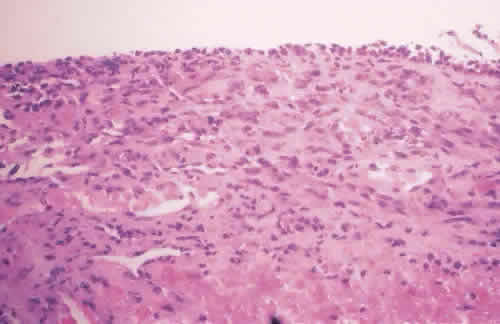
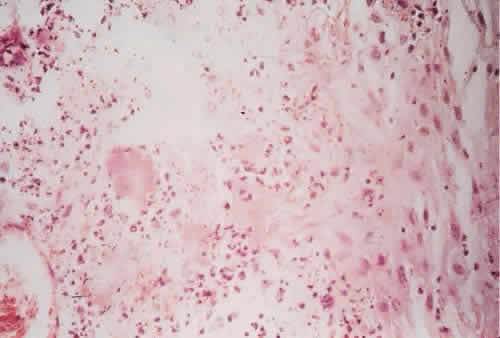
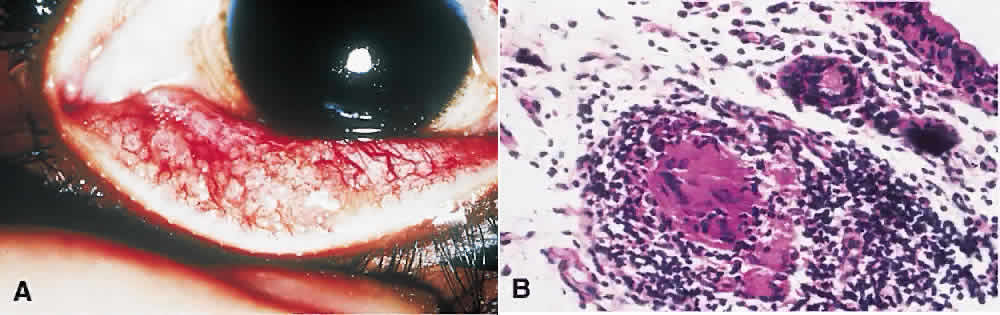
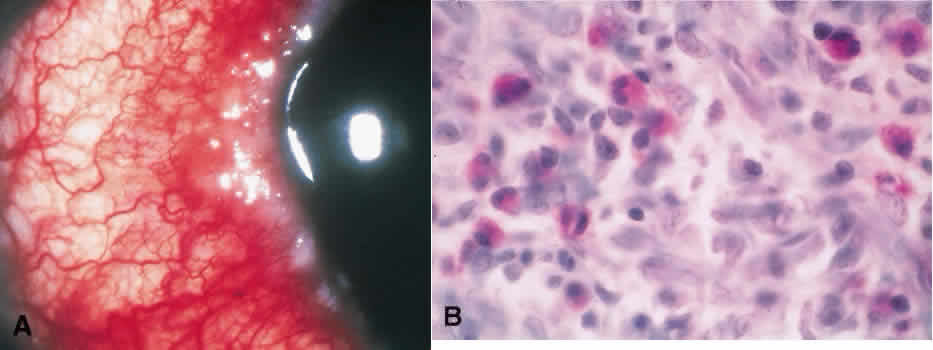
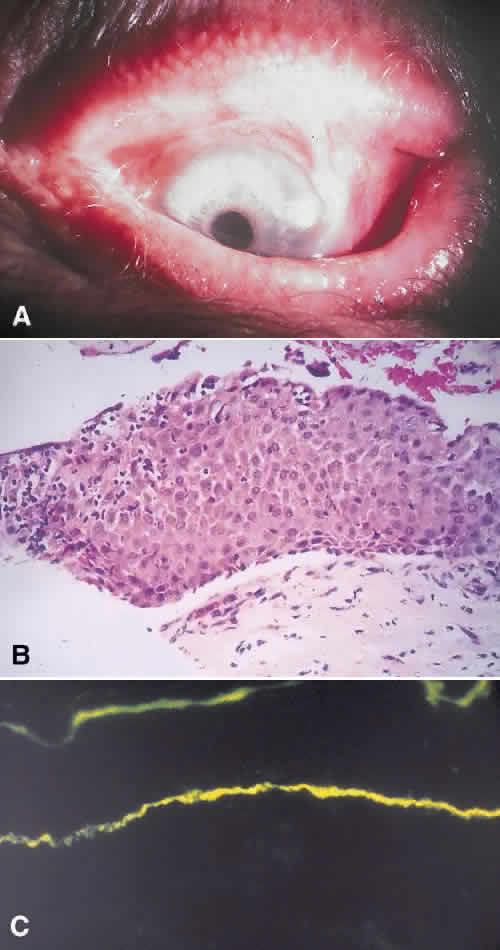
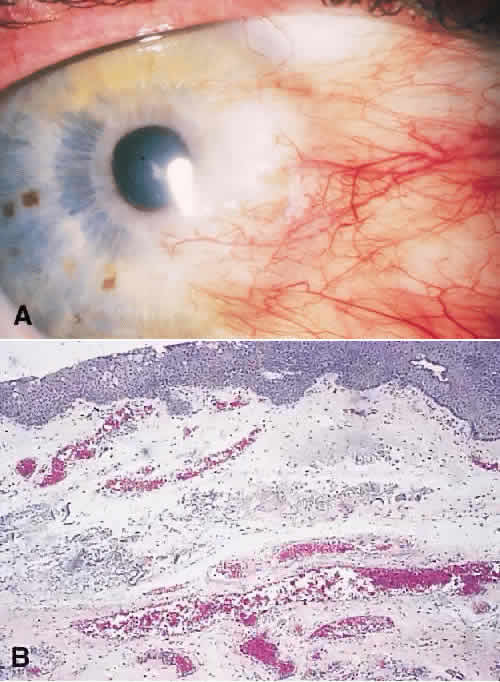
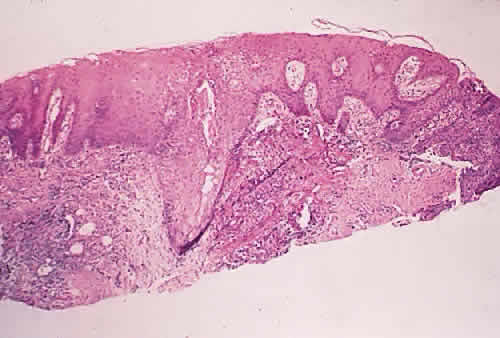
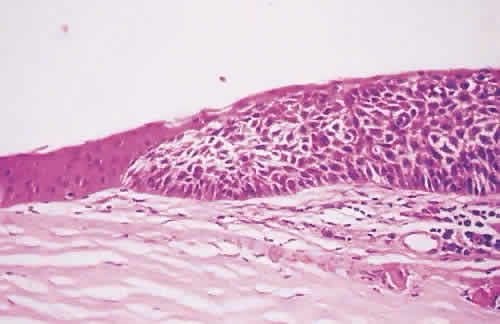
1. Spencer WH, Folberg R: Conjunctiva and melanocytic lesions of the conjunctiva. Ophthalmic
pathology: an atlas and textbook. Philadelphia, WB
Saunders, 1996:38–155. 2. Allansmith MR, Greiner JV, Baird RS: Number of inflammatory cells in normal conjunctiva. Am J Ophthalmol 86:250–259, 1978. 3. Bhan AK, Fujikawa LS, Foster CS: T-cell subsets and Langerhan's cells in normal and diseased conjunctiva. Am J Ophthalmol 94:205–212, 1982. 4. Quigley HA, Kenyon KR: Russell bodies and plasma cells in human conjunctiva. Am J Ophthalmol 76:957–966, 1973. 5. Grossniklaus HE, Green WR, Luckenbach M et al: Conjunctival lesions in adults: A clinical and histopathologic review. Cornea 6:78–116, 1987. 6. Mansour AM, Barber JC, Reinecke RD, Wang FM: Ocular choristomas. Surv Ophthalmol 33:339, 1989. 7. Sugar HS: The oculoauriculovertebral dysplasia syndrome of Goldenhar. Am J Ophthalmol 62:678–682, 1966. 8. Benjamin SN, Allen HF: Classification for limbal dermoid choristomas and branchial arch anomalies: presentation
of an unusual case. Arch Ophthalmol 87:305, 1972. 9. Elsas FJ, Green WR: Epibulbar tumors in childhood. Am J Ophthalmol 79:1001–1007, 1975. 10. Pfaffenbach DD, Green WR: Ectopic lacrimal gland. Int Ophthalmol Clin II 3:149–159, 1971. 11. Boniuk M, Zimmerman LE: Epibulbar osteoma (episcleral osseous choristoma). Am J Ophthalmol 53:290–296, 1962. 12. Dreizen NG, Schachat AP, Shields JA, Augsburger JJ: Epibulbar osseous choristoma. J Pediatr Ophthalmol Strabismus 20:247–249, 1983. 13. Oritz JM, Yanoff M: Epipalpebral conjunctival osseous choristoma. Br J Ophthalmol 63:173–176, 1979. 14. Gupta SP, Saxena RC: Cryptophthalmos. Br J Ophthalmol 46:629–632, 1962. 15. Johnson CC: Developmental abnormalities of the eyelids. Ophthalmol Plast Reconstr Surg 2:219, 1986. 16. Harley RD, Baird HW, Craven EM: Ataxia telangiectasia: report of seven cases. Arch Ophthalmol 77:58–592, 1967. 17. Howard GM, Jakobiec FA, DeVoe AG: Kaposi's sarcoma of the conjunctiva. Am J Ophthalmol 96:95–96, 1978. 18. Shuler JD, Holland GN, Miles SA, Miller BJ, Grossman I: Kaposi sarcoma of the conjunctiva and eyelids associated with the acquired
