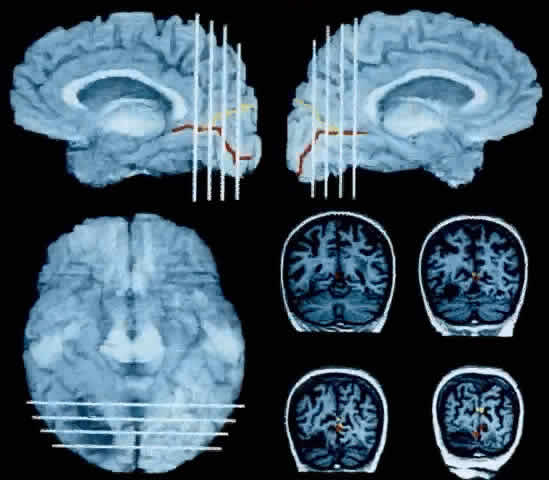
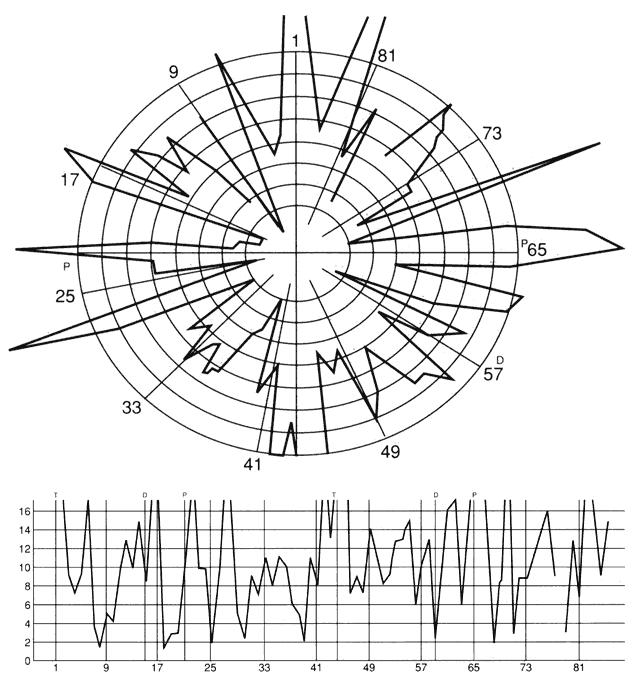
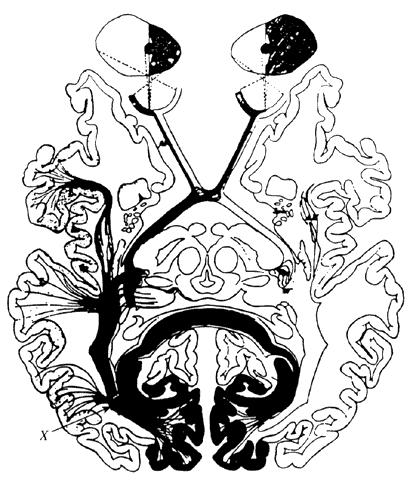
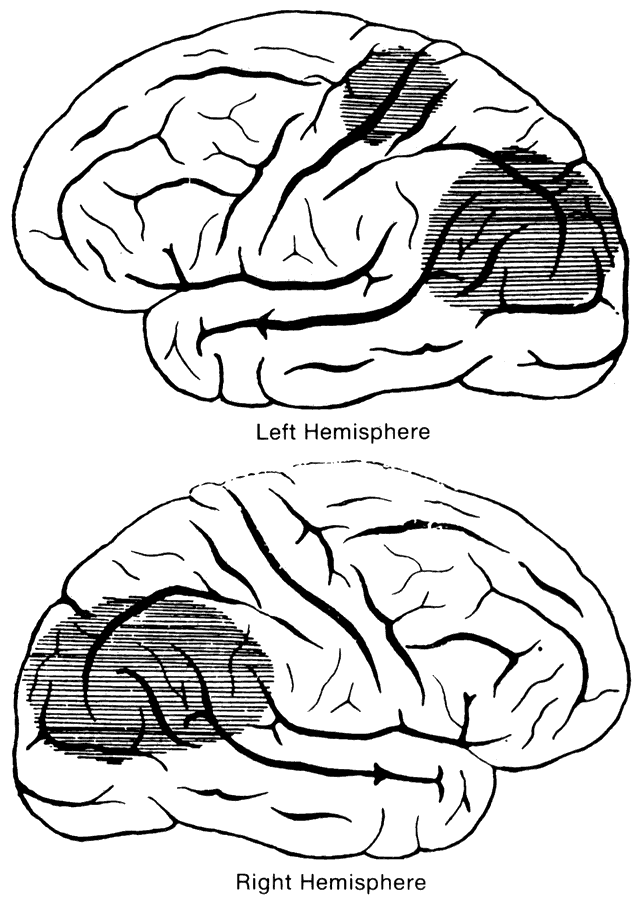
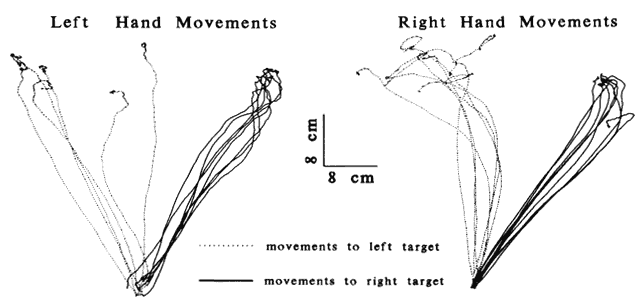
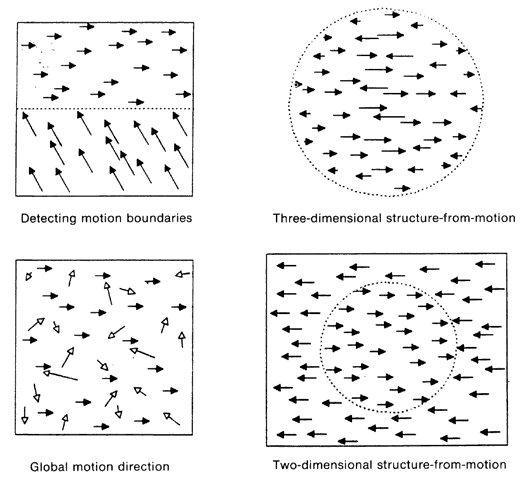
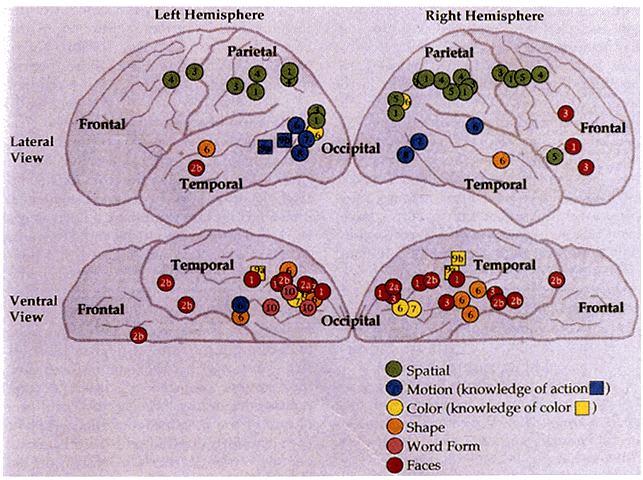
1. Schiller PH, Logothetis NK: The color-opponent and broad-based channels of the primate visual system. Trends Neurosci 11:392, 1990 2. Livingstone MS, Hubel DH: Psychophysical evidence for separate channels for the perception of form, color, movement, and
depth. J Neurosci 7:3416, 1987 3. Felleman DJ, Van Essen DC: Distributed hierarchical processing in the primate cerebral cortex. Cereb Cortex 1:1, 1991 4. Merigan WH, Maunsell JHR: How parallel are the primate visual pathways? Annu Rev Neurosci 16:369, 1993 5. Martin KAC: Parallel pathways converge. Curr Biol 2:555, 1992 6. Lachica EA, Beck PD, Casagrande VA: Parallel pathways in macaque monkey striate cortex: anatomically defined
columns in layer III. Proc Nat Acad Sci USA 89:3566, 1992 7. Ungerleider LG, Mishkin M: Two cortical visual systems. In Ingle DJ, Mansfield
RJW, Goodale MS (eds): The Analysis of Visual Behaviour, pp 549–586. Cambridge, MA, MIT Press, 1982 8. Damasio AR: Disorders of complex visual processing: Agnosias, achromatopsia, Balint's
syndrome, and related difficulties of orientation
and construction. In Mesulam M-M (ed): Principles of Behavioral Neurology, pp 259–288. Philadelphia, FA Davis, 1985 9. Schiller PH, Lee K: The role of the primate extrastriate area V4 in vision. Science 251:1251, 1991 10. Schiller PH: The effects of V4 and middle temporal (MT) area lesions on visual performance
in the Rhesus monkey. Vis Neurosci 10:717, 1993 11. Desimone R, Schein S, Moran J, Ungerleider L: Contour, color and shape analysis beyond the striate cortex. Vision Res 25:441, 1985 12. Fujita I, Tanaka K, Ito M, Cheng K: Columns for visual features of objects in monkey inferotemporal cortex. Nature 360:343, 1992 13. Boussaoud D, Ungerleider LG, Desimone R: Pathways for motion analysis: Connections of the medial superior temporal
and fundus of the superior temporal visual areas in the macaque. J Comp Neurol 296:462, 1990 14. Ungerleider LG, Desimone R: Cortical connections of visual area MT in the macaque. J Comp Neurol 248:190, 1986 15. Logothetis NK, Schall JD: Neuronal correlates of subjective visual perception. Science 256:761, 1989 16. Brodmann K: Vergleichende Lokalisationslehre der Grosshirnrinde in ihren
Printzipien dargestellt auf Grund des Zellenbaues. Leipzig, JA Barth, 1909 17. Inouye T: Die Sehstorungen bei Schussverletzungen der kortikalen Sesphare. Leipzig, Engelmann, 1909 18. Holmes G, Lister WT: Disturbances of vision from cerebral lesions with special reference to
the cortical representation of the macula. Brain 39:34, 1916 19. Horton JC, Hoyt WF: The representation of the visual field in human striate cortex: A revision
of the classic Holmes map. Arch Ophthalmol 109:816, 1991 20. Anderson DR: Testing the Field of Vision. St. Louis, CV Mosby, 1982 21. Smith CG, Richardson WFG: The course and distribution of the arteries supplying the visual (striate) cortex. Am J Ophthalmol 61:1391, 1966 22. Huber A: Homonymous hemianopia after occipital lobectomy. Am J Ophthalmol 54:623, 1962 23. Tootell RBH, Switkes E, Silverman MS, Hamilton SL: Functional anatomy of macaque striate cortex: II. Retinotopic organization. J Neurosci 8:1531, 1988 24. Spalding JMK: Wounds of the visual pathway: Part II. The striate cortex. J Neurol Neurosurg Psychiatry 15:169, 1952 25. Benton S, Levy I, Swash M: Vision in the temporal crescent in occipital infarction. Brain 103:83, 1980 26. Halpern JI, Sedler RR: Traumatic bilateral homonymous hemianopic scotomas. Ann Ophthalmol 12:1022, 1980 27. Sandifer PH: Anosognosia and disorders of body scheme. Brain 69:122, 1946 28. Geschwind N: Disconnexion syndromes in animals and man. Brain 88:17, 1965 29. McDaniel KD, McDaniel LD: Anton's syndrome in a patient with posttraumatic optic neuropathy
and bifrontal contusions. Arch Neurol 48:101, 1991 30. Hartmann JA, Wolz WA, Roeltgen DP, Loverso FL: Denial of visual perception. Brain Cogn 16:29, 1991 31. Teuber HL: Alteration of perception and memory in man. In Weiskrantz L (ed): Analysis
of Behavioural Change. New York, Harper & Row, 1968 32. Pöppel E, Held R, Frost D: Residual visual function after brain wounds involving the central visual
pathways in man. Nature 243:295, 1973 33. Weiskrantz L, Warrington EK, Sanders MD, Marshall J: Visual capacity in the hemianopic field following a restricted occipital
ablation. Brain 97:709, 1974 34. Weiskrantz L: Residual vision in a scotoma: A follow-up study of “form” discrimination. Brain 110:77, 1987 35. Sanders MD, Warrington E, Marshall J, Weiskrantz L: “Blindsight”: Vision in a field defect. Lancet April 20:707, 1974 36. Blythe IM, Kennard C, Ruddock KH: Residual vision in patients with retrogeniculate lesions of the visual
pathways. Brain 110:887, 1987 37. Barbur JL, Ruddock KH, Waterfield VA: Human visual responses in the absence of the geniculo-calcarine projection. Brain 103:905, 1980 38. Barbur JL, Forsyth PM, Findlay JM: Human saccadic eye movements in the absence of the geniculocalcarine projection. Brain 111:63, 1988 39. Weiskrantz L: Outlooks for blindsight: Explicit methodologies for implicit processes. Proc R Soc Lond [B] 239:257, 1990 40. Perenin M-T: Visual functions within the hemianopic field following early cerebral hemidecortication
in man: II. Pattern discrimination. Neuropsychologia 16:697, 1978 41. Stoerig P, Hébner M, Pöppel E: Signal detection analysis of residual vision in a field defect due to a
post-geniculate lesion. Neuropsychologia 23:589, 1985 42. Stoerig P, Cowey A: Increment-threshold spectral sensitivity in blindsight: Evidence for colour
opponency. Brain 114:1487, 1991 43. Campion J, Latto R, Smith YM: Is blindsight an effect of scattered light, spared cortex, and near-threshold
vision? Behav Brain Sci 6:423, 1983 44. Meeres SL, Graves RE: Localization of unseen stimuli by humans with normal vision. Neuropsychologia 28:1231, 1990 45. Stoerig P: Chromaticity and achromaticity: Evidence for a functional differentiation
