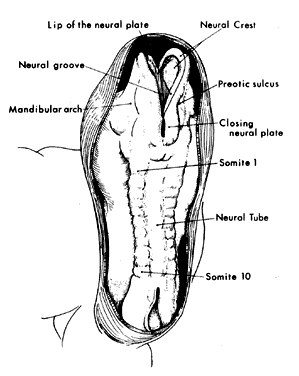
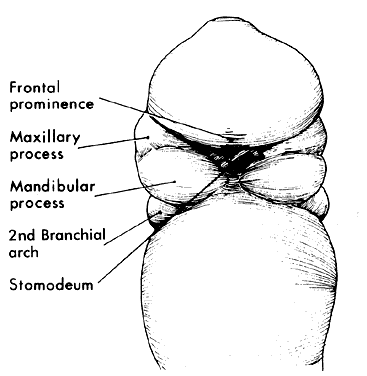
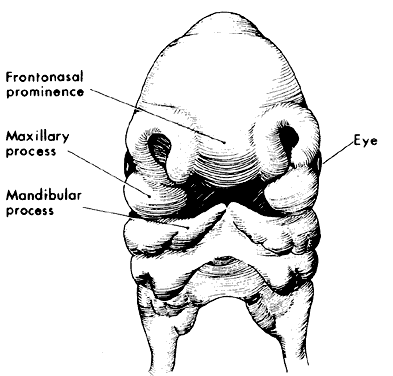
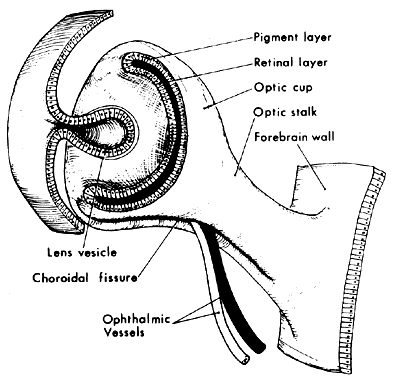
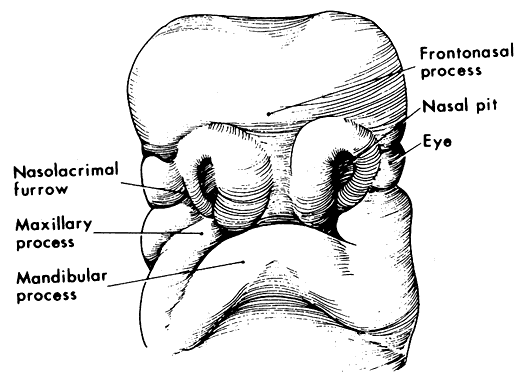
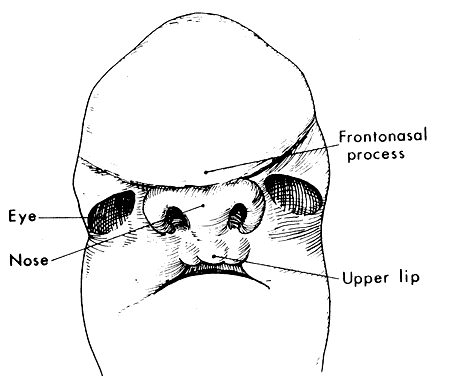
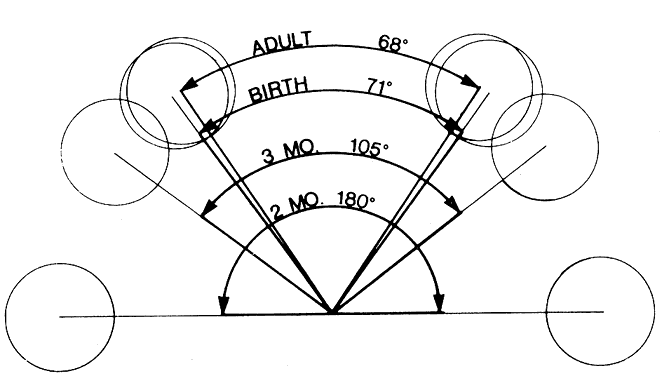
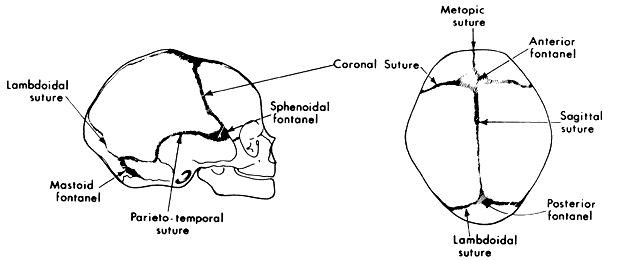
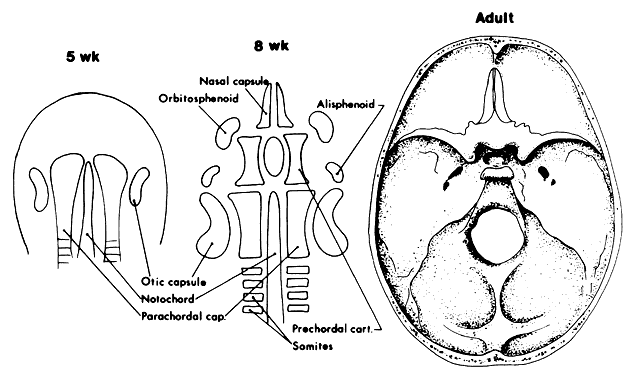
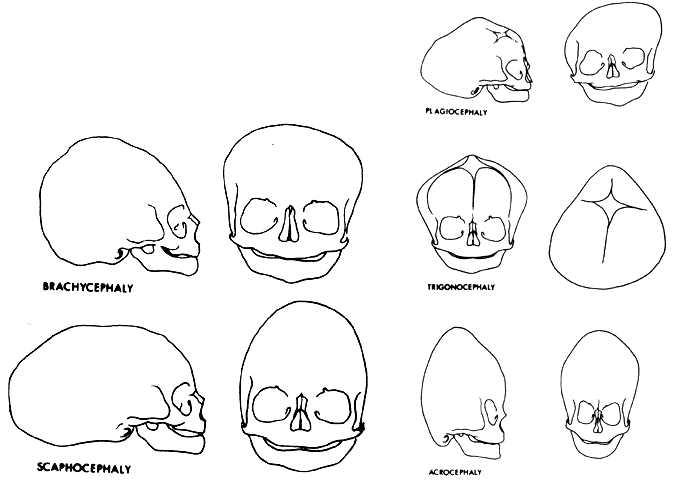
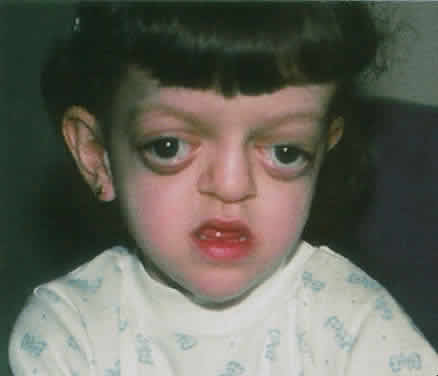
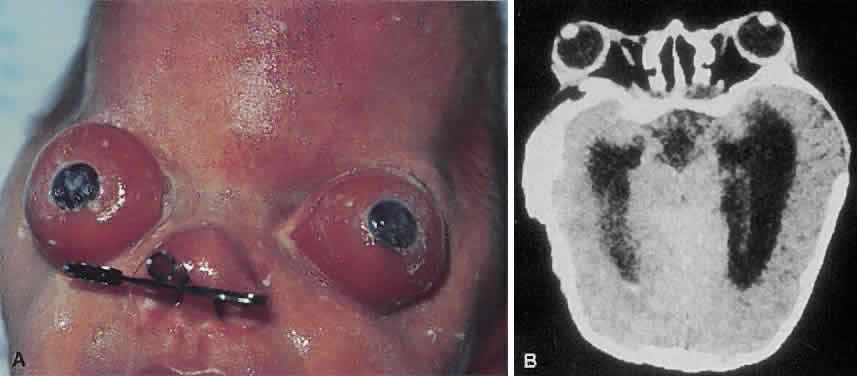
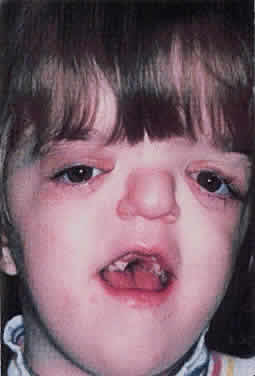
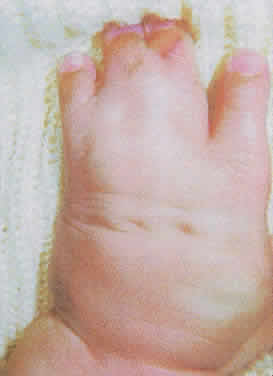
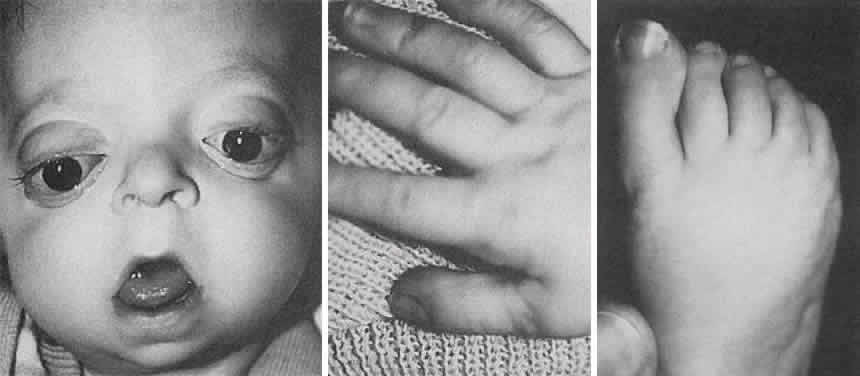
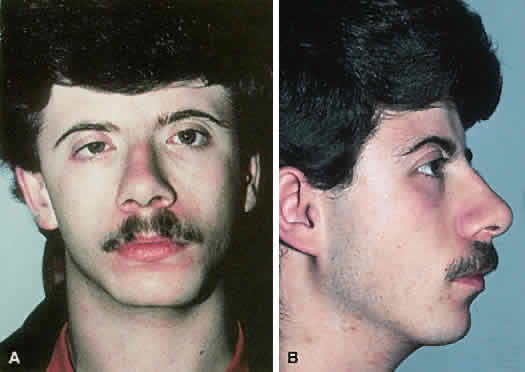
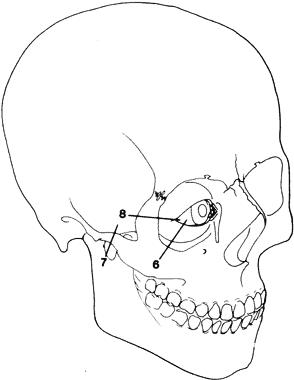
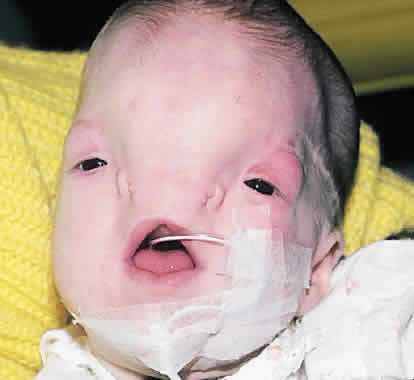
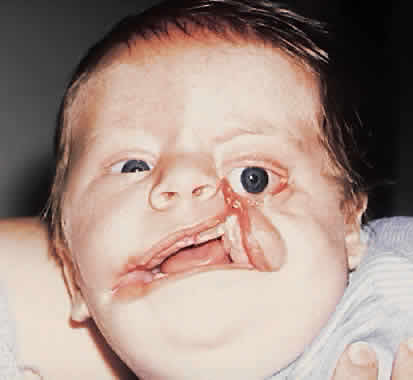
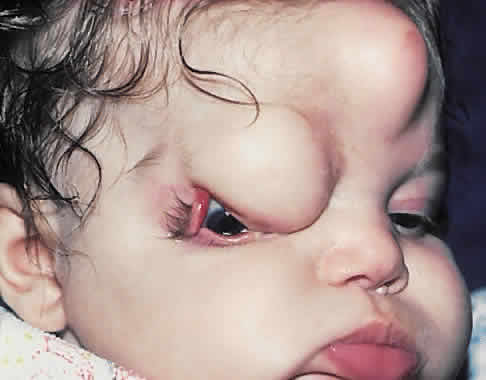
1. Virchow R: Über den Crestinismus, Manenlick in franken, and über pathologische
Schadelformen. Ver Phys Med (Warsburg) 2:230, 1851 2. Jane JA, Edgerto MT, Futrell JW et al: Immediate correction of sagittal synostosis. J Neurosurg 49:705, 1978 3. David JD, Poswillo D, Simpson D: The Craniosynostosis: Causes, Natural
History and Management. New York: Springer, 1982 4. Fries PD, Katowitz JA: Congenital craniofacial anomalies of ophthalmic importance. Surv Ophthalmol 35:87, 1990 5. Cohen MM Jr: Perspectives on craniosynostosis. West J Med 132:507, 1980 6. Cohen MM Jr: Craniosynostosis and syndromes with craniosynostosis: Incidence, genetics, penetrance, variability and new syndrome updating. Birth Defects 15:13, 1979 7. Cohen MM Jr: An etiologic and nosologic overview of craniosynostosis syndromes. Birth Defects 11:137, 1975 8. Moss ML: The pathogenesis of premature cranial synostosis in man. Acta Anat 37:351, 1959 9. Park EA, Powers GF: Acrocephaly and caphocephaly with symmetrically distributed malformations
of the extremities. Am J Dis Child 20:235, 1920 10. Moore GE: Molecular genetic approaches to the study of human craniofacial dysmorphologies. Int Rev Cytol 158: 215, 1995 11. Herman J, Opitz JM: An unusual form of acrocephalosyndactyly. Birth Defects 5:39, 1969 12. Le Merrer M, Ledinot V, Renier D et al: Genetic counseling in craniostenosis: Results of a prospective study performed
with a group of studies on craniofacial malformations. J Genet Hum 36:295, 1988 13. Crouzon O: Dystostose craniofaciale herediture. Bull Mem Soc Med Hosp Paris 33:545, 1912 14. Cohen MM Jr: Frieborg S: Birth prevalence studies of Crouson syndrome: Comparison of direct and
indirect methods. Clin Genet 41:12, 1992 15. Cohen MM: Syndrome with craniosynostosis. In Cohen MM (ed): Craniosynostosis: Diagnosis, Evaluation and Management, pp 413–590. New York: Raven