deficiency syndrome. Arch Ophthalmol 107:858, 1989. 19. Kessing SV: On the conjunctival papillae and follicles. Acta Ophthalmol 44:846–851, 1966. 20. Griener JV, Covington HI, Allansmith MR: Surface morphology of the human upper tarsal conjunctiva. Am J Ophthalmol 83:892–905, 1977. 21. Srinivasan D, Jakobiec FA, Iwamoto T et al: Giant papillary conjunctivitis with ocular prostheses. Arch Ophthalmol 97:892–895, 1979. 22. Greiner JV, Convington HI, Allansmith MR: Surface morphology of giant papillary conjunctivitis in contact lens wearers. Am J Ophthalmol 85:242–252, 1978. 23. Allansmith MR, Korb DR, Greiner JV: Giant papillary conjunctivitis induced by hard or soft contact lens wear: quantitative
histology. Trans Am Acad Ophthalmol Otolaryngol 85:766–778, 1978. 24. Sromovasam BD, Jakobiec FA, Iwamoto T, Devoe G: Giant papillary conjunctivitis with ocular prostheses. Arch Ophthalmol 97:892–895, 1979. 25. Hydayet AA, Riddle PJ: Ligneous conjunctivitis. A clinicopathologic study of 17 cases. Ophthalmology 94:949–959, 1987. 26. Kanai A, Polack FM: Histologic and electron microscope studies of ligneous conjunctivitis. Am J Ophthalmol 72:909–916, 1971. 27. Schuster V, Mingers AM, Seidenspinner S et al: Homozygous mutations in the plasminogen gene of two unrelated girls with
ligneous conjunctivitis. Blood 90:958–966, 1997. 28. Salim AR, Sheikh HA: Trachoma in the Sudan. An epidemiological study. Br J Ophthalmol 59:600–604, 1975. 29. Rapoza PA, Quinn TC, Kiessling LA et al: Epidemiology of neonatal conjunctivitis. Ophthalmology 93:456–461, 1986. 30. Forster RK, Dawson CR, Schachter J: Late follow up of patients with neonatal
inclusion conjunctivitis. Am J Ophthalmol 69:467–472, 1970. 31. ÜmSchachter
J, Dawson CR: Human chlamydial infections. Littleton, MA, PSG