in visual field defects. Brain 110:869, 1987 46. Balliet R, Blood KMT, Bach-Y-Rita P: Visual field rehabilitation in the cortically blind? J Neurol Neurosurg Psychiatry 48:1113, 1985 47. Perenin M-T, Ruel J, Hècaen H: Residual visual capacities in a case of cortical blindness. Cortex 6:605, 1980 48. Perenin M-T, Jeannerod M: Residual function in cortically blind hemifields. Neuropsychologia 13:1, 1975 49. Perenin M-T, Jeannerod M: Visual functions within the hemianopic field following early cerebral hemidecortication
in man: I. Spatial localization. Neuropsychologia 16:1, 1978 50. Meienberg O, Zangemeister WH, Rosenberg M et al: Saccadic eye movement strategies in patients with homonymous hemianopia. Ann Neurol 9:537, 1981 51. Corbetta M, Marzi CA, Tassinari G, Aglioti S: Effectiveness of different task paradigms in revealing blindsight. Brain 113:603, 1990 52. Ptito A, Lepore F, Ptito M, Lassonde M: Target detection and movement discrimination in the blind field of hemispherectomized
patients. Brain 114:497, 1991 53. Bridgeman B, Staggs D: Plasticity in human blindsight. Vision Res 22:1199, 1982 54. Riddoch G: Dissociation of visual perception due to occipital injuries with especial
reference to appreciation of movement. Brain 40:15, 1917 55. Perenin M-T: Discrimination of motion direction in perimetrically blind fields. Neuroreport 2:397, 1991 56. Heide W, Koenig E, Dichgans J: Optokinetic nystagmus, self-motion sensation and their after effects in
patients with occipito-parietal lesions. Clin Vision Sci 5:145, 1990 57. ter Braak JWG, Schenk VWD, van Vliet AGM: Visual reactions in a case of long-lasting cortical blindness. J Neurol Neurosurg Psychiatry 34:140, 1971 58. Pizzamiglio L, Antonucci G, Francia A: Response of the cortically blind hemifields to a moving stimulus. Cortex 20:89, 1984 59. Mestre DR, Brouchon M, Ceccaldi M, Poncet M: Perception of optical flow in cortical blindness: A case report. Neuropsychologia 30:783, 1992 60. Torjussen T: Visual processing in cortically blind hemifields. Neuropsychologia 16:15, 1978 61. Ptito A, Lassonde M, Lepore F, Ptito M: Visual discrimination in hemispherectomized patients. Neuropsychologia 25:869, 1987 62. Hess RF, Pointer JS: Spatial and temporal contrast sensitivity in hemianopia. Brain 112:871, 1989 63. Rafal R, Smith J, Krantz J et al: Extrageniculate vision in hemianopic humans: Saccade inhibition by signals
in the blind field. Science 250:118, 1990 64. Marzi CA, Tassinari G, Agliotti S, Lutzemberger L: Spatial summation across the vertical meridian in hemianopics: A test of
blindsight. Neuropsychologia 24:749, 1986 65. Zihl J: “Blindsight”: Improvement of visually guided eye movements
by systematic practice in patients with cerebral blindness. Neuropsychologia 18:71, 1980 66. Zihl J, Werth R: Contributions to the study of “blindsight”: I. Can stray light
account for saccadic localization in patients with postgeniculate
visual field defects? Neuropsychologia 22:1, 1984 67. Zihl J, Werth R: Contributions to the study of “blindsight”: II. The role of
specific practice for saccadic localization in patients with postgeniculate
visual field defects. Neuropsychologia 22:13, 1984 68. Zihl J, von Cramon D: Restitution of visual function in patients with cerebral blindness. J Neurol Neurosurg Psychiatry 42:312, 1979 69. Zihl J: Recovery of visual function in patients with cerebral blindness. Exp Brain Res 24:159, 1981 70. Bender DB: Electrophysiological and behavioural experiments on the primate pulvinar. Prog Brain Res 75:55, 1988 71. Bender DB: Visual activation of neurons in the primate pulvinar depends on cortex
but not colliculus. Brain Res 279:258, 1983 72. Blythe IM, Bromley JM, Kennard C, Ruddock KH: Visual discrimination of target displacement remains after damage to the
striate cortex in humans. Nature 320:619, 1986 73. Cowey A, Stoerig P: The neurobiology of blindsight. Trends Neurosci 14:140, 1991 74. Weiskrantz L: Behavioural analysis of the monkey's visual nervous system. Proc Royal Soc London B Biol Sci 182:427, 1972 75. Mohler CW, Wurtz RH: Role of striate cortex and superior colliculus in visual guidance of saccadic
eye movements in monkey. J Neurophysiol 40:74, 1977 76. Keating EG: Residual spatial vision in the monkey after removal of striate and preoccipital
cortex. Brain Res 187:271, 1980 77. Zee DS, Tusa RJ, Herdman SJ et al: Effects of occipital lobectomy upon eye movements in primate. J Neurophysiol 58:883, 1987 78. Segraves MA, Goldberg ME, Deng S-Y et al: The role of striate cortex in the guidance of eye movements in the monkey. J Neurosci 7:3040, 1987 79. Gross CG: Contributions of striate cortex and the superior colliculus to visual functions
in area MT, the superior temporal polysensory area and inferior
temporal cortex. Neuropsychologia 29:497, 1991 80. Rodman HR Gross CG, Albright TD: Afferent basis of visual response properties in area MT of the macaque: I. Effects
of striate cortex removal. J Neurosci 9:2033, 1989 81. Girard P, Salin PA, Bullier J: Visual activity in areas V3a and V3 during reversible inactivation of area
V1 in the macaque monkey. J Neurophysiol 66:1493, 1991 82. Girard P, Salin PA, Bullier J: Response selectivity of neurons in area MT of the macaque monkey during
reversible inactivation of area V1. J Neurophysiol 67:1437, 1992 83. Rodman HR, Gross CG, Albright TD: Afferent basis of visual response properties in area MT of the macaque: II. Effects
of superior colliculus removal. J Neurosci 10:1154, 1990 84. Fendrich R, Wessinger CM, Gazzaniga MS: Residual vision in a scotoma: Implications for blindsight. Science 258:1489, 1992 85. Celesia GG, Bushnell D, Toleikis SC, Brigell MG: Cortical blindness and residual vision: Is the “second” visual
system in humans capable of more than rudimentary visual perception? Neurology 41:862, 1991 85a. Merigan WH, Nealey TA, Maunsell JHR: Visual effects of lesions of cortical area V2 in macaques. J Neurosci 13:3180, 1993 86. Braddick O, Atkinson J, Hood B et al: Possible blindsight in infants lacking one cerebral hemisphere. Nature 360:461, 1992 87. Lissauer H: Ein fall von Seelenblindheit nebst einem Bitrag zur Theorie derselben. Arch Psychiatr Nervenkr 2:22, 1890 87a. Bodamer J: Prosopagnosie. Arch Psychiatr Nervenkr 179:6, 1947 88. Benton A: Facial recognition 1990. Cortex 26:491, 1990 89. Damasio AR: Prosopagnosia. Trends Neurosci 8:132, 1985 90. Young AW, Ellis HD: Childhood prosopagnosia. Brain Cogn 9:16, 1989 91. de Haan EHF, Campbell R: A fifteen year follow-up of a case of developmental prosopagnosia. Cortex 27:489, 1991 92. Landis T, Cummings JL, Christen L et al: Are unilateral right posterior lesions sufficient to cause prosopagnosia?: Clinical