Press, 1986 16. Kreiborg S: Crouzon syndrome: A clinical and roentgen cephalometric study. Scand J Plast Reconstr Surg (Suppl) 18:1, 1981 17. Li X, Lewanda AF, Eluma F et al: Two craniosynostotic syndrome loci, Crouzon and Jackson-Weiss, map to chromosome 10q23-q26. Genomics 22:418, 1994 18. Losken HW, Preston RA, Post JC et al: The gene for Crouzon craniofacial
dysostosis maps to chromosome 10q25-q26. Proceedings of the 63rd Annual
Meeting of the American Society of Ophthalmic Plastic and Reconstructive
Surgery, San Diego, CA, September 24–30, 1994 19. White RA, Dowler LL, Angeloni SV et al: Assignment of FGF* to human chromosome 10q25-q26: Mutations in FGF8 may be responsible for some types
of acrocephalosyndactyly linked to this. 20. Preston RA, Post JC, Keats BJ et al: A gene for Crouzon craniofacial dysostosis maps to the long arm of chromosome 10. Nat Genet 7:149, 1994 21. Reardon W, Winter RM, Rutland P et al: Mutations in the fibroblast growth factor receptor 2 gene cause Crouzon
syndrome. Nat Genet 8:98, 1994 22. Jabs EW, Li X, Scott AF et al: Jackson-Weiss and Crouzon syndromes are allelic with mutations in fibroblast
growth factor 2. Nat Genet 8:275, 1994 23. Bentelson TI: The premature synostosis of the cranial sutures. Arch Ophthalmol (Copenh) 5(Suppl):47, 1958 24. Waardenburg J, Franceschetti A, Klein D: Genetics and Ophthalmology, p 222. Vol 1. Springfield, IL: Charles C Thomas, 1961 25. Mermann J, Pallister PD, Opitz JM: Craniosynostosis and craniosynostosis syndromes. Rocky Mt Med J 66:45, 1969 26. Marhac D, Renier D: Craniosynostosis. World J Surg 13:358, 1989 27. Parks MM, Costenbader FO: Craniofacial dysostosis (Crouzon's disease). Am J Ophthalmol 33:77, 1950 28. Anderson FM, Geiger L: Craniosynostosis: A survey of 204 cases. J Neurosurg 22:229, 1965 29. Anderson H, Gomes SP: Craniosynostosis: Review of the literature and indications
for surgery. Acta Paediatr Scand 57:47, 168 30. Atkinson FRB: Hereditary craniofacial dysostosis or Crouzon's disease. Med Press 195:118, 1937 31. Shiller JG. Craniofacial dystosis of Crouzon: A case report and pedigree with emphasis
on heredity. Pediatrics 23: 107, 1959 32. Kreiborg S: Postnatal growth and development of the craniofacial complex
in premature craniosynostosis. In Cohen MM (ed): Craniosynostosis: Diagnosis, Evaluation
and Management, pp 157–189. New York: Raven
Press, 1986 33. Kreiborg S, Pruzandky S: Craniofacial growth in patients with premature craniosynostosis. Scand J Plast Reconstr Surg 15:171, 1981 34. Posnick JC. Craniofacial dysostosis: Staging of reconstruction and management of the