Publishing, 178:1571. 32. MacCallan AF: The epidemiology of trachoma. Br J Ophthalmol 15:369, 1931. 33. Yoneda C, Dawson CR, Daghfous T et al: Cytology as a guide to the presence of chlamydial inclusions in giemsa-stained
conjunctival smears in severe endemic trachoma. Br J Ophthalmol 59:116, 1975. 34. Wear DJ, Malaty RH, Zimmerman LE et al: Cat scratch disease bacilli in the conjunctiva of patients with Parinaud's
oculoglandular syndrome. Ophthalmology 92:1282–1287, 1985. 35. Karcioglu ZA, Brear R: Conjunctival biopsy in sarcoidosis. Am J Ophthalmol 99:68–73, 1985. 36. Nichols CW, Eagle RC, Yanoff M, Merocal NG: Conjunctival biopsy as an aid in the evaluation of the patient with suspected
sarcoidosis. Ophthalmology 87:287–289, 1980. 37. Friedman A, Henkind P: Granuloma pyogenicum of the palpebral conjunctiva. Am J Ophthalmol 71:868–872, 1971. 38. Patten JT, Hyndiuk RA: Granuloma pyogenicum of the conjunctiva. Ann Ophthalmol 7:1588, 1976. 39. Maumenee AE: Keratinization of the conjunctiva. Trans Am Ophthalmol Soc 77:133–143, 1979. 40. Kessing SV: Epithelial cysts in the conjunctiva. Acta Ophthalmol (Copenh) 47:642–655, 1969. 41. Barishak RY, Baruh E, Lazar M: Episcleral traumatic conjunctival inclusion cyst. Br J Ophthalmol 61:299–301, 1977. 42. Dodd MJ, Pusin SM, Green WR: Adult cystinosis. A case report. Arch Ophthalmol 96:1054–1057, 1978. 43. Meredith TA, Wright JD, Gammon JA et al: Ocular involvement in primary hyperoxaluria. Arch Ophthalmol 102:584–587, 1984. 44. Kincaid MC, Green WR, Hoover RE, Schenck PH: Ocular chrysiasis. Arch Ophthalmol 100:1791–1794, 1982. 45. Spencer WH, Garron LK, Conreras F et al: Endogenous and exogenous ocular and systemic silver deposition. Trans Ophthalmol Soc UK 100:171–178, 1980. 46. Fong DS, Frederick AR Jr, Richter CU et al: Adrenochrome deposit. Arch Dermatol 111:1142–1143, 1993. 47. Frazier PD, Wong VG: Cystinosis:histologic and crystallographic examination of crystals in eye
tissues. Arch Ophthalmol 80:87, 1968. 48. Hanna C, Faunfelder FT, Sanchez J: Ultrastructural study of argyrosis of the cornea and the conjunctiva. Arch Ophthalmol 92:18–22, 1974. 49. Corwin ME, Spencer WH: Conjunctival melanin depositions. A side-effect of topical epinephrine