and radiological findings in six additional patients. Cortex 22:243, 1986 93. Bruyer R, Laterre C, Seron X et al: A case of prosopagnosia with some preserved covert remembrance of familiar
faces. Brain Cogn 2:257, 1983 94. de Haan EHF, Young AW, Newcombe F: Face recognition without awareness. Cogn Neuropsychol 4:385, 1987 95. Sergent J, Villemure J-G: Prosopagnosia in a right hemispherectomized patient. Brain 112:975, 1989 96. Sergent J, Poncet M: From covert to overt recognition of faces in a prosopagnosic patient. Brain 113:989, 1990 97. Campbell R, Landis T, Regard M: Face recognition and lipreading: A neurological dissociation. Brain 109:509, 1986 98. Lhermitte F, Chain F, Escourolle R et al: Ötude anatomo-clinique d'un cas de prosopagnosie. Rev Neurol 126:329, 1972 99. Whitely AM, Warrington EK: Prosopagnosia: A clinical, psychological and anatomical study of three
patients. J Neurol Neurosurg Psychiatry 40:395, 1977 100. Damasio AR, Damasio H, van Hoesen GW: Prosopagnosia: Anatomic basis and behavioural mechanisms. Neurology 32:331, 1982 101. Farah MJ: Visual agnosia: Disorders of visual recognition and what they
tell us about normal vision. Cambridge, MIT Press, 1990 102. de Renzi E: Current issues in prosopagnosia. In Ellis HD, Jeeves MA, Newcome
F, Young A (eds): Aspects of Face Processing. Martinus Nijhoff, Dordecht, 1986 103. McNeil JE, Warrington EK: Prosopagnosia: A face-specific disorder. Q J Exp Psychol 46A:1, 1993 104. Malone DR, Morris HH, Kay MC, Levin HS: Prosopagnosia: A double dissociation between the recognition of familiar
and unfamiliar faces. J Neurol Neurosurg Psychiatry 45:820, 1982 105. Levine DN, Warach J, Farah M: Two visual systems in mental imagery: Dissociation of “what” and “where” in imagery disorders due to bilateral posterior
cerebral lesions. Neurology 35:1010, 1985 106. Rizzo M, Hurtig R, Damasio AR: The role of scanpaths in facial recognition and learning. Ann Neurol 22:41, 1987 107. Bauer RM, Verfaellie M: Electrodermal discrimination of familiar but not unfamiliar faces in prosopagnosia. Brain Cogn 8:240, 1988 108. Renault B, Signoret J-L, DeBruille B et al: Brain potentials reveal covert facial recognition in prosopagnosia. Neuropsychologia 27:905, 1989 109. Rizzo M, Corbett JJ, Thompson HS, Damasio AR: Spatial contrast sensitivity in facial recognition. Neurology 36:1254, 1986 110. Bauer RM: Autonomic recognition of names and faces in prosopagnosia: A neuropsychological
application of the guilty knowledge test. Neuropsychologia 22:457, 1984 111. Damasio H, Frank R: Three-dimensional in vivo mapping of brain lesions in humans. Arch Neurol 49:137, 1992 112. Meadows JC: The anatomical basis of prosopagnosia. J Neurol Neurosurg Psychiatry 37:489, 1974 113. Benson DF, Segarra J, Albert ML: Visual agnosia-prosopagnosia. Arch Neurol 30:307, 1974 114. Tyrell PJ, Warrington EK, Frackowiak RSJ, Rossor MN: Progressive degeneration of the right temporal lobe studied with positron
emission tomography. J Neurol Neurosurg Psychiatry 53:1048, 1990 115. Kay MC, Levin HS: Prosopagnosia. Am J Ophthalmol 94:75, 1982 116. Hècaen H, Angelergues R: Agnosia for faces (prosopagnosia). Arch Neurol 7:92, 1962 117. Assal G: Regression des troubles de la reconnaissance des physiognomies et de la
mèmoire topographique chez un malade opèrè d'un
hèmatome intracèrèbral parièto-temporal droit. Rev Neurol 121:184, 1969 118. de Renzi E: Prosopagnosia in two patients with CT scan evidence of damage confined
to the right hemisphere. Neuropsychologia 24:385, 1986 119. Michel F, Perenin M-T, Sieroff E: Prosopagnosie sans hèmianopsie après lèsion unilatèrale
occipito-temporale droite. Rev Neurol 142:545, 1986 120. Michel F, Poncet M, Signoret JL: Les lèsions responsables de la prosopagnosie sont-elles toujours
bilaterales? Rev Neurol 146:764, 1989 121. Landis T, Regard M, Bliestle A, Kleihues P: Prosopagnosia and agnosia for non-canonical views. Brain 111:1287, 1988 122. de Renzi E, Faglioni P, Grossi D, Nichelli P: Apperceptive and associative forms of prosopagnosia. Cortex 27:213, 1991 123. Tranel D, Damasio AR: Knowledge without awareness: An autonomic index of facial recognition by
prosopagnosics. Science 228:1453, 1985 124. Ettlin TM, Beckson M, Benson DF et al: Prosopagnosia: A bihemispheric disorder. Cortex 28:129, 1992 125. Bruyer R: Covert facial recognition in prosopagnosia: A review. Brain Cogn 15:223, 1991 126. McNeil JE, Warrington EK: Prosopagnosia: A reclassification. Q J Exp Psychol 43A:267, 1991 127. Bay E: Disturbances of visual perception and their examination. Brain 76:515, 1953 128. Cohn R, Neumann MA, Wood DH: Prosopagnosia: A clinicopathological study. Ann Neurol 1:177, 1977 129. Hoff H, Potzl O: Über eine optisch-agnostische Störung des ‘Physiognomie-Gedachtnibes’ (Bezeihungen zur Réckbildung ein Wortblindheit. Z Gesamte Neurol Psychiat 159:367, 1937 130. Bruce V, Young A: Understanding face recognition. Br J Psychol 77:305, 1986 131. Levine DN, Calvanio R: Prosopagnosia: A defect in visual configural processing. Brain Cogn 10:149, 1989 132. Rentschler I, Treutwein B, Landis T: Dissociation of local and global processing in visual agnosia. Vision Res 34:963, 1994 133. Pallis CA: Impaired identification of faces and places with agnosia for colours. J Neurol Neurosurg Psychiatry 18:218, 1955 134. Korner F, Regli F, Haynal A: Eine durch Farbinnstörung, Prosopagnosie und Orienterungs-störung
charakterisierte visuelle Agnosie. Arch Psychiatr NervKrankh 209:1, 1967 135. Meadows JC: Disturbed perception of colours associated with localized cerebral lesions. Brain 97:615, 1974 136. MacKay G, Dunlop JC: The cerebral lesions in a case of complete acquired colour-blindness. Scott Med Surg J 5:503, 1899 137. Rizzo M, Smith V, Pokorny J, Damasio AR: Color perception profiles in central achromatopsia. Neurology 43:995, 1993 138. Lenz G: Zwei Sectionsfalle doppelseitiger zentrale Farbenhemianopsie. Z Gesamte Neurol Psychiat 71:135, 1921 139. Sacks O: The case of the colour-blind painter. In: An Anthropologist on
