing factor. Uveal malignant melanoma has a definite predilection for Europeans who have blue or lightly pigmented eyes.34, 35, 36 In a recent study in Florida, the risk of uveal melanoma in non-Hispanic white men was 72 times that in black men.29 Uveal melanoma also is less common in Latin and Native Americans29,37 and Asians. The incidence of uveal melanoma in the United States is 25 times greater than that in Taiwan, that is, 7 per million versus 0.28 per million.38 The prognosis of melanoma in black and white patients is similar. Tumors in black patients typically show more necrosis and heavy pigmentation.39
Caucasian patients who have congenital ocular or oculodermal melanocytosis (nevus of Ota) are especially at risk to develop uveal malignant melanoma.40 It has been estimated that about one in 400 white patients with oculodermal melanocytosis will develop uveal melanoma in his or her lifetime.41 This risk is about 25 times greater than the risk in unaffected patients. Melanomas have developed in young patients with Ota's nevus.33,42–44 Ocular and oculodermal melanocytosis are relatively common in Asians45 but do not appear to predispose them to melanoma. Several African Americans with nevus of Ota who developed uveal melanoma have been reported.46,47
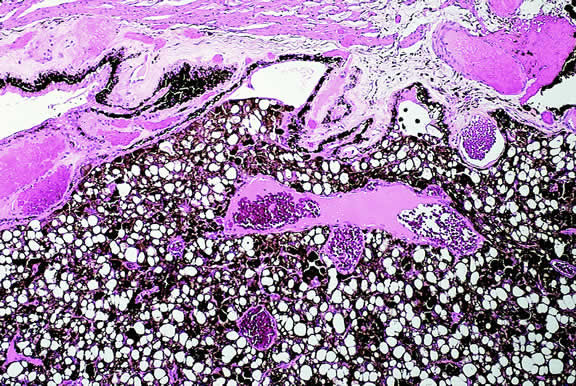
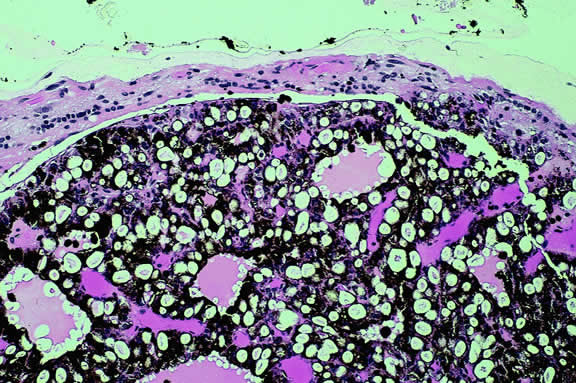
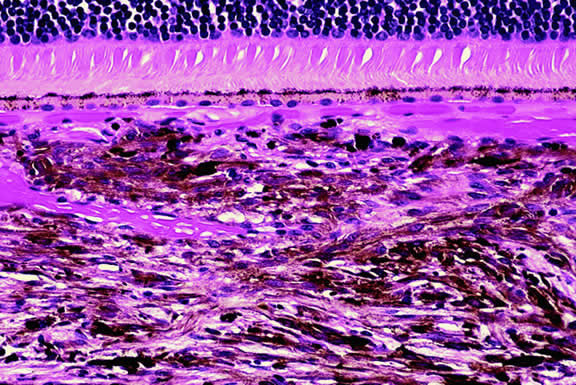
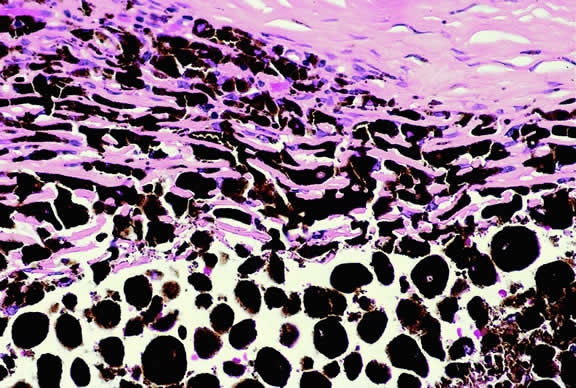
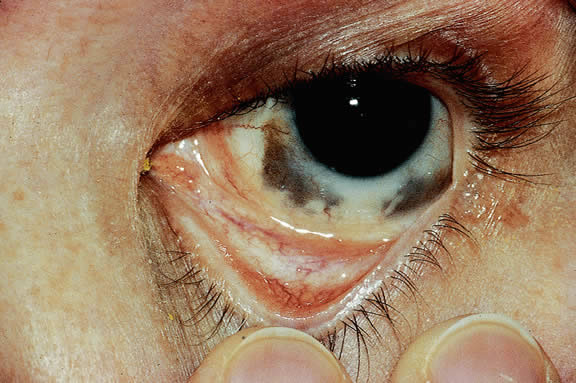
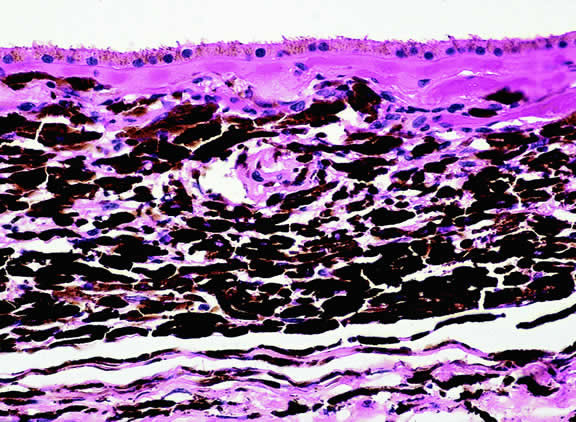
The substrate for the development of melanoma in white patients with ocular melanocytosis is a diffuse nevus that affects all, or part, of the uveal tract. Clinically, the nevus typically is evident as hyperchromic heterochromia iridum, patchy slate gray epibulbar pigmentation, and a darker aspect of the fundus compared with the fellow eye (Fig. 8). A sector of uvea is affected in some patients, however, and the nevus may spare the iris. The uvea in congenital melanocytosis is thickened by an increased number of heavily pigmented nevus cells similar to those found in melanocytomas (Fig. 9). RPE abnormalities such as drusen often develop on the surface of the thickened choroid.48 The sclera also contains patchy foci of pigmentation. The slate gray conjunctival pigment reflects the presence of dendritic melanocytes on the epibulbar tissues deep to the conjunctiva. Nevus of Ota does not predispose to conjunctiva melanoma. However, patients rarely may develop melanomas of the orbit49–51 and even leptomeninges.1,52,53 White patients who have congenital ocular or oculodermal melanocytosis should be followed periodically because of the increased risk of uveal melanoma.54
|
|
Uveal melanoma occasionally arises from localized uveal nevi, but the estimated incidence of malignant transformation is quite low. Nevus-like cells observed histopathologically at the base of many uveal melanomas probably are a compression artifact and do no represent a pre-existing nevus.55 Cases of uveal melanoma have been reported in patients with von Recklinghausen's neurofibromatosis type I42,56–59 and the dysplastic nevus or familial atypical mole-melanoma syndrome.59–63 The tumor's propensity for light-eyed individuals and the exposed inferior part of the iris suggests that exposure to ultraviolet light could be a predisposing factor.35,36 “Conjugal melanoma,” that is, the occurrence of tumors in both husband and wife, may be coincidental but could be indicative of the role of shared environmental factors.64 An increased incidence of uveal melanoma has been reported in a group of chemical workers.65
Uveal melanoma can arise in patients who have a rare paraneoplastic syndrome called benign diffuse uveal melanocytic proliferation (BDUMP) syndrome.66–69 Unknown factors produced by a nonocular cancer stimulate diffuse proliferation of benign appearing nevoid or spindle cells throughout the uveal tract of both eyes. Often occult, the primary tumor is typically a poorly differentiated ovarian or uterine carcinoma in women or lung carcinoma in men. Patients typically present with rapid visual loss and usually have cataracts. Fundus examination shows multiple pigmented and nonpigmented placoid nodules and reddish patches in the choroid, which show striking early hyperfluorescence during intravenous fluorescein angiography (IVFA), as well as serous retinal detachment.67 One patient presented with nonocular pigmentation typical of the Peutz-Jeghers syndrome.68
Clinical Features
CLINICAL PRESENTATION.
The clinical signs and symptoms of uveal malignant melanoma depend largely on the location of the tumor and the extent of the disease when the patient initially seeks medical attention. More than a century ago, Fuchs70 described four characteristic stages in the natural history of uveal melanoma. These include a relatively asymptomatic stage; a symptomatic stage of glaucoma and/or inflammation; a stage of extraocular growth; and finally, distant metastasis. Today, most patients are encountered in the early stages of the disease. Although metastatic disease ensues in entirely too many patients regardless of therapy, metastatic disease is almost never evident when the patient initially presents or is treated.71
Totally asymptomatic tumors occasionally are detected on a routine ophthalmoscopic examination. However, most choroidal melanomas present with painless visual loss. In most cases, visual loss is caused by serous or solid detachment of the retina. In rare instances the pathway of light to the fovea is physically blocked by the mushrooming apex of an actively growing, anteriorly located tumor. Ciliary body melanomas often do not produce retinal detachment and, therefore, may remain relatively asymptomatic for some time. Ciliary body melanomas cause loss of vision by directly encroaching on the visual axis or by impinging on the lens causing visually significant distortion, subluxation, or secondary cataract formation. Peripheral melanomas also can cause decreased vision by inducing cystoid macular edema. Dilated episcleral and conjunctival vessels called sentinel vessels often mark the site of an underlying ciliary body tumor.