therapy. Arch Ophthalmol 69:317–321, 1963. 50. Messmer E, Font RL, Sheldon G, Murphy D: Pigmented conjunctival cysts following tetracycline minocycline therapy. Histochemical
and electron microscopic observations. Ophthalmology 90:1462–1467, 1983. 51. Brothers DM, Hidayat AA: Conjunctival pigmentation associated with tetracycline medication. Ophthalmology 88:1212–1215, 1981. 52. Impera PS, Lazarus HM, Lass JH: Ocular complications of systemic cancer therapy. Surv Ophthalmol 34:209, 1989. 53. Lloyd WC: Ophthalmology pathology with clinical correlations. Joseph W. Sassani, ed. Philadelphia, Lippincott-Raven, 1997:11–62. 54. Braude LS, Chandler JW: Atopic corneal disease. Int Ophthalmol Clin 24:145–156, 1984. 55. Foster CS, Calonge M: Atopic keratoconjunctivitis. Ophthalmology 97:992–1000, 1990. 56. Brown SI, Shahinian L Jr: Diagnosis and treatment of ocular rosacea. Ophthalmology 85:779–786, 1978. 57. Mondino BJ: Cicatricial pemphigoid and erythema multiforme. Ophthalmology 97:939, 1990. 58. Foster CS: Cicatricial pemphigoid. Trans Am Ophthalmol Soc 84:527, 1986. 59. Anderson SR, Jensen OA, Kristensen EB, Norm MS: Benign mucous membrane pemphigoid III. Biopsy. Acta Ophthalmol (Copenh) 52:455–463, 1974. 60. Mondino BJ, Brown SI: Ocular cicatricial pemphigoid. Ophthalmology 88:95–100, 1981. 61. Furey N, West C, Andrews T et al: Immunofluorescent studies of ocular cicatricial pemphigoid. Am J Ophthalmol 80:825–831, 1975. 62. Orfanos CE, Schaumburg-Lever G, Leveer WF: Dermal and epidermal types of erythema multiforme: a histopathologic study
of 24 cases. Arch Dermatol 109:682–688, 1974. 63. Smith RS Farrell R, Bailey T: Keratomalacia. Surv Ophthalmol 20:213–219, 1975. 64. Sjögren H, Bloch K: Keratoconjunctivitis sicca and the Sjögren syndrome. Surv Ophthalmol 16:147–159, 1971. 65. Eagle RC: Eye pathology: an atlas and basic text. Philadelphia, WB Saunders, 1999:47,72–73. 66. Cameron ME: Histology of pterygium: an electron microscopic study. Br J Ophthalmol 67:604–608, 1983. 67. Hogan MJ, Alvarado J: Pterygium and pinguecula: electron microscopic study. Arch Ophthalmol 78:174–186, 1967. 68. Blodi FC, Apple DJ: Localized conjunctival amyloidosis. Am J Ophthalmol 88:346–350, 1979 69. Smith ME, Zimmerman LE: Amyloidosis of the eyelid and conjunctiva. Arch Ophthalmol 75:42–50, 1976. 70. Sandgren O: Ocular amyloidosis, with special reference to the hereditary forms with