Mars. New York, Alfred A Knopf, 1995 140. Critchley M: Acquired anomalies of colour perception of central origin. Brain 88:711, 1965 141. Victor JD, Maiese K, Shapley R et al: Acquired central dyschromatopsia: Analysis of a case with preservation
of color discrimination. Clin Vision Sci 4:183, 1989 142. Heywood CA, Cowey A, Newcombe F: Chromatic discrimination in a cortically blind observer. Eur J Neurosci 3:802, 1991 143. Ogden JA: Visual object agnosia, prosopagnosia, achromatopsia, loss of visual imagery, and
autobiographical amnesia following recovery from cortical blindness: Case
M.H. Neuropsychologia 31:571, 1993 144. Green GJ, Lessell S: Acquired cerebral dyschromatopsia. Arch Ophthalmol 95:121, 1977 145. Paulson HL, Galetta SL, Grossman M, Alavi A: Hemiachromatopsia of unilateral occipitotemporal infarcts. Am J Ophthalmol 118:518, 1994 146. Albert ML, Reches A, Silverberg R: Hemianopic colour blindness. J Neurol Neurosurg Psychiatry 38:546, 1975 147. Kölmel HW: Pure homonymous hemiachromatopsia: Findings with neuro-ophthalmologic examination
and imaging procedures. Eur Arch Psychiatry Neurol Sci 237:237, 1988 148. Damasio A, Yamada T, Damasio H et al: Central achromatopsia: Behavioral, anatomic, and physiologic aspects. Neurology 30:1064, 1980 149. Zeki SM: A century of cerebral achromatopsia. Brain 113:1721, 1990 150. Pearlman AL, Birch J, Meadows JC: Cerebral color blindness: An acquired defect in hue discrimination. Ann Neurol 5:253, 1979 151. Lueck CJ, Zeki S, Friston KJ et al: The colour center in the cerebral cortex of man. Nature 340:386, 1989 152. de Renzi E, Spinnler H: Impaired performance in colour tasks in patients with hemispheric damage. Cortex 3:194, 1967 153. Aldrich MS, Vanderzant CW, Alessi AG et al: Ictal cortical blindness with permanent visual loss. Epilepsia 30:116, 1989 154. Freedman L, Costa L: Pure alexia and right hemiachromatopsia in posterior dementia. J Neurol Neurosurg Psychiatry 55:500, 1992 155. Lawden MC, Cleland PG: Achromatopsia in the aura of migraine. J Neurol Neurosurg Psychiatry 56:708, 1993 156. Heywood CA, Wilson B, Cowey A: A case study of cortical colour “blindness” with relatively
intact achromatic discrimination. J Neurol Neurosurg Psychiatry 50:22, 1987 157. Wyszecki G, Stiles WS: Color science: Concepts and methods, quantitative
data and formulae. New York, John Wiley & Sons, 1982 158. Kaiser PK, Boynton RM: Human color vision, 2nd ed. Optical Society of America, USA, 1996 159. Land EH: Recent advances in retinex theory. Vision Res 26:7, 1986 160. Land EH, Hubel DH, Livingstone MS et al: Colourgenerating interactions across the corpus callosum. Nature 303:616, 1983 161. Zeki SM: Colour coding in the cerebral cortex: The reaction of cells in monkey visual
cortex to wavelengths and colours. Neuroscience 9:741, 1983 162. Zeki SM: Colour coding in the cerebral cortex: The responses of wavelength-selective
and colour-coded cells in monkey visual cortex to changes in wavelength
composition. Neuroscience 9:767, 1983 163. Schein SJ, Desimone R: Spectral properties of V4 neurons in the macaque. J Neurosci 10:3369, 1990 164. Heywood CA, Cowey A: On the role of cortical area V4 in the discrimination of hue and pattern
in macaque monkeys. J Neurosci 7:2601, 1987 165. Heywood CA, Gadotti A, Cowey A: Cortical area V4 and its role in the perception of color. J Neurosci 12:4056, 1992 166. Wild HM, Butler SR, Carden D, Kulikowski JJ: Primate cortical area V4 important for colour constancy but not wavelength
discrimination. Nature 313:133, 1985 167. Dean P: Visual cortex ablation and thresholds for successively presented stimuli
in Rhesus monkeys: II. Hue. Exp Brain Res 35:69, 1979 168. Holmes G: Pure word blindness. Folia Psychiatr Neurol Neurochir Neerl 53:279, 1950 169. Geschwind N, Fusillo M: Color-naming defects in association with alexia. Arch Neurol 15:137, 1966 170. Oxbury JM, Oxbury SM, Humphrey NK: Varieties of colour anomia. Brain 92:847, 1969 171. de Vreese LP: Two systems for color-naming defects: Verbal disconnection versus colour
imagery disorder. Neuropsychologia 29:1, 1991 172. Davidoff JB, Ostergaard AL: Colour anomia resulting from weakened short-term colour memory. Brain 107:415, 1984 173. Kinsbourne M, Warrington EK: Observations on colour agnosia. J Neurol Neurosurg Psychiatry 27:296, 1964 174. Black SE, Behrmann M: Localization in alexia. In: Localization and Neuroimaging
in Neuropsychology, pp 331–376. New York, Academic Press, 1994 175. Galaburda AM, Rosen GD, Drislane FW, Livingstone MS: Physiological and anatomical evidence for a magnocellular defect in developmental
dyslexia (abstr). Soc Neurosci 17:20, 1991 176. Livingstone MS, Rosen GD, Drislane FW, Galaburda AM: Physiological and anatomical evidence for magnocellular defect in developmental
dyslexia. Proc Natl Acad Sci USA 88:7943, 1991 177. Dejerine J: Sur un cas de cecite verbale avec agraphie, suive d'autopsie. CR Societè du Biologie 43:197, 1891 178. Dejerine J: Contributions a l'ètude anatomopathologique et clinique des differentes
varietes de cecite verbale. Memoires de la Societè Biologique 44:61, 1892 179. Damasio AR, Damasio H: The anatomic basis of pure alexia. Neurology 33:1573, 1983 180. Binder JR, Mohr JP: The topography of callosal reading pathways: A case control analysis. Brain 115:1807, 1992 181. Bub DN, Black SE, Howell J: Word recognition and orthographic context effects in a letter-by-letter
reader. Brain Lang 36:357, 1989 182. Coslett HB, Saffran EM, Greenbaum S, Schwartz H: Reading in pure alexia. Brain 116:21, 1993 183. Greenblatt SH: Alexia without agraphia or hemianopia. Brain 96:307, 1973 184. Vincent FM, Sadowsky CH, Saunders RL, Reeves AG: Alexia without agraphia, hemianopia, or color-naming defect: A disconnection
syndrome. Neurology 27:689, 1977 185. Henderson VW, Friedman RB, Teng EL, Weiner JM: Left hemisphere pathways in reading: Inferences from pure alexia without
hemianopia. Neurology 35:962, 1985 186. Iragui V, Kritchevsky M: Alexia without agraphia or hemianopia in parietal infarction. J Neurol Neurosurg Psychiatry 54:841, 1991 187. de Renzi E, Zambolin A, Crisi G: The pattern of neuropsychological impairment associated with left posterior