Vitreous hemorrhage is an extremely rare cause of visual loss in uveal melanoma because the retina serves as a barrier that prevents the blood from entering the vitreous. Retinal perforation is a requisite for vitreous hemorrhage. This occurs most often when highly elevated tumors are situated near the optic disc or ora serrata where the retina is attached. Perforation of the retina also can lead to extensive seeding of the vitreous and the inner surface of the retina.72 In rare instances, malignant epiretinal membranes of seeded tumor cells can produce a pseudoretinitis pigmentosa-like picture.73
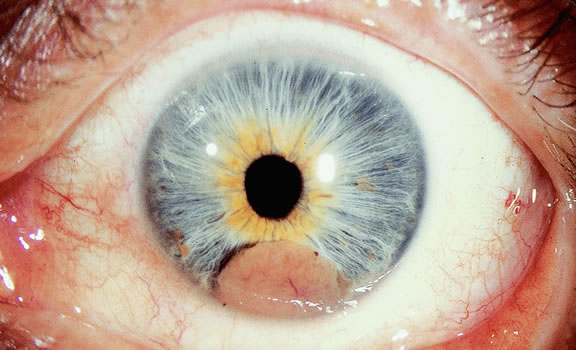
The ready visibility of many iris melanomas leads to their diagnosis at an earlier age and a smaller size (Fig. 10). Iris melanoma typically is discovered when a patient or his or her physician detects a focal pigmented blemish. Higher grade iris melanomas, or ciliary body tumors that invade the anterior chamber, can seed the iris and anterior chamber causing acquired hyperchromic heterochromia iridum (Fig. 11). Secondary glaucoma often accompanies the change in iris color. Not infrequently, the underlying diagnosis may be overlooked and the patient is treated for glaucoma, especially when a melanoma is nonpigmented or confined to the periphery of the iris or the trabecular meshwork.
|
|
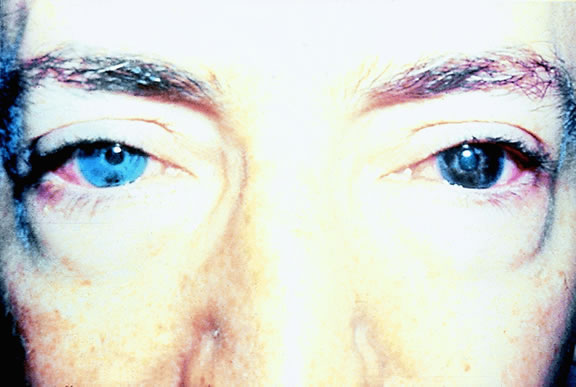
Most eyes with untreated uveal melanoma eventually develop glaucoma.74,75 Anterior tumors, that is, high-grade iris melanomas or ciliary body melanomas that invade the anterior chamber, typically cause secondary open-angle glaucoma by seeding or directly infiltrating the trabecular meshwork (Fig. 12). The latter often occurs in a circumferential or ring fashion. Posterior melanomas generally cause secondary angle closure glaucoma by stimulating iris neovascularization or by causing pupillary block. The pupil is blocked when large tumors or tumors that have caused highly elevated total retinal detachments displace the lens iris diaphragm forward (Fig. 13). The same mechanism also occurs in eyes with other neoplasms that typically are associated with high bullous retinal detachments such as uveal metastases, choroidal hemangiomas, and exophytic retinoblastomas. In such cases, severe pain often prompts a patient with a blind eye to seek medical care. More than a century ago, it initially was emphasized that a glaucomatous eye with a total retinal detachment harbors an intraocular tumor, typically a melanoma, until proven otherwise. Imaging studies such as ultrasonography are mandatory before therapy in such cases, especially if evisceration of a blind painful eye is contemplated instead of enucleation.
|
|
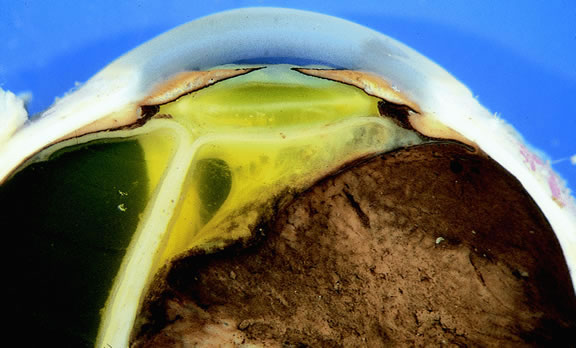
Rarely, patients with uveal melanoma may present with extraocular extension of their tumors.48,76,77 Epibulbar foci from ciliary body melanomas that have grown out of the eye through anterior emissarial canals or Axenfeld nerve loops can be confused clinically with conjunctival melanomas, but they are not attached to the conjunctival epithelium (Fig. 14). Although some posterior melanomas infiltrate directly through the sclera, transscleral extension typically occurs along the emissarial canals of vessels or nerves (Fig. 15). Rarely a posterior nodule of extrascleral extension develops that is much larger than the primary intraocular part of the tumor that spawned it.48,77,78 Such patients may present with ocular proptosis. In exceptional circumstances, patients with chronically neglected tumors, which have filled and destroyed the eye, will present with an orbital mass.
|
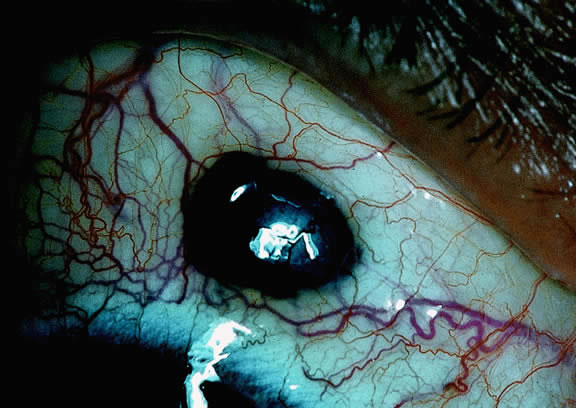
|
Necrotic melanomas may present with severe orbital and adnexal inflammation that can mimic infectious cellulitis or idiopathic orbital inflammation.48 Extensive necrosis typically is found in eyes with rapidly growing high-grade tumors. Marked elevation of intraocular pressure in eyes with secondary glaucoma may cause infarction of the tumor and other intraocular structures.
Unsuspected melanomas occasionally are discovered when blind painful eyes with opaque media are examined pathologically. Before the advent of ultrasonography as many as 10% of blind painful eyes were said to harbor previously undiagnosed tumors.79 The possibility of a tumor should always be entertained when cataract is unilateral or asymmetric. It is quite embarrassing to diagnose a melanoma during cataract surgery.
CLINICAL SIGNS.
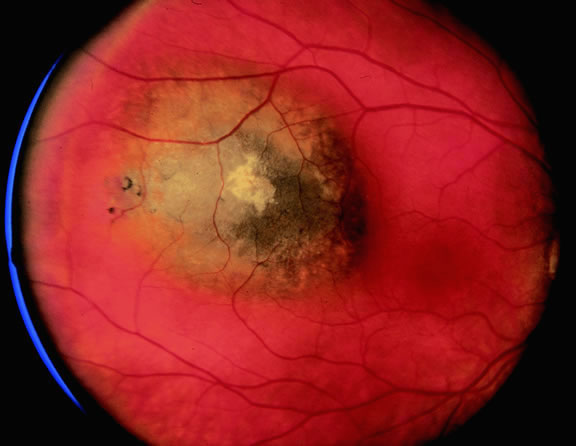
Ophthalmoscopic examination of a choroidal melanoma typically reveals a sessile or dome-shaped pigmented mass located deep to the retina (Figs. 16, 17, and 18). There often is an associated secondary nonrhegmatogenous serous detachment of the retina. Orange pigment may be located on the surface of smaller lesions.80,81 Tumors vary in pigment content and may be heavily pigmented or totally amelanotic. The differentiation of a totally amelanotic melanoma from uveal metastasis, hemangioma, lymphoma, or osteoma may be difficult. Compared with metastasis, melanoma usually is more highly elevated and drusen and RPE proliferation more evident on its surface. Ophthalmoscopy also typically shows well-defined vessels within melanoma.48 Ancillary studies such as fluorescein angiography and ultrasonography may be necessary to make the diagnosis.