midfacial deformity. Neurosurg Clin North Am 2:683, 1991 35. Kolar JC, Munro IS, Farkas LG: Patterns of dysmorphology in Crouzon syndrome: An anthropometric study. Cleft Palat J 25:235, 1988 36. Bergstrom L, Weblett LM, Hemenway WG: Otologic manifestations of acrocephalosyndactyly. Arch Otolaryngol 96: 177, 1972 37. Mathur JS, Nema HV, Metra KS et al: Apert's anomaly a transition. Acta Ophthalmol 46:1041, 1968 38. Waardenburg J, Franceschetti A, Klein D: Genetics and Ophthalmology, p 123. Vol 1. Springfield, IL: Charles C Thomas, 1961 39. Perlman JM, Zaidman GW: Bilateral keratoconus in Crouzon's syndrome. Cornea 13:80, 1994 40. Miller MT: Ocular abnormalities in craniofacial malformations. Int Ophthalmol Clin 24:143, 1984 41. Hertle RW, Wuinn GE, Minguini M et al: Visual loss in patients with craniofacial synostosis. J Pediatr Ophthalmol Strabismus 28:344, 1991 42. Whitaker LA, Katowitz JA: Facial anomalies involving the nasolacrimal apparatus. In
Tessier P, Callahan A, Mustarde J et al (eds): Symposium
on Plastic Surgery in the Orbital Region. St Louis: CV Mosby, 1974 43. Whitaker LA, Katowitz JA: Canthal and nasolacrimal apparatus problems. In
Tessier P, Callahan A (eds): Symposium on Pediatric Surgery. St Louis: CV
Mosby, 1974 44. Whitaker LA, Katowitz JA, Randall P: the nasolacrimal apparatus in congenital facial anomalies. J Maxillofac Surg 2:59, 1974 45. Francois J, Bacskulin J: External congenital fistula of the lacrimal sac. Ophthalmologica 159:249, 1969 46. Whitaker LA, Katowitz JA, Jacobs WE: Ocular adenexal problems in craniofacial deformities. J Maxillofac Surg 7:55, 1979 47. Choy AE, Margolis S, Breinin GM et al: Analysis of preoperative and postoperative
extraocular muscle function in surgical translocation of bony
orbits: A preliminary report. In Converse JM (ed): Symposium on Diagnosis
and Treatment of Craniofacial Anomalies, p 128. St Louis: CV Mosby, 1979 48. Diamond GR, Whitaker LA: Ocular motility in craniofacial reconstruction. Plast Reconstr Surg 73:31, 1984 49. Diamond GR, Katowitz JA, Whitaker LA et al: Ocular alignment after craniofacial reconstruction. Am J Ophthalmol 90:248, 1980 50. Apert E: De l'acrocephalosyndactylie. Bull Mem Soc Med Hop Paris 243:1310, 1906 52. Weaton SW: Two specimens of congenital cranial deformities in infarcts associated
with fusion of fingers and toes. Trans Pathol Soc Lond 45:238, 1894 53. Blank CE: Apert's syndrome (a type of aerocephalosyndactyly): Observations on
a British series of thirty-nine cases. Ann Hum Genet 24:151, 1960 54. Bull MJ, Escobar V, Bixler D et al: Phenothpe definition and recurrence risk in the acrocephalosyndactyly syndrome. Birth Defects 15:65, 1979 55. Eastman JR, Escobar V, Bixler D: Linkage analysis in dominant acrocephalosyndactyly. J Med Genet 15:292, 1978 56. Gramey D, Farriaux JP: A dominant syndrome associated with polysyndactyly, spatula thumb, facial