vitreous involvement. Surv Ophthalmol 40:173, 1995. 71. Aronson SB, Shaw R: Corneal crystals in multiple myeloma. Arch Ophthalmol 61:541–546, 1959. 72. Pinkerton RMH, Robertson DM: Corneal and conjunctival changes in dysproteinemia. Invest Ophthalmol 7:357–364, 1969. 73. Benjamin I, Taylor H, Spindler J: Orbital and conjunctival involvement in multiple myeloma. Am J Clin Pathol 63:811, 1975. 74. Erie JC, Campbell RJ, Liesegang TJ: Conjunctival and corneal intraepithelial and invasive neoplasia. Ophthalmology 93:176–183, 1986. 75. Dark AJ, Streeten BW: Preinvasive carcinoma of the cornea and conjunctiva. Br J Ophthalmol 64:506–514, 1980. 76. Char DH: The management of lid and conjunctival malignancies. Surv Ophthalmol 24:679–689, 1980. 77. Spinak M, Friedman AH: Squamous cell carcinoma of the conjunctiva. Value of exfoliative cytology
in diagnosis. Surv Ophthalmol 21:354–355, 1977. 78. Tseng SCG: Staging of conjunctival squamous metaplasia by impression cytology. Ophthalmology 1985;92:728–733. 79. Zimmerman LE: The cancerous, precancerous, and pseudo-cancerous lesions
of the cornea and conjunctiva. In: Rycroft PV, ed. Corneoplastic surgery: proceedins
of the Second International Corneoplastic Conference. Oxford, Pergamon
Press, 1969:547. 80. Dykstra PC, Dykstra BA: The cytologic diagnosis of carcinoma and related lesions of the ocular
conjunctiva and cornea. Trans Am Acad Ophthalmol Otolaryngol 73:979, 1969. 81. Iliff WJ, Marback R, Green R: Invasive squamous cell carcinoma of the conjunctiva. Arch Ophthalmol 93:119, 1975. 82. Doxanas MT, Green WR: Sebaceous gland carcinoma. Review of 40 cases. Arch Ophthalmol 102:245–249, 1984. 83. Margo CE, Stern AL, Stern GA: Intraepithelial sebaceous carcinoma of the conjunctiva and skin of the