cerebral artery infarcts. Brain 110:1099, 1987 188. Albert ML, Yamadori A, Gardner H, Howes D: Comprehension in alexia. Brain 96:317, 1973 189. Coslett HB, Saffran EM: Evidence for preserved reading in pure alexia. Brain 112:327, 1989 190. Caplan LR, Hedley-White T: Cuing and memory dysfunction in alexia without agraphia: A case report. Brain 97:251, 1974 191. Warrington EK, Shallice T: Word-form dyslexia. Brain 103:99, 1980 192. Behrmann M, Black SE, Bub DN: The evolution of pure alexia: A longitudinal study of recovery. Brain Lang 39:405, 1990 193. Silver FL, Chawluk JB, Bosley TM et al: Resolving metabolic abnormalities in a case of pure alexia. Neurology 38:731, 1988 194. Stommel EW, Friedman RJ, Reeves AG: Alexia without agraphia associated with spleniogeniculate infarction. Neurology 41:587, 1991 195. Vaina LM: Functional segregation of color and motion processing in the human visual
cortex: Clinical evidence. Cereb Cortex 5:555, 1994 196. Greenblatt SH: Localization of lesions in alexia. In Kertesz A (ed): Localization
in Neuropsychology, p 323–356. New York, Academic Press, 1983 197. Gazzaniga MS, Freedman H: Observations on visual processes after posterior callosal section. Neurology 23:1126, 1973 198. Castro-Caldas A, Salgado V: Right hemifield alexia without hemianopia. Arch Neurol 41:84, 1984 199. Binder JR, Lazar RM, Tatemichi TK et al: Left hemiparalexia. Neurology 42:562, 1992 200. Benson DF: Alexia. In Bruyn GW, Klawans HL, Vinken PJ (eds): Handbook of
Clinical Neurology, pp 433–455. New York, Elsevier, 1985 201. Kawahata N, Nagata K: Alexia with agraphia due to the left posterior inferior temporal lobe lesion: Neuropsychological
analysis and its pathogenetic mechanisms. Brain Lang 33:296, 1988 202. Benson DF, Brown J, Tomlinson EB: Varieties of alexia: Word and letter blindness. Neurology 21:951, 1971 203. Benson DF: The third alexia. Arch Neurol 34:327, 1977 204. Kirkham TH: The ocular symptomology of pituitary tumors. Proc R Soc Med 65:517, 1972 205. Shallice T, Warrington EK: The possible role of selective attention in acquired dyslexia. Neuropsychologia 15:31, 1977 206. Levine DN, Calvanio R: A study of the visual defect in verbal alexia-simultanagnosia. Brain 101:65, 1978 207. Behrmann M, Moscovitch M, Black SE, Mozer M: Perceptual and conceptual factors in neglect dyslexia: Two contrasting
case studies. Brain 113:1163, 1990 208. Bálint R: Seelenlahmung des ‘Schauens’, optische Ataxie, räumliche
Storung der Aufmerksamkeit. Monatschrift für Psychiatrie und Neurologie 25:51, 1909 209. Wolpert T: Die Simultanagosie. Zeischrift für gesamte Neurologie und Psychiatrie 93:397, 1924 210. Holmes G: Disturbance of vision by cerebral lesions. Br J Ophthalmol 2:353, 1918 210a. Hécaen H, de Ajuriaguerra J: Bálint's syndrome (psychic paralysis of visual fixation) and
its minor forms. Brain 77:373, 1954 211. Luria AR: Disorders of ‘simultaneous perception’ in a case of bilateral
occipitoparietal brain injury. Brain 82:437, 1959 212. Luria AR, Pravdina-Vinarskaya EN, Yarbus AL: Disturbances of ocular movement in a case of simultanagnosia. Brain 86:219, 1962 213. Rizzo M: Bálint's syndrome and associated visuospatial disorders. In
Kennard C (ed): Bailliere's International Practice and
Research, pp 415–437. Philadelphia, WB Saunders, 1993 214. Ball KK, Roenker DL, Bruni JR: Developmental changes in attention and visual
search throughout adulthood. In JT Enns (ed): The Development of
Attention: Research and Theory, pp 489–507. New York, Elsevier
NorthHolland, 1990 215. Moran J, Desimone R: Selective attention gates visual processing in the extrastriate cortex. Science 229:782, 1985 216. Spitzer J, Desimone R, Moran J: Increased attention enhances both behavioral and neuronal performance. Science 240:338, 1988 217. Corbetta M, Miezin FM, Dobmeyer S et al: Selective and divided attention during visual discriminations of shape, color
and speed: Functional anatomy by positron emission tomography. J Neurosci 11:2383, 1991 218. Damasio AR, Benton AL: Impairment of hand movements under visual guidance. Neurology 29:170, 1979 219. Zihl J, von Cramon D, Mai N: Selective disturbance of movement vision after bilateral brain damage. Brain 106:313, 1983 220. Montero J, Pena J, Genis D et al: Bálint's syndrome: Report of four cases with watershed parieto-occipital
lesions from vertebrobasilar ischemia or systemic hypotension. Acta Neurol Belg 82:270, 1982 221. Hijdra A, Meerwaldt JD: Bálint's syndrome in a man with border-zone infarcts caused
by atrial fibrillation. Clin Neurol Neurosurg 86:51, 1984 222. Schnider A, Landis T, Regard M: Bálint's syndrome in subacute HIV encephalitis. J Neurol Neurosurg Psychiatry 54:822, 1991 223. Hof PR, Bouras C, Constantinidis J et al: Selective disconnection of specific visual association pathways in cases
of Alzheimer's disease presenting the Bálint's syndrome. J Neuropathol Exp Neurol 49:168, 1990 224. Mendez MF, Mendez MA, Martin R et al: Complex visual disturbances in Alzheimer's disease. Neurology 40:439, 1990 225. Tranel D: Assessment of higher-order visual function. Curr Opin Ophthalmol 5:29, 1994 226. Dubner R, Zeki SM: Response properties and receptive fields of cells in an anatomically defined
region of the superior temporal sulcus in the monkey. Brain Res 35:528, 1971 227. Maunsell JHR, van Essen DC: Functional properties of neurons in middle temporal visual area of the
macaque monkey: I. Selectivity for stimulus direction, speed, and orientation. J Neurophysiol 49:1127, 1983 228. Felleman DJ, Kaas JH: Receptive field properties of neurons in middle temporal visual area (MT) of
owl monkeys. J Neurophysiol 52:488, 1984 229. Albright TD: Form-cue invariant motion processing in primate visual cortex. Science 255:1141, 1992 230. Lagae L, Raiguel S, Orban GA: Speed and direction selectivity of macaque middle temporal neurons. J Neurophysiol 69:19, 1993 231. van Essen DC, Maunsell JHR, Bixby JL: The middle temporal visual area in the macaque: Myeloarchitecture, connections, functional
properties and topographic organization. J Comp Neurol 199:293, 1981 232. Maunsell JHR, van Essen DC: The connections of the middle temporal visual area (MT) and their relationship