abnormalities, and mental retardation: A particular form of Noack's
acrocephalodacrylia. J Genet Hum 19:299, 1971 57. Lemaitre A, Van Reck J: Genetic aspects of craniofacial dystosis, the Crouzon disease and the Apert-Crouzon
disease. Acta Stomatol Belg 73:279, 1976 58. Sanchez HM, De Negrotti TC: Variable expression in Pfeiffer syndrome. J Med Genet 18:73, 1981 59. Erickson JD, Cohen MM Jr: A study of parental age effects on the occurrence of fresh mutations for
the Apert syndrome. Ann Hum Genet 38:89, 1974 60. Smith CAB: Note on the estimation of parental age effects. Ann Hum Genet 35:337, 1972 61. Wilkie AO, Starey SF, Oldridge M et al: Apert syndrome results from localized mutations of FGFR2 and is allelic
with Crouzon syndrome. Nat Genet 9:165, 1995 62. Park WJ, Theda C, Maestri Ne et al: Analysis of phenotypic features and
FGFR2 mutations in Apert syndrome. Am J Hum Genet 57:321, 1995 63. Diamond GR, Katowitz JA, Whitaker LA et al: Variations in extraocular muscle and structure in craniofacial dystosis. Am J Ophthalmol 90:416, 1980 64. Cuttone JM, Brazis PT, Miller MT et al: Absence of the superior rectus muscle in Apert's syndrome. J Pediatr Ophthalmol Strabismus 16:349, 1979 65. Galli Ml: Apert syndrome does not equal mental retardation. J Pediatr 89:691, 1976 66. Murcovic JA, Posnick JC, Drake JM et al: Hydrocephalus in the Apert syndrome: A retrospective review. Pediatr Neurosurg 1963:151, 1993 67. Nowtzel MJ, Marsh JL, Palkes H et al: Hydrocephalus and mental retardation in craniocynostosis. J Pediatr 107:885, 1985 68. Cohen MM, Kreigorg S: Unusual cranial aspects of Apert syndrome. J Craniofac Genet Dev Bus 14:48, 1994 69. Ferraro NF: Dental, orthodontic, and one/maxillofacial evaluation and treatment in
Apert syndrome. Clin Plast Surg 18:291, 1991 70. Cohen MM Jr: Cardiovascular anomalies in Apert type acrocephalosyndactyly. Birth Defects 8:239, 1982 71. Stegger C: The acneiform eruption of Apert's syndrome is not acne vulgaris. Am J Dermatopathol 6:213, 1984 72. Marden PM, Smith DW, McDonald MJ: Congenital anomalies in newborn infant including minor variations. J Pediatr 64:358, 1964 73. McGill T: Otolaryngologic aspects of Apert syndrome. Clin Plast Surg 18:309, 1991 74. Gorlin RJ, Cohen MM, Levin LS: Syndromes of the Head and Neck. New York: Oxford
University Press, 1990 75. Mah J, Kasser J, Upton J: The foot in Apert syndrome. Clin Plast Surg 18:391, 1991 76. Green SM: Pathological anatomy of the hands in Apert's syndrome. J Hand Surg 7:450, 1982 77. Fogh-Anderson P: Rare clefts of the face. Acta Univ Scand 129:275, 1965 78. Carpenter G: Two sisters showing malformations of the skull and other congenital abnormalities. Rep Soc Study Dis Child Lond 1:110, 1901 79. Temtamy SA: Carpenter's syndrome: Acrocephalopolysyndactyly. An autosomal recessive