|
|
|

Many choroidal melanomas have a characteristic mushroom or collar button configuration that results when the tumor breaks through Bruch's membrane (Figs. 19, 20, 21, and 22). In such cases there is often secondary choroidal, subretinal, or vitreous hemorrhage. Dilated, intrinsic vessels often are evident in the mushrooming dome of nonpigmented melanomas. The vessels are obscured in more pigmented tumors.48
|
|
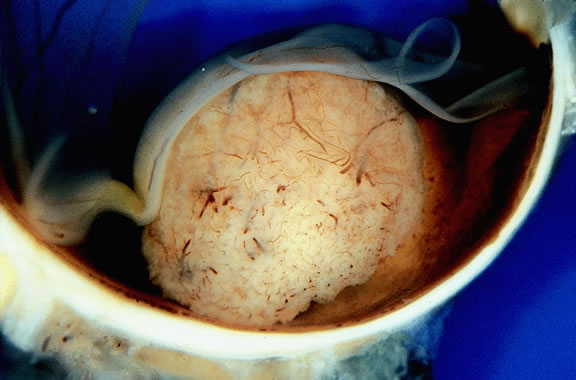
|
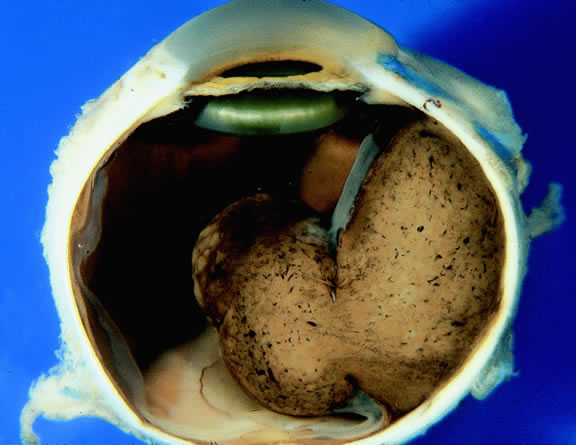
|
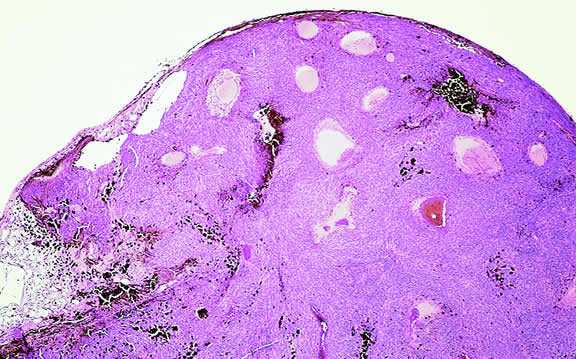
Although the diagnosis of melanoma often can be made by careful indirect ophthalmoscopy, ancillary studies such as intravenous fluorescein angiography, indocyanine green angiography, ultrasonography, computed tomography, magnetic resonance imaging, the radioactive phosphorus uptake test, and fine-needle aspiration biopsy (FNAB) occasionally are required to support or confirm the diagnosis.48,54
The characteristic fluorangiographic features of uveal melanoma include mottled hyperfluorescence in the vascular filling phases and diffuse late staining of the mass and surrounding subretinal fluid. A characteristic double circulation in which both the retinal vessels and the choroidal vessels in the tumor are readily evident may be shown by larger amelanotic tumors, particularly those that have perforated Bruch's membrane. The prominent blood vessels seen in amelanotic mushroom-shaped melanomas may impart a pseudoangiomatous appearance to the lesion.48
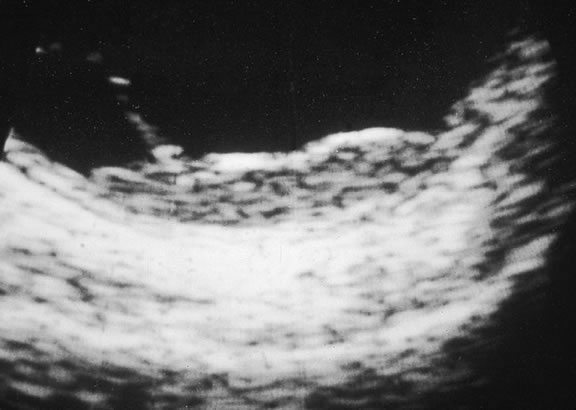
Ultrasonography can support the diagnosis of ciliary body or choroidal melanoma and is a particularly helpful way of disclosing tumors in eyes with opaque media.54 Ultrasonographic studies can alsodemonstrate areas of extrascleral extension. A scan ultrasonography shows a high internal spike and low internal reflectivity. B scan ultrasonography shows characteristic acoustic hollowness and choroidal excavation (Fig. 23). Hemangiomas and metastases usually appear acoustically solid because they contain multiple acoustic interfaces.
|
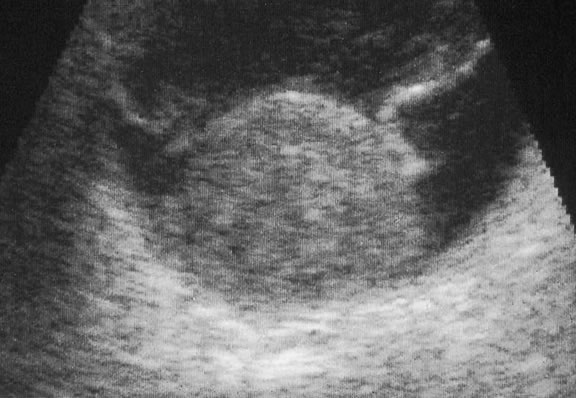
Transvitreal FNAB occasionally is performed if the diagnosis remains uncertain after routine tests and the choice of therapy requires an accurate diagnosis.82,83 Potential candidates for FNAB include the patient who has a history of breast cancer and presents with a solitary amelanotic choroidal tumor that could be a primary amelanotic melanoma or the patient who is thought to have a choroidal metastasis but has no history of cancer.83
Gross Pathology
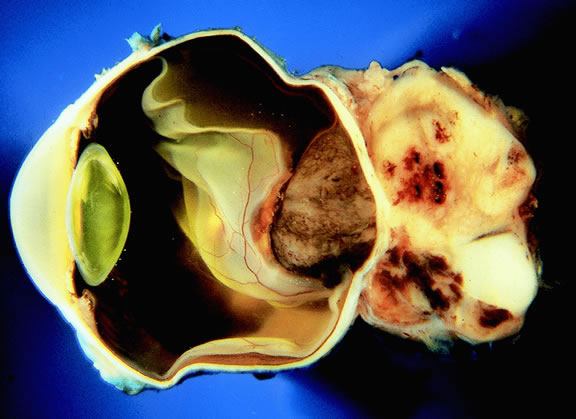
Choroidal melanomas initially arise in the stroma of the choroid. In early cases, the profile of the sectioned tumor is oval or almond-shaped, and its tissue usually appears relatively cohesive after fixation (Fig. 24). Although some tumors diffusely infiltrate the uvea, most uveal melanomas are relatively well-circumscribed tumors with distinct margins. In many cases the growing melanoma perforates Bruch's membrane and enters the subretinal space where its apex typically assumes a spherical shape that often is likened to a mushroom or collar button (see Figs. 19 to 22.). Dilated vessels often are found in the mushrooming head of the tumor because the ends of Bruch's membrane exert a compressive cinch-like effect on the waist of the tumor (see Fig. 22). Rupture of Bruch's membrane was present in 87.7% of 1527 large- or medium-sized melanomas examined in the COMS.84 Retinal invasion was present in nearly half (49.1%), and tumor cells were found in the vitreous body in one fourth.
|
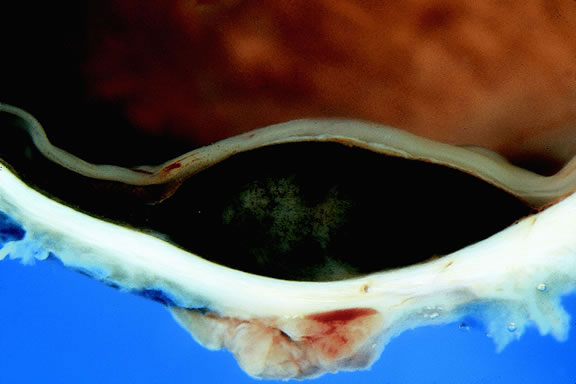
Uveal melanomas vary markedly in their pigment content. Some tumors are totally amelanotic; others are maximally pigmented. In some instances, distinct clones of tumor cells that vary in pigment content are evident on gross examination or even clinically (Fig. 25). A friable or granular appearance of the cut surface of a melanoma noted during gross examination may be indicative of necrosis or a tumor rich in poorly cohesive epithelioid cells. Melanomas that contain cystic cavities occasionally are encountered.85
|
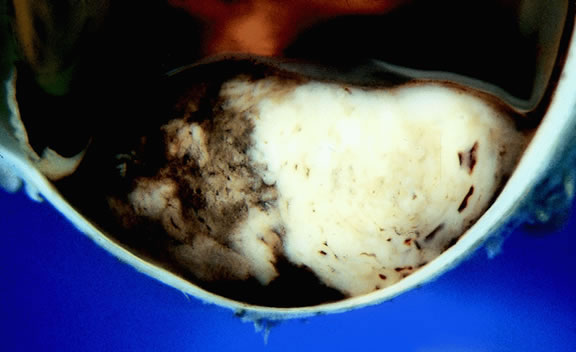
About 3% of melanomas have a diffuse growth pattern. These relatively flat tumors grow laterally without thickening the choroid.86,87 Most are about 2 mm in thickness. Diffuse melanomas often are of mixed cell type and, compared with localized lesions, are more apt to infiltrate the sclera and invade the optic nerve or orbit (Fig. 26). Delayed diagnosis or misdiagnosis is common.
|
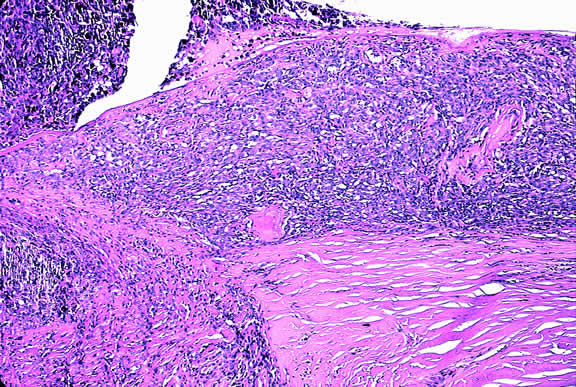
Choroidal melanomas are classified as small, medium, or large based on the largest tumor diameter (LTD). Small choroidal melanomas are 10 mm in diameter or less and appear as focal discoid or oval areas of choroidal thickening. Medium-sized melanomas measure 11 to 15 mm, and large tumors are more than 15mm in largest basal tumor diameter. Larger tumors are more likely to have ruptured through Bruch's membrane.