eyelid. Ophthalmology 99:227, 1992. 84. Knowles DM, Jakobiec FA, McNally L, Burke JS: Lymphoid hyperplasia and malignant lymphoma occurring in the ocular adnexa (orbit, conjunctiva, and eyelids). Hum Pathol 21:959, 1990. 85. Feinberg A, Spraul CW, Holden JT, Grossniklaus HE: Conjunctival lymphocytic infiltrates associated with Epstein-Barr Virus. Ophthalmology 107:159–163, 2000. 86. Jakobiec FA, Folberg R, Iwamoto T: Clinicopathologic characteristics of premalignant and malignant melanocytic
lesions of the conjunctiva. Ophthalmology 96:147–166, 1989. 87. Guille(c)n FJ, Albert DM, Mihm MC Jr: Pigmented melanocytic lesions of the conjunctiva: a new approach to their
classification. Pathology 17:175–280, 1985. 88. Folberg R, McLean IW: Primary acquired melanosis and melanoma of the conjunctiva: terminology, classification
and biologic behavior. Hum Pathol 17:652–655, 1986. 89. Folberg R, McLean IW, Zimmerman LE: Conjunctival melanosis and melanoma. Ophthalmology 91:673–678, 1984. 90. Folberg R, McLean IW, Zimmerman LE: Primary acquired melanosis of the conjunctiva. Hum Pathol 16:129–135, 1985. 91. Zimmerman LE: The histogenesis of conjunctival melanomas: the first Algernon
B Reese lecture. In Jakobiec FA, ed. Ocular and Adnexal Tumors. Birmingham, AL, Aesculapius Publishing Company, 1978:600–630. 92. Folberg R, McLean IW, Zimmerman LE: Conjunctival acquired melanosis ad
malignant melanoma. Ophthalmology 1984;91:673–678, 1984. 93. Liesegang TJ, Campbell RJ: Mayo Clinic experience with conjunctival melanomas. Arch Ophthalmol 98:1385–1389, 1980. 94. Helm CJ: Melanoma and other pigmented lesions of the ocular surface. In: Focal
points: clinical modules for ophthalmologists, vol 14. San Francisco, American
Academy of Ophthalmology, 1996:11. 95. Silvers D, Jakobiec FA, Freeman T et al: Melanoma of the conjunctiva: a
clinicopathologic study. In: Jakobiec FA, ed. Ocular and adnexal tumors. Birmingham, AL, Aesculapius Publishing Company, 1978:583–599. 96. Crawford JB: Conjunctival melanomas: prognostic factors. A review and an analysis of
a series. Trans Am Ophthalmol Soc 78:467–502, 1980. 97. Folberg R, McLean IW, Zimmerman LE: Malignant melanoma of the conjunctiva. Hum Pathol 16:136–143, 1985. 98. Albert DM, Jakobiec FA, eds. Principles and practice of ophthalmology, 2nd
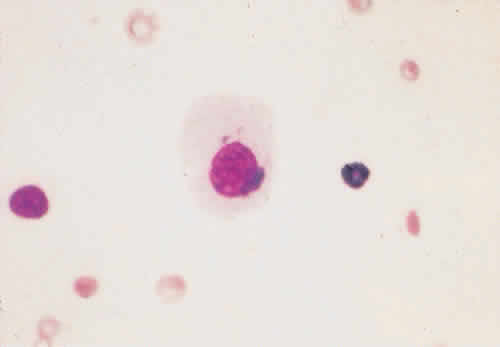
ed. Philadelphia, WB Saunders, 1994:286–289, 2132–2134. |