syndrome. J Pediatr 69: 111, 1964 80. Eaton AP, Sommer A, Kontres SB et al: Carpenter's syndrome: Acrocephalopolysyndactyly type II. Birth Defects 10:249, 1974 81. Der Kaloustain VM, Sinno AA, Nassar SI: Acrocephalopolysyndactyly type II (Carpenter's syndrome). Am J Dis Child 124:716, 1972 82. Robinson LK, James HE, Mubarak SJ et al. Carpenter syndrome: Natural history and clinical spectrum. Am J Med Genet 20:461, 1985 83. Frias JC, Felman AH, Rosenbloom AL et al. Normal intelligence in two children
with Carpenter's syndrome. Am J Med Genet 2:191, 1978 84. Cohen DM, Green JG, Miller J et al: Acrocephalopolysyndactyly type II: Carpenter syndrome: Clinical spectrum
and attempt at unification with Goodman and Summit syndromes. Am J Med Genet 28:311, 1987 85. Nissen SJ, Turner S, Ray S: An unusual presentation of Carpenter syndrome. Contemp Orthop 27:129, 1993 86. Pfeiffer RA: Dominant erbliche Akrocephalosyndaktylie. Z Kinderheilk 90: 301, 1964 87. Cohen MM: Pfeiffer syndrome update, clinical subtypes, and guidelines for differential
diagnosis. Am J Med Genet 45:300, 1993 88. Vanes J, Logan F: Pfeiffer's type acrocephalosyndactyly in two families. J Med Genet 19:289, 1982 89. Schell B, Hehr A, Feldman GJ et al: Mutations in FGFR1 and FGFR2 cause familial and sporadic Pfeiffer's
syndrome. Hum Med Genet 4:323, 1995 90. Navey Y, Freedman A: Pfeiffer's syndrome: Report of a family in review of the literature. J Med Genet 13:227, 1976 91. Kreiborg S, Cohen MM: A severe case of Pfeiffer syndrome associated with stub thumb on the maternal
side of the family. J Craniofac Genet Dev Biol 13:73, 1993 92. Saldino RM, Seinbech HL, Epstein CJ: Familial acrocephalosyndactyly (Pfeiffer syndrome). AJR Am J Roentgenol 116:609, 1972 93. Chotzen F: Eine eigenartige familiars Entwicklungsstorung (Akrocephalosyndaktylie: Dystosis
cranifacialis und Hyperteloismus). Monatsschr Kinderheilk 55:97, 1932 94. Saethre H. Ein Beitrag zum turmschadel Problem. Pathogenese, Erblichkeit
und Symptomatologie. Stsch Z Nervenheilk 117:533, 1931 95. Lewanda AF, Green ED, Weissenbach J et al: Evidence that the Saethre-Chotzen syndrome locus lies between D75664 and
D75507, by genetic analysis and detection of a microdeletion in a patient. Am J Hum Genet 55:1195, 1994 96. Van Merwerden L, Rose CS, Reardon W et al: Evidence for locus heterogeneity in acrocephalosyndactyly: A refined localization
for the Saethre-Chotzen syndrome locus on distal chromosome