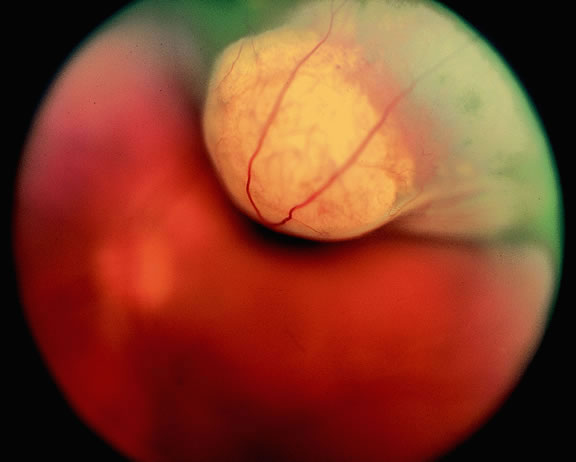
In the COMS, medium-sized tumors were located more posteriorly than large tumors.84 Presumably, more anteriorly situated ciliary body tumors are larger when they are first detected because they remain hidden behind the iris, cause late retinal detachment, and hence remain asymptomatic. Ciliary body melanomas (Figs. 27 to 29) are less common than choroidal melanomas and tend to be more spherical in shape. As noted previously, ciliary body melanomas can cause unilateral cataract, lens molding, or subluxation (see Fig. 28). In addition to causing secondary glaucoma by angle seeding or circumferential infiltration, ciliary body melanomas that invade the periphery of the anterior chamber often displace the iris root centrally causing a solid iridodialysis. Heterochromia iridum also is possible if there is extensive chamber seeding. Ciliary body tumors that perforate the iris initially may be misdiagnosed as a small iris tumor that in reality is the “tip of an iceberg” (see Fig. 29).
|
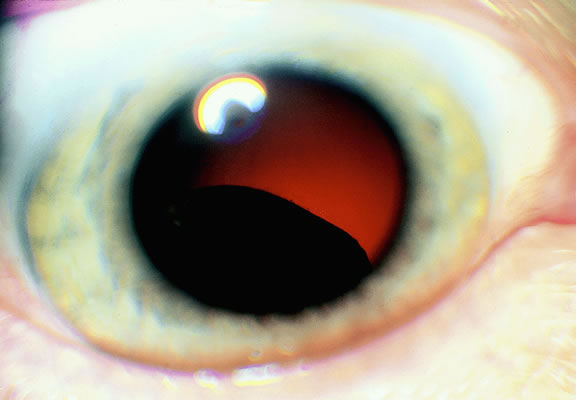
|
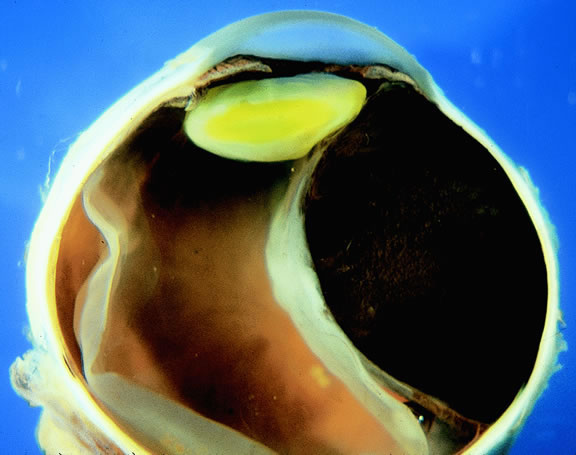
|
Retinal detachment is the most common cause of visual loss in eyes with posterior melanoma. Initially solid, the detachment becomes exudative as serous fluid accumulates. Visual loss initially may be caused by induced hyperopia as a subfoveal tumor displaces the retina anteriorly. The exudative detachment initially is localized; fluid accumulates in the subretinal space tented up by the growing tumor. Failure to find a retinal hole in a patient with retinal detachment should always raise the possibility of a choroidal tumor or some other choroidal process known to produce an exudative detachment. Total retinal detachments are found in some eyes with choroidal melanomas, generally those with larger or neglected tumors (see Fig. 13). Melanoma-bearing eyes with total retinal detachments often have secondary closed-angle glaucoma caused by pupillary block or iris neovascularization.74,75 Clumps of orange pigment may be seen in the subretinal space overlying the tumor (see Fig. 40).
|
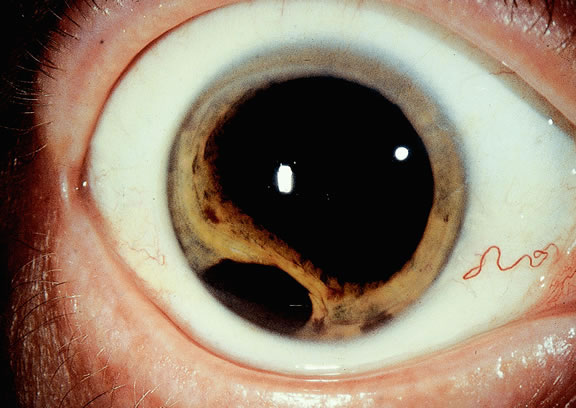
During gross examination, the external surface of an eye harboring a melanoma should be inspected carefully for signs of extraocular extension. The latter may be evident as an epibulbar nodule or a vortex vein that is dilated and heavily pigmented. Tumor infiltration of scleral emissarial canals may be evident on the cut surface of the globe at the base of the tumor.
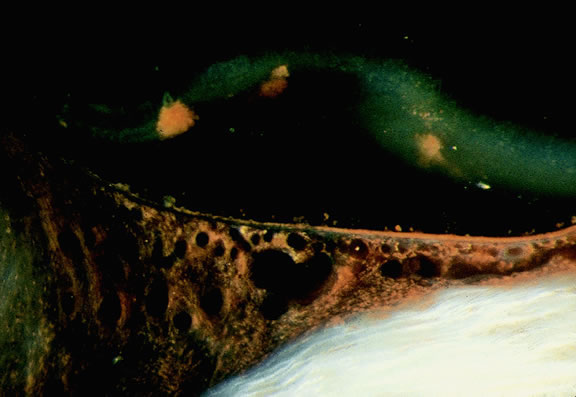
Treatment effects are evident in eyes whose tumors have been treated previously with radioactive plaque brachytherapy or transpupillary thermotherapy (TTT).88,89 Prior plaque therapy often causes prominent areas of diffuse chorioretinal depigmentation and atrophy. TTT scars usually are white and sharply demarcated like a coloboma and often are rimmed by heavily pigmented foci of RPE hyperplasia.
Histopathology
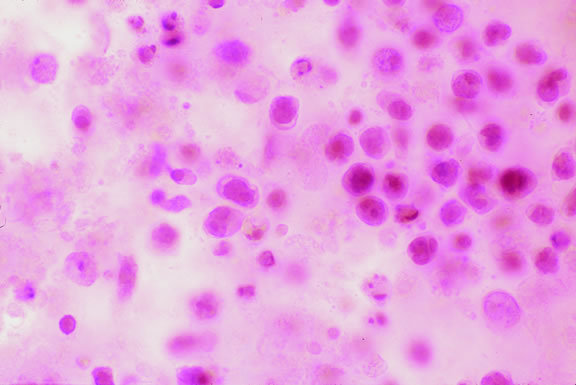
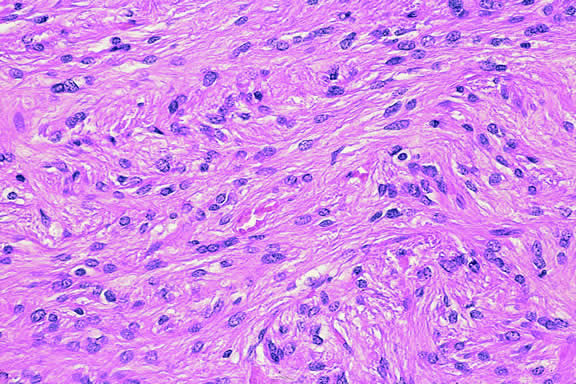
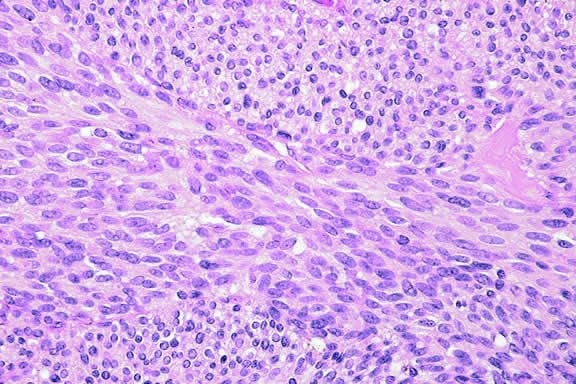
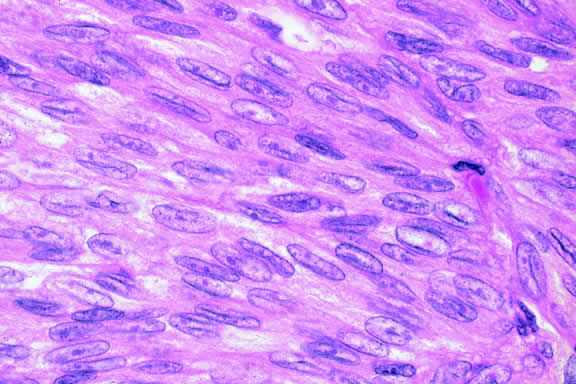
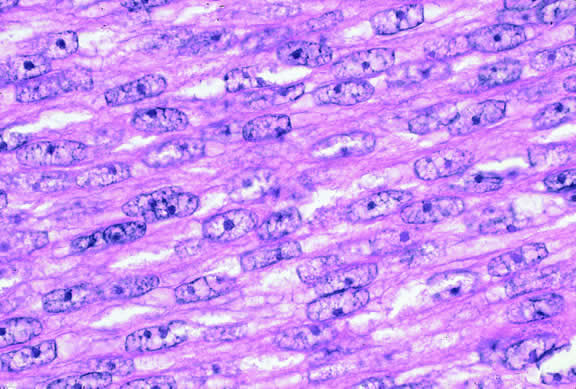
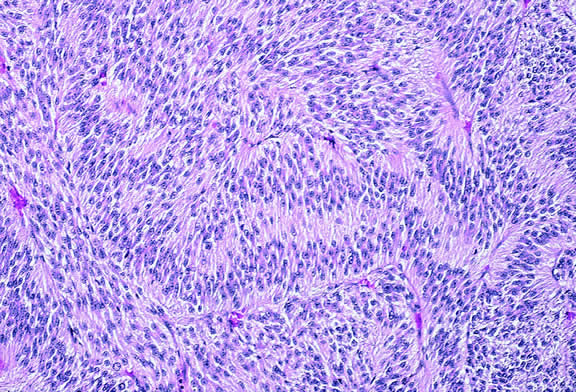
The cells comprising uveal melanoma constitute a biologic spectrum comprising bland spindle A melanoma cells at one end and wildly anaplastic epithelioid cells at the other. The term spindle cell is derived from the fusiform or spindled configuration of the cells' cytoplasmic outline. They are bipolar in shape, and many have long tapering processes that occasionally are highlighted when individual pigmented cells are seen in a largely amelanotic tumor. Spindle cells grow in a syncytial fashion forming interweaving fascicles of parallel oriented cells (Fig. 30). The cells can be pigmented or nonpigmented. Two types of spindle cells are recognized; spindle A and spindle B. These are distinguished by their nuclear characteristics. Spindle A nuclei are tapering ovals or cigar-shaped and have finely dispersed chromatin (Fig. 31). If a nucleolus is present, it usually is inconspicuous. Many spindle A cells have a longitudinally oriented chromatin stripe that actually is caused by a fold in the nuclear membrane. The nuclei of spindle B cells have distinct nucleoli and coarser chromatin and tend to be plumper and more oval in shape (Fig. 32).