to a cortical hierarchy in the macaque monkey. J Neurosci 3:2563, 1983 233. Tanaka K, Sugita Y, Moriya M, Saito H-A: Analysis of object motion in the ventral part of the medial superior temporal
area of the macaque visual cortex. J Neurophysiol 69:128, 1993 234. Tanaka K, Saito H-A: Analysis of motion of the visual field by direction, expansion/contraction, and
rotation cells clustered in the dorsal part of the medial superior
temporal area of the macaque monkey. J Neurophysiol 62:626, 1989 235. Duffy CJ, Wurtz RH: Sensitivity of MST neurons to optic flow stimuli: I. A continuum of response
selectivity to large-field stimuli. J Neurophysiol 65:1329, 1991 236. Duffy CJ, Wurtz RH: Response of monkey MST neurons to optic flow stimuli with shifted centers
of motion. J Neurosci 15:5192, 1995 237. Graziano MSA, Andersen RA, Snowden RJ: Tuning of MST neurons to spiral motions. J Neurosci 14:54, 1994 238. Colby CL, Duhamel J-R, Goldberg ME: Ventral intraparietal area of the macaque: Anatomic location and visual
response properties. J Neurophysiol 69:902, 1993 239. Bruce CJ, Desimone R, Gross CG: Visual properties of neurons in a polysensory area in superior temporal
sulcus of the macaque. J Neurophysiol 46:369, 1981 240. Oram MW, Perrett DI, Hietanen JK: Directional tuning of motion-sensitive cells in the anterior superior temporal
polysensory area of the macaque. Exp Brain Res 97:274, 1993 241. Perrett DI, Smith PAJ, Mistlin AJ et al: Visual analysis of body movements by neurons in the temporal cortex of
the macaque monkey: A preliminary report. Behav Brain Res 16:153, 1985 242. Sáry G, Vogels R, Orban G: Cue-invariant shape selectivity of macaque inferior temporal neurons. Science 260:995, 1993 243. Sáry G, Vogels R, Kovcs G, Orban GA: Responses of monkey inferior temporal neurons to luminance-, motion-, and
texture-defined gratings. J Neurophysiol 73:1341, 1995 244. Snowden RJ, Treue S, Erickson RG, Andersen RA: The response of area MT and V1 neurons to transparent motion. J Neurosci 11:2766, 1991 245. Qian N, Andersen RA: Transparent motion perception as detection of unbalanced motion signals: II. Physiology. J Neurosci 14:7367, 1994 246. Qian N, Andersen RA, Adelson EH: Transparent motion perception as detection of unbalanced motion signals: I. Psychophysics. J Neurosci 14:7357, 1994 247. Movshon JA, Adelson EH, Gizzi MS, Newsome WT: The analysis of moving visual
patterns. In Chagas C, Gattass R, Gross CG (eds): Study Group on
Pattern Recognition Mechanisms. Vatican City, Pontifica Academia Scientiarum, 1985 248. Rodman HR, Albright TD: Single-unit analysis of pattern-motion selective properties in the middle
temporal visual area (MT). Exp Brain Res 75:53, 1989 249. Bradley DC, Qian N, Andersen RA: Integration of motion and stereopsis in middle temporal cortical area of
macaques. Nature 373:609, 1995 250. Olavarria JF, DeYoe EA, Knierim JJ et al: Neural responses to visual texture patterns in middle temporal area of
the macaque monkey. J Neurophysiol 68:164, 1992 251. Born RT, Tootell RBH: Segregation of global and local motion processing in primate middle temporal
visual area. Nature 357:497, 1992 252. Allman J, Miezin F, McGuinness E: Direction- and velocity-specific responses from beyond the classical receptive
field in the middle temporal area (MT). Perception 14:105, 1985 253. Tanaka K, Hikosaka K, Saito H-A et al: Analysis of local and wide-field movements in the superior temporal visual
areas of the macaque monkey. J Neurosci 6:134, 1986 254. Newsome WT, Wurtz RH, Dürsteler MR, Mikami A: Deficits in visual motion processing following ibotenic acid lesions of
the middle temporal visual area of the macaque monkey. J Neurosci 5:825, 1985 255. Dürsteler MR, Wurtz RH, Newsome WT: Directional pursuit deficits following lesions of the foveal representation
within the superior temporal sulcus of the macaque monkey. J Neurophysiol 57:1262, 1987 256. Dürsteler MR, Wurtz RH: Pursuit and optokinetic deficits following chemical lesions of cortical
areas MT and MST. J Neurophysiol 60:940, 1988 257. Schiller PH, Lee K: The effects of lateral geniculate nucleus, area V4, and middle temporal (MT) lesions
on visually guided eye movements. Vis Neurosci 11:229, 1994 258. Newsome WT, Parè EB: A selective impairment of motion perception following lesions of the middle
temporal visual area (MT). J Neurosci 8:2201, 1988 259. Yamasaki DS, Wurtz RH: Recovery of function after lesions in the superior temporal sulcus in the
monkey. J Neurophysiol 66:651, 1991 260. Cowey A, Marcar VL: The effect of removing superior temporal cortical areas in the macaque
monkey: I. Motion discrimination using simple dots. Eur J Neurosci 4:1219, 1992 261. Marcar VL, Cowey A: The effect of removing superior temporal cortical areas in the macaque
monkey: II. Motion discrimination using random textured displays. Eur J Neurosci 4:1228, 1992 262. Britten KH, Newsome WT, Saunders RC: Effects of inferotemporal cortex lesions on form-from-motion discrimination
in monkeys. Exp Brain Res 88:292, 1992 263. Salzman CD, Britten KH, Newsome WT: Cortical microstimulation influences perceptual judgements of motion direction. Nature 346:174, 1990 264. Salzman CD, Murasugi CM, Britten KH, Newsome WT: Microstimulation in visual area MT: Effects on direction discrimination
performance. J Neurosci 12:2331, 1992 265. Murasugi CM, Salzman CD, Newsome WT: Microstimulation in visual area MT: Effects of varying pulse amplitude
and frequency. J Neurosci 13:1719, 1993 266. Newsome WT, Britten KH, Movshon JA: Neuronal correlates of a perceptual decision. Nature 341:52, 1989 267. Celebrini S, Newsome WT: Neuronal and psychophysical sensitivity to motion signals in extrastriate
area MST of the macaque monkey. J Neurosci 14:4109, 1994 268. Nakayama K: Biological image motion processing: A review. Vision Res 25:625, 1985 269. Zeki S: Cerebral akinetopsia (visual motion blindness): A review. Brain 114:811, 1991 270. Rizzo M, Nawrot M, Zihl J: Motion and shape perception in cerebral akinetopsia. Brain 118:1105, 1995 271. Zihl J, von Cramon D, Mai N, Schmid C: Disturbance of movement vision after bilateral posterior brain damage: Further
evidence and follow-up observations. Brain 114:2235, 1991 272. Hess RH, Baker CL Jr, Zihl J: The “motion-blind” patient: Low-level spatial and temporal