up—and exclusion of Jackson-Weiss syndrome from craniosynostosis
loci on up and 5q. Am J Hum Genet 54:669, 1994 97. Reid C, McMorrow LE, McDonald-McGinn DM et al: Saethre-Chotzen syndrome with familial translocation at chromosome 7, p. 22. Am J Med Genet 47:637, 1993 98. Rearden W, McManus SP, Summers D et al: Cytogenetic evidence that the Saethre-Chotzen gene maps to 7p.212. Am J Med Genet 47:633, 1993 99. Franks V et al: Saethre-Chotzen syndrome with additional abnormalities in a Mexican family. Birth Defects 13:241, 1977 100. Evans CA, Christiasen RL: Cephalic malformations in Saethre-Chotzen syndrome. Radiology 121:399, 1976 101. Pantke OA, Cohen MM Jr, Witkop CJ et al: The Saethre-Chotzen syndrome. Birth Defects 11:190, 1975 102. Kopysc Z, Stanska M, Ryzko J et al: The Saethre-Chotzen syndrome with partial bifid of the distal phalanges
of the great toes. Hum Genet 56:195, 1980 103. Bianchi E, Arico M, Podesta AF et al: A family with the Saethre-Chotzen syndrome. Am J Med Genet 22:649, 1985 104. Prudzansky S, Pashayan H, Kreiborg S et al: Roentgen cephalometric studies of the premature craniofacial synostosis: Report
of a family with the Saethre-Chotzen syndrome. In Bergsma (ed): Malformation
Syndromes. Amsterdam: Excerpta Medica for the National
Foundation—March of Dimes. Birth Defects 11:226, 1975. 105. Aase JM, Smith DW: Facial asymmetry and abnormalities of palms and ears: A dominantly inherited
developmental syndrome. J Pediatr 76:928, 1970 106. Kawamotu MK: The kaleidoscopic world of rare craniofacial clefts: Order out of chaos (Tessier
classification). Clin Plast Surg 3:529, 1976 107. Cohen MM: Syndromes with cleft lip and cleft palate. Cleft Palate J 15:306, 1978 108. Morin J, Hio J, Anderson J et al: A study of growth in the interorbital region. Am J Ophthalmol 152:13, 1966 109. Lund OE: Combination of ocular and cranial malformations with craniofacial dysplasia. Ophthalmologica 152: 13, 1966 110. Burian F: Median clefts of the nose. Acta Chir Plast 2: 180, 1960 111. Savenero-Rosselli G: Developmental pathology of the fact and the dysraphic syndromes: An essay
of interpretation based on experimentally produced congenital defects. Plast Reconst Surg 11:36, 1953 112. Franceschetti A, Their CJ: Symptomenkomplex in Rahmen der mandibulofazialen Dysplazion. Klin Monatsbl Augenheilkd 135:391, 1959 113. Tessier P: Anatomical classification of facial, craniofacial, laterofacial clefts. J Maxillofac Surg 4: 69, 1976 114. Dursy E. Zur entwicklungs Geschichte des Kopfes des Menschen und der heheren
Wirbelt geire. Tubingen: Verlag der H. Lauppschen-Buchhandlung, 1869 115. His W:. Die Entwicklung der Menschiliche und Thierischer Physiognomen. Arch