|
|
|
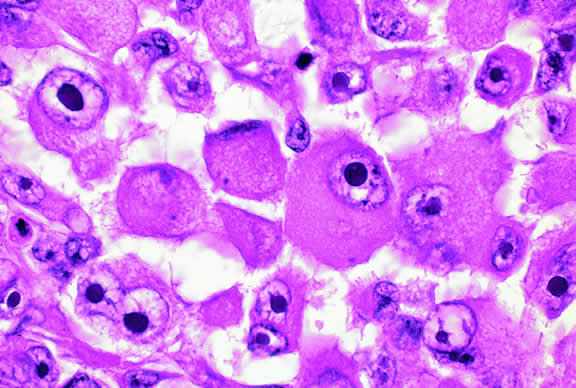
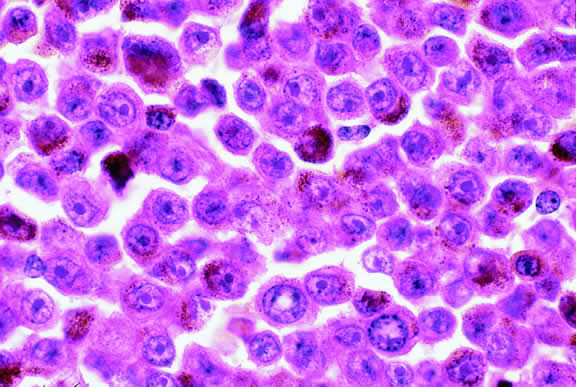
Epithelioid melanoma cells comprise the poorly differentiated end of the cytologic spectrum. Melanomas that contain epithelioid cells have a poorer prognosis. The term epithelioid meaning epithelial-like reflects the fanciful resemblance of the tumor cells to the cells of simple epithelia. Epithelioid cells have abundant cytoplasm and are often polygonal in shape (Fig. 33). They have distinct cytoplasmic margins, are poorly cohesive, and do not grow as a syncytium. The nuclei of epithelioid cells are usually round or oval and often appear vesicular because of margination or clumping of the chromatin along the inner side of the nuclear membrane. Epithelioid melanoma cells also have prominent nucleoli that are often large and reddish purple. Variants of epithelioid cells include relatively uniform small epithelioid cells (Fig. 34) and bizarre tumor giant cells that may appear wildly anaplastic (Fig. 35).
|
|
|
Melanoma cells should be classified by their nuclear characteristics. Spindle-shaped cells that have epithelioid nuclei occasionally are encountered; such cells are classified as epithelioid. In recent years, the term intermediate cell has been used increasingly. Intermediate cells are cells that have nuclear characteristics that are intermediate between spindle B and epithelioid. For example, one might apply the term intermediate cell to a spindle B cell that has a nucleus that is somewhat large and has a fairly prominent nucleolus.
Occasionally, the spindle cells in a melanoma are arranged in a radial fashion around vessel or fibrovascular septa (vasocentric pattern), or the nuclei form rows that resemble the Verocay bodies or Antoni A pattern seen in schwannoma (Verocay pattern). Melanomas are called fascicular if these patterns dominate5 (Fig. 36). Fascicular melanoma was a category in Callender's initial classification90 that was dropped from McLean's 1983 modification.5,91
|
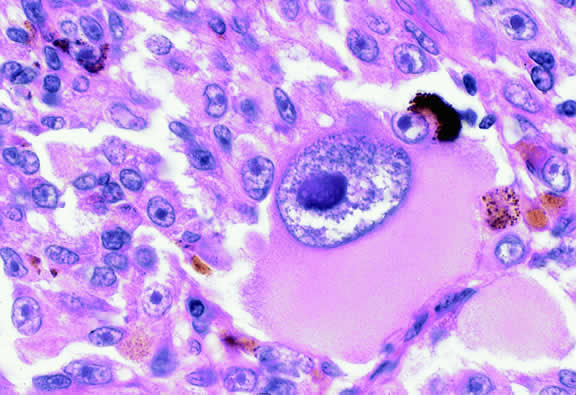
Varying degrees of necrosis may be found (Figs. 37 and 38). Necrosis tends to be more prominent in rapidly growing high-grade tumors or tumors that have had prior brachytherapy. The necrosis may be patchy and focal or may involve extensive parts or even all of the tumor. Aggregates of melanophages typically are found in the necrotic areas. Total infarction of the tumor (and other intraocular structures) may occur in eyes with severe secondary closed-angle glaucoma. As mentioned previously, melanocytoma is prone to spontaneous necrosis. The latter diagnosis should always be considered when a totally necrotic, heavily pigmented tumor is found.
|
|
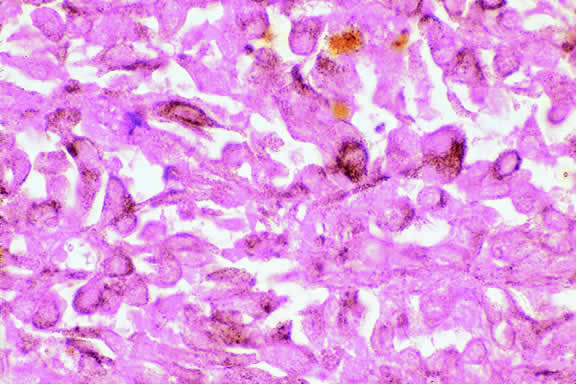
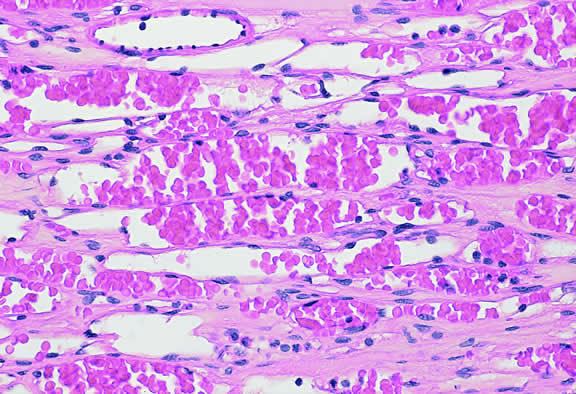
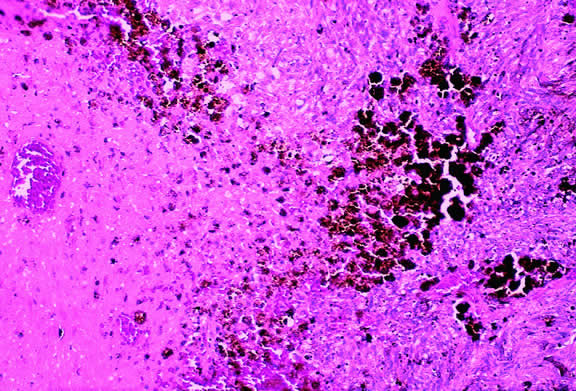
Choroidal melanomas produce abnormalities in the overlying retinal pigment epithelium including atrophy, hyperplasia, and the formation of drusen and drusenoid material.92 The overlying retina often shows photoreceptor loss and may develop cystoid edema. The latter tends to be more common in slower growing lesions, especially choroidal hemangiomas. After Bruch's membrane has ruptured, the vessels located in the mushrooming head of the tumor are often quite prominent, reflecting vascular stagnation caused by the compression at the waist of the tumor (see Fig. 22). Aggregates of macrophages that have ingested periodic acid-Schiff (PAS)-positive lipofuscin pigment and melanin from the damaged retinal pigment epithelium can be found in the subretinal fluid (Figs. 39 to 41). These are evident ophthalmoscopically as clumps of orange pigment that serve as a clinical marker for an actively growing neoplasm.80,81
|
|
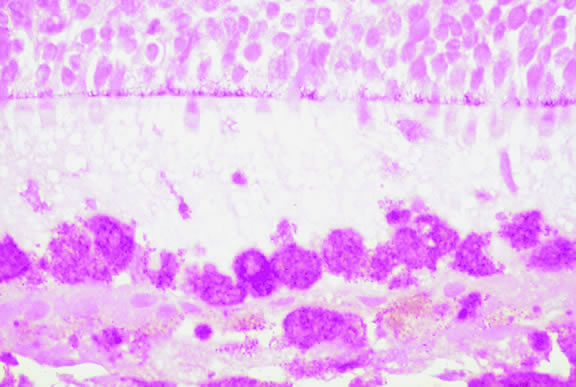
Melanocytic tumors of the uvea are classified into four groups on the basis of cytology. Tumors composed entirely of spindle A cells or even blander nevus cells are classified as spindle cell nevi. Tumors composed of a mixture of malignant spindle A and spindle B cells are called spindle melanomas. Melanomas of mixed cell type contain a mixture of spindle and epithelioid melanoma cells (Fig. 42). Some laboratories specify the predominant cell type found in a mixed cell melanoma, for example, reporting mixed cell, predominantly spindle if only a few epithelioid cells are present. Epithelioid melanomas are composed predominantly of epithelioid cells. They are relatively rare and have the poorest prognosis. Most medium- and large-sized melanomas contain a mixture of spindle and epithelioid cells. In the COMS histopathology study, 86% of the posterior melanomas were classified as mixed cell type, 8% were of spindle cell type, and 5% were epithelioid.84 The association between cytology and mortality is known as the Callender classification.90 (See later section on prognostic factors.)