filters. J Neurosci 9:1628, 1989 273. Baker CL Jr, Hess RF, Zihl J: Residual motion perception in a “motion-blind” patient, assessed
with limited-lifetime random dot stimuli. J Neurosci 11:454, 1991 274. Plant GT, Laxer KD, Barbaro NM et al: Impaired visual motion perception in the contralateral hemifield following
unilateral posterior cerebral lesions in humans. Brain 116:1303, 1993 275. Greenlee MW, Lang HJ, Mergner T, Seeger W: Visual short-term memory of stimulus velocity in patients with unilateral
posterior brain damage. J Neurosci 15:2287, 1995 276. Barton JJS, Sharpe JA, Raymond JE: Retinotopic and directional defects in motion discrimination in humans
with cerebral lesions. Ann Neurol 37:665, 1995 277. Vaina LM: Selective impairment of visual motion interpretation following lesions
of the right occipito-parietal area in humans. Biol Cybern 61:347, 1989 278. Regan D, Giaschi D, Sharpe JA, Hong XH: Visual processing of motion-defined form: Selective failure in patients
with parietotemporal lesions. J Neurosci 12:2198, 1992 279. Nawrot M, Rizzo M, Damasio H: Motion perception in humans with focal cerebral lesions (abstr). Invest Ophthalmol Vis Sci 34 (suppl): 1231, 1993 280. Critchley M: Types of visual perseveration: “Paliopsia” and “illusory
visual spread.” Brain 74:267, 1951 281. Bender MB, Feldman M, Sobin AJ: Palinopsia. Brain 91:321, 1968 282. Bender MB: Polyopia and monocular diplopia of cerebral origin. Arch Neurol Psychiatry 54:323, 1945 283. Michel EM, Troost BT: Palinopsia: Cerebral localization with computed tomography. Neurology 30:887, 1980 284. Kinsbourne M, Warrington EK: A study of visual perseveration. J Neurol Neurosurg Psychiatry 26:468, 1963 285. Cummings JL, Syndulko K, Goldberg Z, Treiman DM: Palinopsia reconsidered. Neurology 32:444, 1982 286. Meadows JC, Munro SSF: Palinopsia. J Neurol Neurosurg Psychiatry 40:5, 1977 287. Young WB, Heros DO, Ehrenberg BL, Hedges TR: Metamorphopsia and palinopsia: Association with periodic lateralizing epileptiform
discharges in a patient with malignant astrocytoma. Arch Neurol 46:820, 1989 288. Swash M: Visual perseveration in temporal lobe epilepsy. J Neurol Neurosurg Psychiatry 42:569, 1979 289. Blythe IM, Bromley JM, Ruddock KH et al: A study of systematic visual perseveration involving central mechanisms. Brain 106:661, 1986 290. Purvin VA: Visual disturbances secondary to clomiphene citrate. Arch Ophthalmol 113:482, 1995 291. Friedman DI, Hu EH, Sadun AA: Neuro-ophthalmic complications of interleukin 2 therapy. Arch Ophthalmol 109:1679, 1991 292. Hughes MS, Lessell S: Trazodone-induced palinopsia. Arch Ophthalmol 18:399, 1990 293. Marneros A, Korner J: Chronic palinopsia in schizophrenia. Psychopathology 26:236, 1993 294. Gates TJ, Stagno SJ, Gulledge AD: Palinopsia posing as psychotic depression. Br J Psychiatry 153:391, 1988 295. Jacome DE: Palinopsia and bitemporal visual extinction on fixation. Ann Ophthalmol 17:251, 1985 296. Lepore FE: Spontaneous visual phenomena with visual loss: 104 patients with lesions
of retinal and neural afferent pathways. Neurology 40:444, 1990 297. Kölmel HW: Complex visual hallucinations in the hemianopic field. J Neurol Neurosurg Psychiatry 48:29, 1985 298. Schultz G, Melzack R: The Charles Bonnet syndrome: ‘Phantom visual images.’ Perception 20:809, 1991 299. Teunisse RJ, Cruysberg JRM, Verbeek A, Zitman FG: The Charles Bonnet syndrome: A large prospective study in the Netherlands. Br J Psychiatry 166:254, 1995 300. Kölmel HW: Coloured patterns in hemianopic fields. Brain 107:155, 1984 301. Plant GT: A centrally generated coloured phosphene. Clin Vis Sci 1:161, 1986 302. Anderson SW, Rizzo M: Hallucinations following occipital lobe damage: The pathological activation
of visual representations. J Clin Exp Neurol 16:651, 1994 303. Weinberger LM, Grant FC: Visual hallucinations and their neuro-optical correlates. Arch Ophthalmol 23:166, 1940 304. Cogan DG: Visual hallucinations as release phenomenon. Albrecht von Graefe's Arch Klin Exp Ophthalmol 188:139, 1973 305. Lance JW: Simple formed hallucinations confined to the area of a specific visual
field defect. Brain 99:719, 1976 306. Siatkowski RM, Zimmer B, Rosenberg PR: The Charles Bonnet syndrome: Visual perceptive dysfunction in sensory deprivation. J Clin Neuro Ophthalmol 10:215, 1990 307. Heron W: The pathology of boredom. Sci Am 196:52, 1957 308. Adair DK, Keshaven MS: The Charles Bonnet syndrome and grief reaction. Am J Psychiatry 145:895, 1988 309. Cole MG: Charles Bonnet hallucinations: A case series. Can J Psychiatry 37:267, 1992 310. Schultz G, Melzack R: Visual hallucinations and mental state: A study of 14 Charles Bonnet syndrome
hallucinators. J Nerv Ment Dis 181:639, 1993 311. Bhatia MS, Khastgir U, Malik SC: Charles Bonnet syndrome. Br J Psychiatry 161:409, 1992 312. Schedlack KJ, McDonald WM, Laskowitz DT, Krishnan KRR: Geniculocalcarine hyperintensities on brain magnetic resonance imaging
associated with visual hallucinations in the elderly. Psychiatry Res 54:283, 1994 313. Fisman M: Musical hallucinations: Report of two unusual cases. Can J Psychiatry 36:609, 1991 314. Penfield W, Perot P: The brain's record of auditory and visual experience. Brain 86:595, 1963 315. Foerster O: The cerebral cortex in man. Lancet 2:309, 1931 316. Rousseau M, Debrock D, Cabaret M, Steinling M: Visual hallucinations with written words in a case of left parietotemporal
lesion. J Neurol Neurosurg Psychiatry 57:1268, 1994 317. Cohen L, Verstichel P, Pierrot-Deseilligny C: Hallucinatory vision of a familiar face following right temporal damage. Neurology 42:2052, 1992 318. Panayiotopoulos CP: Elementary visual hallucinations in migraine and epilepsy. J Neurol Neurosurg Psychiatry 57:1371, 1994 319. Lance JW, Smee RI: Partial seizures with visual disturbance treated by radiotherapy of cavernous
hemangioma. Ann Neurol 26:782, 1989 320. Gastaut H: A new type of epilepsy: Benign partial epilepsy of childhood with occipital
spike-waves. Clin Electroencephalogr 13:13, 1982 321. Lance JW, Anthony M: Some clinical aspects of migraine: A prospective survey of 500 patients. Arch Neurol 15:356, 1966 322. Plant GT: The fortification spectra of migraine. Br Med J 293:1613, 1986 323. Richards W: The fortification illusions of migraine. Sci Am 224:89, 1971 324. Grüsser O-J: Migraine phosphenes and the retino-cortical magnification factor. Vision Res 35:1125, 1995 325. Fox PT, Mintun MA, Raichle ME et al: Mapping human visual cortex with positron emission tomography. Nature 323:806, 1986 326. Sereno MI, Dale AM, Reppas JB et al: Borders of multiple visual areas revealed by functional magnetic resonance