Anat Entwicklungs Gesch 5:384, 1892 116. Johnston MC: The neural crest in abnormalities of the face and brain. Birth Defects 11:416, 1975 117. Wetson JA: A radioautographic analysis of the migration and localization of trunk
neural crest cells in the chick. Dev Biol 6:279, 1963 118. Vermeij-Keers C, Mazzola RF, Van der Meulen JC et al: Cerebrocraniofacial and craniofacial malformations: An embryologic analysis. Cleft Palate J 20:128, 1983 119. Vermeij-Keers C, Poelmann RE: The neural crest: A study in cell degeneration and the improbability of
cell migration in mouse embryos. Neth J Zool 30:74, 1990 120. Dixon MF, Read AP, Donnai D et al: The gene for Treacher-Collins syndrome maps to the long arm of chromosome 5. Am J Hum Genet 49:17, 1991 121. Dixon MJ, Dixon J, Houseal T et al: Narrowing the position of the Treasher-Collins syndrome locus to a small
interval between three new microsatellite markers at 5q32-33, 1. Am J Hum Genet 52:907, 1993 122. Neel JV: A study of major congenital defects in Japanese infants. Am J Hum Genet 10:398, 1958 123. Montnenegro MA, Palomino H et al: The influence of earthquake-induced stress on human facial clefting and
simulation in mice. Arch Oral Biol 40:33, 1995 124. Erdelyi R: The influence of toxoplasmosis on the incidence of congenital facial malformations: Preliminary
report. Plast Reconstr Surg 20:306, 1957 125. Leck I, Hay S, Witte JI et al: Malformations recorded on birth certificates following A2 influenza epidemics. Public Health Rep 84:971, 1969 126. Erickson JD, Oakley JP: Seizure disorder in mothers of children with orofacial clefts: A case-control
study. J Pediatr 84:244, 1974 127. Fries MTL, Kuller JA, Norton T et al: Facial features of infants exposed prenatally to cocaine. Teratology 48:413, 1993 128. Granstrom G, Kullaa-Mikkohen A: Experimental craniofacial malformations induced by retinoids and resembling
branchial arch syndromes. Scand J Plast Reconstr Surg Hand Surg 24:3, 1990 129. Hansman C: Growth of intraorbital distance and skull thickness as observed in roentgenographic
measurements. Radiology 86:87, 1966 130. Mazzola RF: Congenital malformations in the frontonasal area: Their pathogenesis and
classification. Clin Plast Surg 3:573, 1976 131. David DJ, Mahatumarat C, Cooter RD: Hemifacial microsomia: A multisystem classification. Plast Reconstr Surg 80:525, 1987 132. Kawamoto HF Jr: Incidence, pathology and classification of orbital clefts
and the pathology of orbital hypertelorism. In Converse JM et al (eds): Symposium
or Diagnosis and Treatment of Craniofacial Anomalies, p 164. St