|
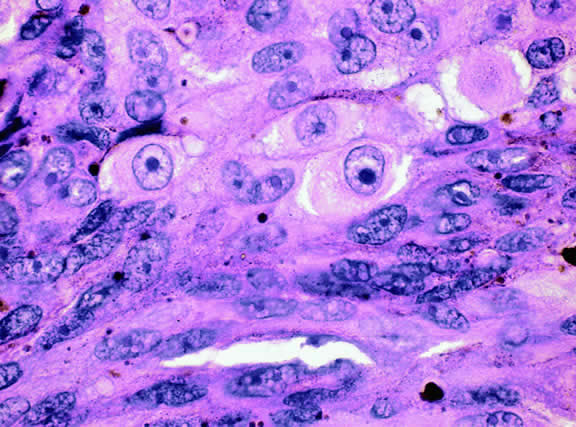
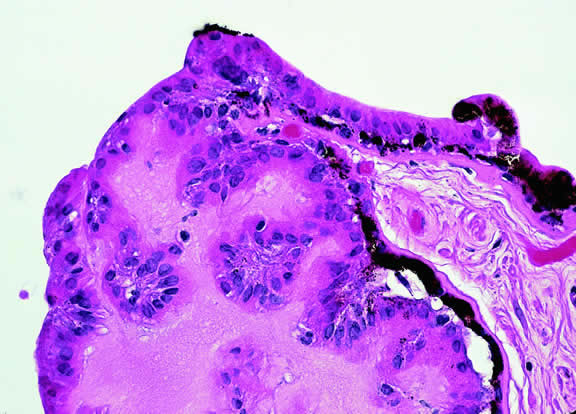
Iris melanoma is relatively rare, constituting between 4% to 15% of uveal melanomas in various series.93 The studies with higher reported incidences may reflect inclusion of nevi. Iris melanomas differ in some respects from tumors of the posterior segment. Most are low-grade spindle cell tumors (Fig. 43). However, iris melanomas with epithelioid cells occasionally are encountered (Fig. 44). Many pigmented iris tumors actually are nevi,94 and relatively few of these enlarge when observed. Several studies have shown that the risk of enlargement in 5 years is only 5% to 6% after a subset of promptly treated lesions is excluded.95,96 One study showed that iris tumors are likely to be considered melanomas and be treated promptly if the basal diameter is greater than 3 mm, pigment dispersion or prominent tumor vascularity are present, the intraocular pressure is elevated, or there are tumor-related symptoms.95 The mean age of patients with iris melanomas is about 10 years younger (age 43 years) than the age of patients with posterior segment melanomas.97 The prognosis of iris melanoma is also relatively favorable compared with tumors of the posterior segment. Shields and coworkers97 studied 169 patients with histologically confirmed iris melanoma and found that distant metastases developed in 5% at 10-years follow-up. The relatively small size of most iris melanomas probably is a major factor in their good prognosis. The prognosis of iris melanomas actually may be similar to posterior segment tumors of similar size and cell type. Diffuse iris melanomas that cause heterochromia iridis and secondary glaucoma are a rare but clinically important group of iris tumors.98,99 Many diffuse iris melanomas are higher grade tumors that contain epithelioid cells, which are poorly cohesive and prone to aqueous dispersal. Glaucoma surgery should be avoided in such cases. It invariably fails and puts patients at greater risk for extraocular extension and metastasis.100
|
|
Prognostic Factors
About one half of patients with choroidal and ciliochoroidal malignant melanomas die from their tumors.101,102 Because the eye and orbit lack lymphatics, uveal melanoma spreads via the bloodstream. Metastasis to the liver occur most often; more than 90% of cases with metastatic melanoma have liver metastases, and they are the first metastases detected in 80%. Other common sites of metastatic uveal melanoma include the lung (24%) and bone (16%).103 Multiple sites are found in 87%. Unfortunately, once distant metastases are manifest clinically, therapy generally is ineffective, and more than 50% of patients who have metastatic uveal melanoma die within 1 year. In recent years, there has been an effort to identify prognostic factors that could identify patients at high risk for metastatic disease who hopefully might benefit from prophylactic chemotherapy or immunotherapy.104
STANDARD HISTOPATHOLOGIC FACTORS.
The association between the cytologic characteristics of uveal melanoma (cell type) and mortality was initially reported in 1931 by Major George Russel Callender90 who examined a series of cases on file in the Registry of Ophthalmic Pathology at the Army Medical Museum in Washington, DC. Callender reported that melanomas were composed of two types of spindle cells that he designated spindle A and B and less differentiated epithelioid cells. He found that tumors that contained epithelioid cells had a poorer prognosis.
Ian McLean and his coworkers at the Armed Forces Institute of Pathology (AFIP) modified Callender's original classification in 1978. Spindle A and spindle B melanomas, which formed separate categories in Callender's original classification, were combined into the single category of spindle melanoma.105 Very low-grade spindle cell tumors classified as spindle cell nevi. McLean also introduced the concept of intermediate cells, that is, cells with nuclear characteristics that were intermediate between spindle B and epithelioid.
The presence or absence of epithelioid cells is an extremely important prognostic factor in uveal melanoma. In 1982, McLean and coworkers102 reviewed a series of 3432 cases of malignant melanoma of the choroid and ciliary body on file in the AFIP's Registry of Ophthalmic Pathology and found that 56% were mixed cell tumors composed of a mixture of spindle and epithelioid cells. The 15-year mortality of patients with melanomas of mixed cell type was three times that of patients whose tumors were composed solely of spindle cells. Tumor size, measured as LTD, was also highly correlated with mortality.
Cell type remains one of prognostic mainstays of surgical pathologists because assessment is relatively rapid and requires no special stains or equipment. However, application of the Callender classification unquestionably has limitations. First, determination of cell type is highly subjective and diagnostic accuracy of can vary with the expertise and experience of the pathologist. A masked study showed that even experienced ophthalmic pathologists disagree about their classification of individual tumor cells.106 Secondly, melanoma cells constitute a continuous biologic spectrum that includes extremely bland spindle A melanoma cells at one end and highly anaplastic epithelioid cells at the other. Despite this, only three categories—spindle, mixed, or epithelioid—are available for the classification of a given tumor, and tumors in a single category, for example, mixed cell type, can vary significantly in their apparent degree of differentiation.
MORPHOMETRY AND NUCLEOLAR PARAMETERS.
The limitations of Callender's classification prompted a search for more objective and reliable criteria for the histopathologic assessment of the malignant potential of uveal melanomas. Gamel and coworkers107–109 performed morphometric analysis on series of tumors with known survival and showed that certain nucleolar parameters, most notably the inverse of the standard deviation of the area of the nucleolus, were useful predictors of death from metastatic melanoma. Using the combination of nucleolar parameters and LTD, Gamel and colleagues108 were able to correctly predict the clinical course of 88% of cases. The technique subsequently was applied to 340 cases with known outcome from two independent laboratories, and successfully subdivided patients into groups that suffered a sixfold difference in mortality.109,110
Gamel's laboratory subsequently developed another simpler objective method of nucleolar assessment based on the measurement of the 10 largest nucleoli (MTLN).111 This was more reproducible than cell type and had substantial cost and labor advantages over the original computer-controlled system required to measure the standard deviation of nucleolar area (SDNA). The authors also used a digital filar micrometer to measure nucleoli. The latter method for measuring MTLN required less than 11 minutes per case, compared with an average of 28 minutes for the original method.112 The technique compared favorably with LTD, cell type, and SDNA in the prognostic assessment of a large series of patients with melanoma.113 Other researchers have had less success with MTLN, probably because of technical factors.114,115
Nucleolar parameters also have been assessed with the silver-stained nucleolar organizer region (AgNOR) technique that examines nucleolar organizing regions that have been stained with colloidal silver azotate.116–119 Spindle, mixed, and epithelioid melanomas were found to have progressively larger mean AgNOR values.117 and the COMS group showed that malignant lesions have higher mean AgNOR counts (4.347) than benign nevi119 (1.855). Automated image capture and analysis has been performed on melanomas stained with the AgNOR technique.116
The assessment of nucleolar parameters is one of the most accurate ways of predicting survival after enucleation using morphologic data contained within routine histologic slides. Unfortunately, the technique has not been widely adopted because it is labor-intensive and relatively time-consuming and requires special expertise and equipment.