imaging. Science 268:889, 1995 327. Ungerleider LG: Functional brain imaging studies of cortical mechanisms for memory. Science 270:769, 1995 328. Miezin FM, Fox PT, Raichle ME, Allman JM: Localized responses to low contrast moving random dot patterns in human
visual cortex monitored with positron emission tomography (abstr). Soc Neurosci 13:631, 1987 329. Zeki S, Watson JDG, Lueck CJ et al: A direct demonstration of functional specialization in human visual cortex. J Neurosci 11:641, 1991 330. Watson JDG, Myers R, Frackowiak RSJ et al: Area V5 of the human brain: Evidence from a combined study using positron
emission tomography and magnetic resonance imaging. Cereb Cortex 3:79, 1993 331. Dupont P, Orban GA, de Bruyn B et al: Many areas in the human brain respond to visual motion. J Neurophysiol 72:1420, 1994 332. de Jong BM, Shipp S, Skidmore B et al: The cerebral activity related to the visual perception of forward motion
in depth. Brain 117:1039, 1994 333. Tootell RBH, Reppas JB, Kwong KK et al: Functional analysis of human MT and related visual cortical areas using
magnetic resonance imaging. J Neurosci 15:3215, 1995 334. McCarthy G, Spicer M, Adrignolo A et al: Brain activation associated with visual motion studied by functional magnetic
resonance imaging in humans. Human Brain Mapping 2:234, 1995 335. Tootell RBH, Reppas JB, Dale AM et al: Visual motion after-effect in human cortical area MT revealed by functional
magnetic resonance imaging. Nature 375:139, 1995 336. Probst T, Plendl H, Paulus W et al: Identification of the visual motion area (area V5) in the human brain by
dipole source analysis. Exp Brain Res 93:345, 1993 337. Beckers G, Zeki S: The consequences of inactivating areas V1 and V5 in visual motion perception. Brain 118:49, 1995 338. Beckers G, Hömberg V: Cerebral visual motion blindness: transitory akinetopsia induced by transcranial
magnetic stimulation of human area V5. Pro R Soc Lond [Biol] 249:173, 1992 339. Hotson J, Braun D, Herzberg W, Boman D: Transcranial magnetic stimulation of extrastriate cortex degrades human
motion direction discrimination. Vision Res 34:2115, 1994 340. Tootell RBH, Hamilton SL, Silverman MS: Topography of cytochrome oxidase activity in owl monkey cortex. J Neurosci 5:2786, 1985 341. Tootell RBH, Taylor JB: Anatomical evidence for MT and additional cortical visual areas in humans. Cereb Cortex 5:39, 1995 342. DeYoe EA, Hockfield S, Garren H, van Essen DC: Antibody labelling of functional subdivisions in visual cortex: CAT-301 immunoreactivity
in striate and extrastriate cortex of the macaque monkey. Vis Neurosci 5:67, 1990 343. Clarke S, Miklossy J: Occipital cortex in man: Organization of callosal connections, related
myelo- and cytoarchitecture, and putative boundaries of functional visual
areas. J Comp Neurol 298:188, 1990 344. Clarke S: Modular organization of human extrastriate visual cortex: Evidence from
cytochrome oxidase pattern in normal and macular degeneration cases. Eur J Neurosci 6:725, 1994 345. Lueck CJ, Zeki S, Friston KJ et al: The colour center in the cerebral cortex of man. Nature 340:386, 1989 346. Gulyás B, Heywood CA, Popplewell DA et al: Visual form discrimination from color or motion cues: functional anatomy
by positron emission tomography. Proc Natl Acad Sci USA 91:9965, 1994 347. Gulyás B, Roland PE: Cortical fields participating in form and colour discrimination in the
human brain. Neuroreport 2:585, 1991 348. Martin A, Haxby JV, Lalonde FM et al: Discrete cortical regions associated with knowledge of color and knowledge
of action. Science 270:102, 1995 349. Allison T, Begleiter A, McCarthy G et al: Electrophysiological studies of color processing in human visual cortex. Electroencephalogr Clin Neurophysiol 88:343, 1993 350. Sergent J, Ohta S, MacDonald B: Functional neuroanatomy of face and object processing. A positron emission
tomography study. Brain 115:15, 1992 351. Grady CL, Maisog JM, Horwitz B et al: Age-related changes in cortical blood flow activation during visual processing
of faces and location. J Neurosci 14:1450, 1994 352. Haxby JV, Ungerleider LG, Horwitz B et al: Face encoding and recognition in the human brain. Proc Natl Acad Sci USA 93:922, 1996 353. Haxby JV, Horwitz B, Ungerleider LG et al: The functional organization of human extrastriate cortex: a PET-rCBF study
of selective attention to faces and locations. J Neurosci 14:6336, 1994 354. Kapur N, Friston KJ, Young A et al: Activation of human hippocampal formation during memory for faces: a PET
study. Cortex 31:99, 1995 355. Puce A, Allison T, Gore JC, McCarthy G: Face-sensitive regions in human extrastriate cortex studied by functional
MRI. J Neurophysiol 74:1192, 1995 356. McIntosh AR, Grady CL, Ungerleider LG et al: Network analysis of cortical visual pathways mapped with PET. J Neurosci 14:655, 1994 357. Dissociation of pathways for object and spatial vision: a PET study in
humans. Neuroreport 6:1865, 1995 358. Corbetta M, Shulman GL, Miezin FM, Petersen SE: Superior parietal cortex activation during spatial attention shifts and
visual feature conjunction. Science 270:802, 1995 359. Petersen SE, Corbetta M, Miezin FM, Shulman GL: PET studies of parietal
involvement in spatial attention: comparison of different task types. Can
J Exp Psychol 48:319, 1994This work was supported by NIH grant No. PO
NS 19632 and by a Medical Research Council of Canada Fellowship. | 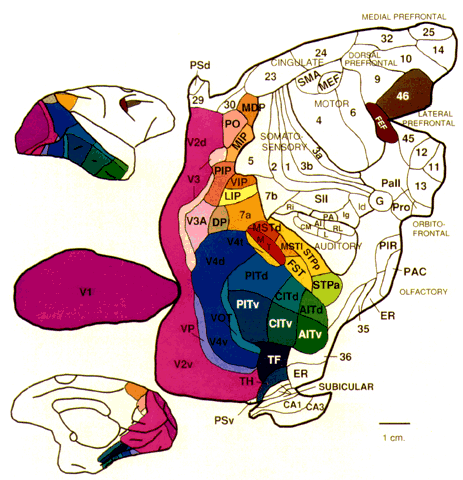
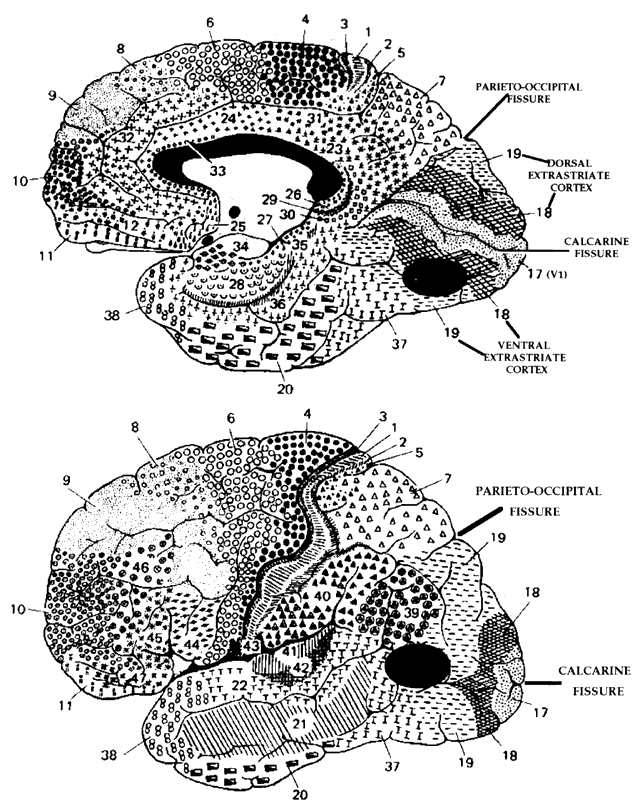
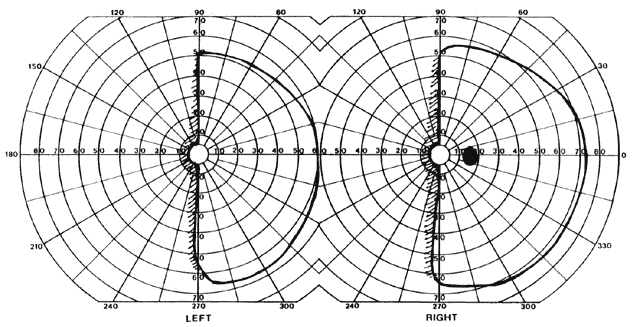
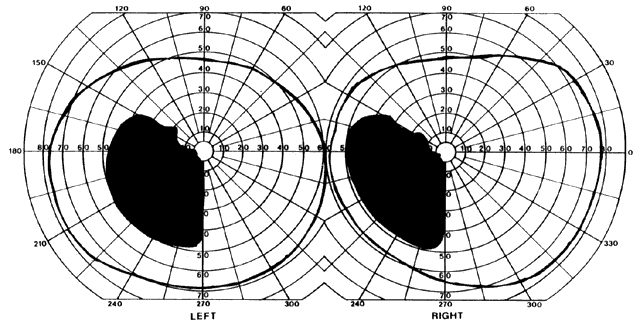
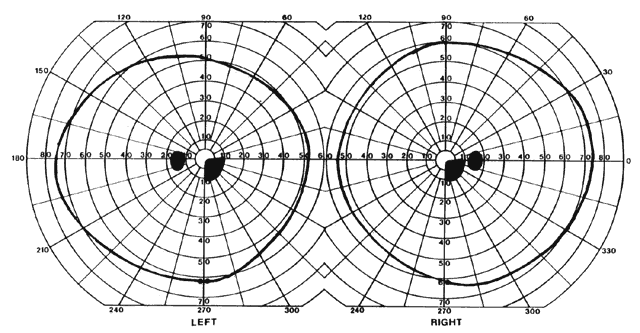
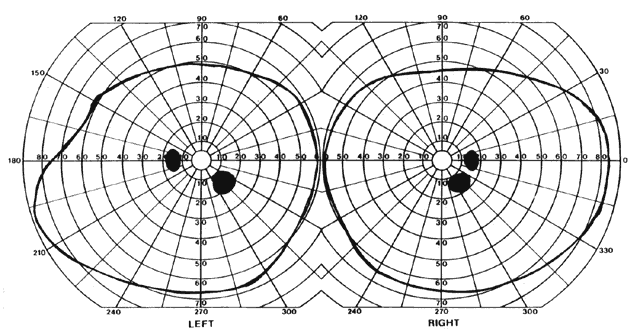
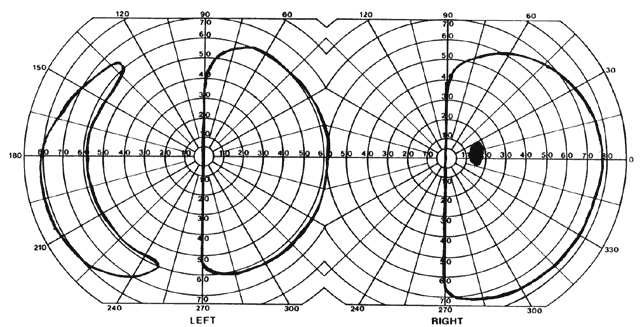
 = perception), means “not knowing.” Associative agnosia is an impairment of recognition in which “normal percepts are stripped
of their meanings.
= perception), means “not knowing.” Associative agnosia is an impairment of recognition in which “normal percepts are stripped
of their meanings.