Other attempts to make the assessment of cell type more objective and quantitative include counting the number of epithelioid cells and intermediate cells in 40 high-power fields (HPFs).110,120 The mitotic activity of uveal melanoma is routinely assessed by counting the number of mitotic figures in 40 HPFs.
MICROVASCULAR PATTERNS.
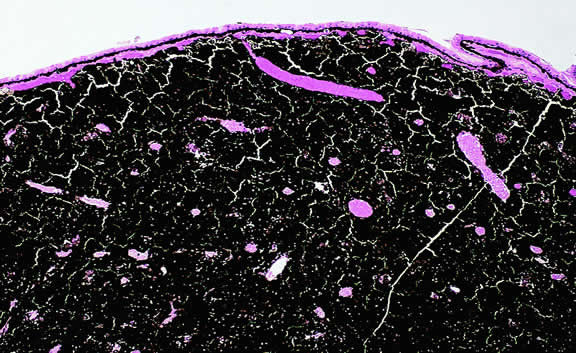
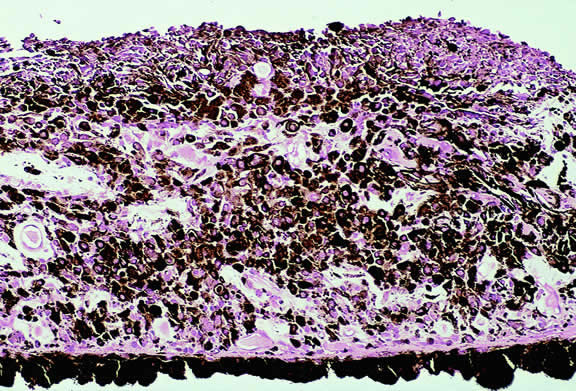
Certain microvascular patterns within uveal melanomas have been shown to be prognostic indicators for death from metastatic melanoma.121 Folberg and associates identified nine morphologic patterns of tumor vessels in eyes removed for ciliary body or choroidal melanoma and designated them: (1) normal, (2) silent, (3) straight, (4) parallel, (5) parallel with cross-linking, (6) arcs, (7) arcs with branching, (8) loops, and (9) networks. The presence of vascular loops and microvascular networks composed of back-to-back loops that encircle microdomains of tumor are strongly associated with death from metastatic melanoma122 (Fig. 45). Prognostic vascular patterns appear to be consistent throughout the depth of a tumor, and the cross-sectional area occupied by prognostic microvascular patterns also has prognostic value.123,124 Additional studies from Folberg's laboratory have compared the microcirculation architecture of nevi and melanomas125 and examined the relationship between microvascular architecture and the aggressive behavior of ciliary body melanomas.126 Attempts to detect prognostically significant microcirculatory patterns in vivo using noninvasive imaging techniques including ultrasonography127,128 and confocal angiography with fluorescein or indocyanine green have been made.129,130 A study of 496 posterior uveal melanomas at the AFIP confirmed that the presence of loops did indicate poor outcome but was not as good a prognostic indicator as the mean diameter of the largest nucleoli, cell type, or tumor size.131
|
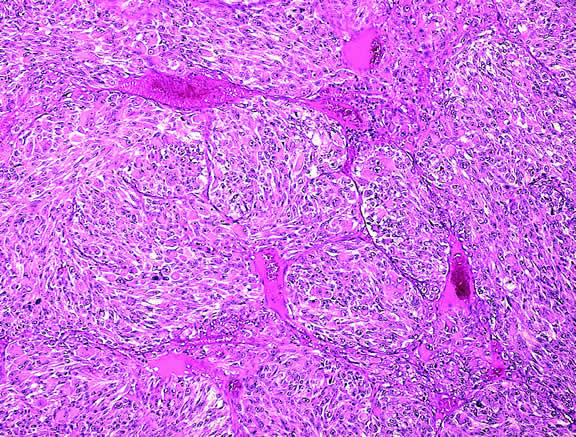
Microvascular density is another prognostic factor based on tumor vessels.132 Less subjective than the assessment of microvascular patterns, this technique involves counting tumor vessels, which have been highlighted by immunostaining for endothelial markers like CD34, in the area of highest vessel density.
Blood-filled spaces within melanomas called venous lakes also may have an adverse effect on survival.133 Venous lakes are not lined by vascular endothelial cells. Folberg and colleagues134 at the University of Iowa Cancer Center recently have emphasized that highly invasive uveal melanoma cells are able to generate vascular channels that are not lined by endothelial cells. These vascular channels appear to be able to facilitate tumor perfusion independent of tumor angiogenesis.
MITOTIC ACTIVITY AND THE CELL CYCLE.
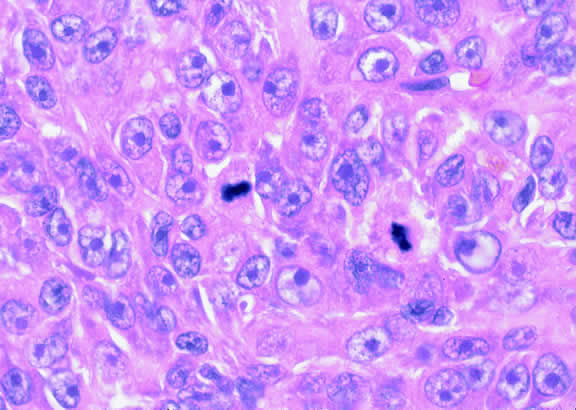
Ocular pathologists routinely assess the mitotic activity (Fig. 46) of uveal melanoma by counting the number of mitotic figures in 40 high power (“high dry”) microscopic fields. Forty fields are counted because most uveal melanomas contain relatively few mitoses, that is, only 5 or 10 per 40 HPFs. Not unexpectedly, patients whose tumors have more mitoses have a poorer prognosis.
|
Other techniques have been used to more accurately assess the proliferative (cell cycling) capability of uveal melanomas. These include labeling cells with bromodeoxyuridine (BrdUrd-labeling index)85,135–137 and the use of immunohistochemistry with monoclonal antibodies such as Ki-67 and PC-10 that recognize antigens that are markers for cellular proliferation.114,138
The rate of cellular proliferation also can be assessed by using flow cytometry to measure the fraction of cells in the S phase of the cell cycle when a cell's deoxyribonucleic acid (DNA) content doubles before division. A significant correlation has been found between a high S-phase fraction and epithelioid cells and large tumors.139 Spindle cell tumors tend to have lower cell turnover rates.140
DNA CONTENT AND PLOIDY.
Flow cytometric analysis has been used to assess the effect of aneuploidy in melanoma cells on survival, with varying results.141–146 Most studies have been retrospective analyses of archival tissue. A prospective study that used fresh tissue showed that the estimation of DNA index or %SPF (S phase fraction) by flow cytometry was of little value in predicting survival from uveal melanomas after a minimum follow-up of 5 years.142 A study that used microspectrophotometry to measure DNA content from microscopic slides showed that the SDNA was a better predictor of metastasis than DNA content.147
ONCOGENES.
The hallmark of most, if not all, malignant neoplasms is an aberrant regulation of cellular proliferation caused by the overexpression of dominant oncogenes such as myc or the subversion of recessive tumor suppressor genes like the retinoblastoma gene and p53. A number of investigators have examined the expression of oncogenes and proliferation-associated proteins such as cyclin D1, p53, p16, c-myc, and bcl-2 in uveal melanoma.137,148–154 In one study, high expression of c-myc oncoprotein actually correlated with improved outcome in uveal melanoma. This contrasts with cutaneous melanoma, in which high levels of nuclear c-myc expression have been found to correlate with poor outcome in primary and metastatic disease.148,155 The expression of cyclin D1 in uveal melanoma appears to be associated with a more aggressive course and histologically unfavorable disease.149,150
CHROMOSOMAL AND CYTOGENETIC ABNORMALITIES.
The identification of nonrandom cytogenetic abnormalities in a specific type of neoplasm may serve to identify the location of genes that are involved in the molecular pathogenesis of that lesion. For example, the occurrence of retinoblastoma in patients with a deletion of the long arm of chromosome 13 (13q- syndrome) initially suggested that the retinoblastoma gene was located on the 13th chromosome.156 Although the pathogenesis of uveal melanoma remains uncertain, recent studies have suggested that cytogenetic analysis of uveal melanoma may provide prognostic information. Many uveal melanoma cells harbor recurrent nonrandom clonal abnormalities involving chromosomes 3, 6, and 8. These cytogenetic changes include monosomy 3, trisomy 8, and structural or numerical abnormalities of chromosome 6.59,157 Monosomy 3, which is present in approximately 60% of cases, is the most frequent karyotypic abnormality.158 Monosomy 3 has been shown to be a significant predictor of poor prognosis in uveal melanoma.159,160 Another study indicated that abnormalities of chromosomes 3 and 8 were associated with a poor prognosis, but only when these two chromosomal abnormalities were present together.158 Chromosomal abnormalities have been identified using fluorescence in situ hybridization (FISH) with an alpha-satellite probe for chromosome 3 on uveal melanoma imprints.161
OTHER MOLECULAR MARKERS.
Cell adhesion mechanisms and molecules theoretically are involved in the development and establishment of metastases. Adhesion molecules are thought to play an important role in the detachment of cells from the primary tumor, the interaction of tumor cells with the extracellular matrix, and the adhesion of circulating tumor cells to vascular endothelium. Anastassiou and colleagues162 showed that the loss of ICAM-1 expression by melanoma cells was associated with an increased risk of metastasis within the first 5 years after diagnosis, but NCAM and VCAM-1 expression was not. VLA-2, VLA-3, and alpha (v) integrin receptors also had no prognostic value.163
Proteases such as plasminogen activators (PAs) and metalloproteinases are involved in degradation of the extracellular matrix and other tissue barriers during tumor development. DeVries assessed the expression and distribution of various components of the PA system and the presence of PA enzyme in 45 freshly frozen primary uveal melanomas with known follow-up. Preliminary data suggest that the expression of urokinase PA (u-PA) correlates with the occurrence of metastasis. Metastatic melanoma also expresses tissue-type PA and plasminogen activator inhibitors.164
Vaisanen and coworkers165 showed that melanomas that expressed matrix metalloproteinase-2 (MMP-2) had a dismal prognosis: The 5-year overall survival rate for MMP-2-positive cases was 49% compared with 86% for MMP-2 negative cases. Patients with MMP-2-positive nonspindle cell uveal melanomas were at high risk of metastatic disease; only 38% survived for 5 years.
The expression of epidermal growth factor receptor (EGFR) has been correlated with the development of metastases in various malignancies. In one small study, EGFR expression by melanoma cells correlated significantly with death resulting from metastatic disease. Five of six patients whose tumors were immunoreactive for EGFR (83%) died because of metastases, compared with 2 (17%) of 12 patients whose tumors did not express EGFR expression.166
Primary melanomas that express cytokeratins CK 8 and 18 are more likely to metastasize.167 These cytokeratins, which are detected by monoclonal antibody CAM 5.2, occur more often in mixed cell melanomas. Cytokeratin expression may contribute to the epithelioid phenotype of melanoma cells because these intermediate filaments are normal cytoskeletal constituents of epithelial cells.
Hendrix and coworkers169 have shown that so-called interconverted melanoma cells that express CK 8 and 18 as well as vimentin, the intermediate filament typically found in melanoma cells, are six times more invasive in an in vitro system, compared with uveal melanoma cells that express solely vimentin.168 The interconverted cells have receptors for hepatocyte growth factor/scatter factor (HGF/SF) (also known as the c-met proto-oncogene). HGF/SF may play a role in local invasion and targeted dissemination to the liver.
IMMUNOLOGIC FACTORS.
In 1978, Zimmerman and colleagues170 hypothesized that enucleation of an eye containing a malignant melanoma might accelerate the dissemination of tumor cells. Although Zimmerman's hypothesis has never been proved or disproved, it is now believed that many uveal melanomas continuously shed tumor cells into the circulation. Host immunologic responses undoubtedly are a major factor in determining which patients develop overt metastatic disease and which do not.
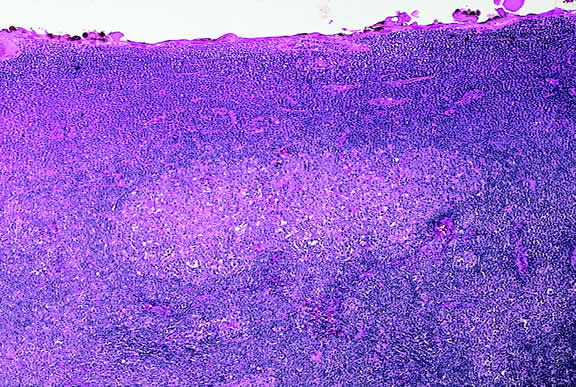
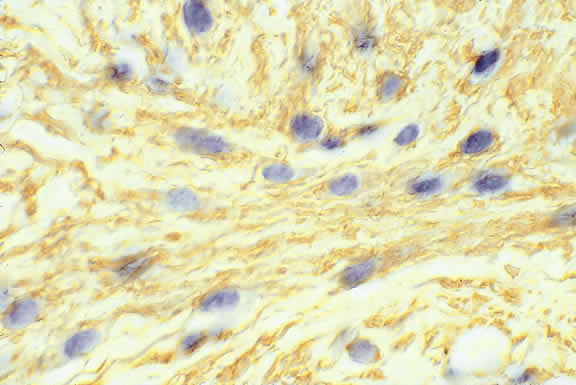
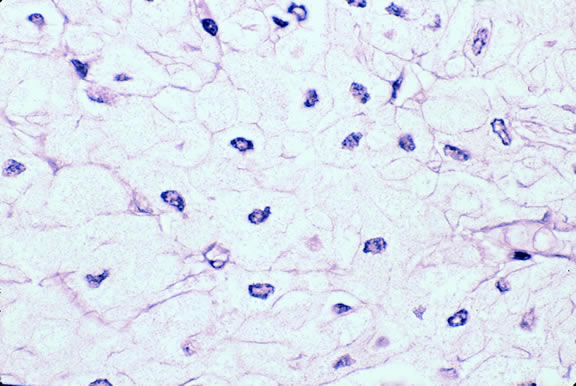
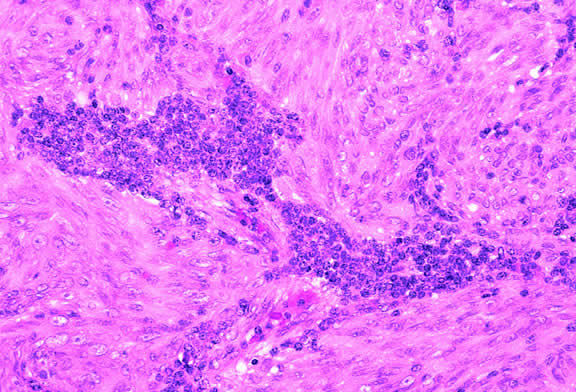
Lymphocytic infiltration of the tumor is one facet of the host response that can be assessed during the routine histopathologic examination (Fig. 47). In most parts of the body, a lymphocytic response to a malignant tumor generally is associated with an improved prognosis. In uveal melanoma, however, the presence of tumor infiltrating lymphocytes is associated with decreased survival.171 De la Cruz and coworkers used light microscopy to examine 1078 cases of uveal melanoma with known survival and found that 12.4% harbored 100 or more lymphocytes per 20 high-power (× 400) microscopic fields. The survival rate at 15 years was 36.7% for patients in the high lymphocytic group and 69.6% for patients in the low lymphocytic group. This seemingly counterintuitive observation is explained by the fact that extraocular dissemination of tumor cells is a requisite for stimulation of a T lymphocyte-mediated immune response. Ocular antigens are processed primarily in the spleen because the eye lacks lymphatics. Using immunohistochemistry, Whelchel and colleagues172 confirmed that lymphocytic infiltration is associated with death from metastasis and showed that T lymphocytes were the predominant cell type in about three fourths of the tumors.
|
Genetic determinants such as the genes for human leukocyte antigen (HLA) antigens also appear to be important in the development and clinical behavior of uveal melanoma. An association between the presence of HLA-B40 and death resulting from metastasis of uveal melanoma has been reported.173,174 High expression of class I HLA antigen HLA-B significantly correlates with the presence of epithelioid cells in the tumor, and high expression of HLA-A and -B are associated with decreased survival. Patients had improved survival when their tumors did not express HLA-A and -B. These data suggest that the low expression of HLA class I on micrometastases shed from uveal melanomas facilitates their removal from the systemic circulation and prevents the development of metastases. These findings support a protective role for natural killer cells in the development of metastatic disease.
Therapy
Most posterior melanomas are treated by enucleation or radiotherapy, typically plaque brachytherapy.48,54,175,176 Irradiation therapy with beams of protons or helium ions is performed at a few centers.177–179 Larger tumors or tumors that have caused secondary glaucoma generally are enucleated. Medium-sized tumors can be treated with brachytherapy. The COMS has shown that the mortality rates after I-125 plaque therapy and enucleation are similar.180 Another arm of the COMS study showed that pre-enucleation external beam radiotherapy of large choroidal melanomas does not improve survival.181 Although plaque brachytherapy conserves eyes, radiation retinopathy or papillopathy occurs commonly leading to loss of useful vision.89,182 Almost half of treated eyes have 20/200 vision 3 years after therapy.182 Some smaller tumors can be locally resected by partial lamellar sclerouvectomy. Today, this technique generally is performed on iridociliary tumors. Small relatively thin tumors of the posterior choroid can be treated with transpupillary thermotherapy (TTT), a infrared diode laser therapy that kills tumors cells by slowly heating them.89 A sandwich technique combining TTT and plaque brachytherapy has been used prevent tumor recurrence from cells sheltered in scleral canals at the base of a tumor.
If a pigmented lesion of the iris is thought to be a melanoma based on documented growth or other clinical factors, it can be locally excised by iridectomy, or by iridocyclectomy if there is focal angle and ciliary body involvement. Enucleation is often necessary when an iris melanomas has caused glaucoma or is unresectable.98 Enucleation also may be done after histopathologic examination has shown that a previously resected tumor is a high-grade lesion.98 Plaque brachytherapy occasionally is used to treat unresectable iris melanomas.54