Louis: CV Mosby, 1979. 133. Ortiz-Monasterio F, Fuente Del Campo A, Dimopulos A: Nasal Clefts. Ann Plast Surg 18:377, 1987 134. Kawamoto HF: Rare craniofacial clefts. In McCarthy J (ed): Plastic Surgery. Vol 4. Philadelphia: WB Saunders, 1990 135. Mann I: A theory of the embryology of oxycephaly. Trans Ophthalmol Soc UK 55:279, 1935 136. Fuente del Campo A, Elizendo MM, Arnaud E. Treacher-Collins syndrome (mandibulofacial
dysostosis). Clin Plast Surg 21:613, 1994 137. Rogers BO: The surgical treatment of mandibulofacial dystosis (Berry syndrome, Treacher-Collins
syndrome, Franceschetti-Zwahlen-Klein syndrome). Clin Plast Surg 3:653, 1976 138. Rovin S, Dachi SF, Borenstein DB et al: Mandibulofacial dysostosis, a familial study of five generations. J Pediatr 65:215, 1964 139. Rogers BO: Berry-Treacher-Collins syndrome: A review of 200 cases. Br J Plast Surg 17:109, 1964 140. Franceschetti A, Kline D: The mandibulofacial dysostosis: A new hereditary syndrome. Acta Ophthalmol 27:144, 1949 141. Hurwitz P: Mandibulofacial dysostosis. Arch Ophthalmol 57:69, 1954 142. Rogers BO: Mandibulofacial dysostosis: Treacher-Collins syndrome. In Converse
JM et al (eds): Symposium on Diagnosis and Treatment of Craniofacial
Anomalies, pp 423–432. St Louis: CV Mosby, 1979 143. Hertle RW, Ziylan S, Katowitz JA: Ophthalmic features and visual prognosis in the Treacher-Collins syndrome. Br J Ophthalmol 77:642, 1993 144. Harrison SH: Treacher-Collins syndrome. Br J Plast Surg 3:282, 1951 145. Stovin JJ, Lyon JA, Clemmens RL: Mandibulofacial dysostosis. Radiology 74:225, 1960 146. Berry GA: Note on a congenital defect (?coloboma) of the lower lid. R Lond Ophthalmol Hosp Rep 12:255, 1889 147. Wilson TG: A case of unilateral mandibulofacial dystosis associated with agenesis
of the homolateral lung. J Laryngol Otol 72:238, 1959 148. Dixon MJ, Dixon J, Raskova D et al: Genetic and physical mapping of the Treacher-Collins syndrome locus: Refinement
of the localization to chromosome 5q 32-332. Hum Molec Genet 1:249, 1992 149. Loftus SK, Edwards SJ, Scherpbier-Heddema T et al: A combined genetic and radiation hybrid map surrounding the Treacher-Collins
syndrome locus on chromosome 5q. Hum Mol Genet 2:1785, 1993 150. Jabs EW, Lix F, Lovett M et al: Genetic and physical mapping of the Treacher-Collins syndrome locus with
respect to loci in the chromosome 5q3 region. Genomics 18:7, 1993 151. Marres HA, Cremers CW, Dixon MJ et al: The Treacher-Collins syndrome. A clinical, radiological and genetic linkage
study on two pedigrees. Arch Otolaryngol Head Neck Surg 121:509, 1995 152. Harstadius S: The Neural Crest, p 176. London: Oxford University Press, 1950 153. Morels O: Zur Pathogenese der Missbildugen des 1 Visceralbogens unter desunderer
Berucksichtigung der Dysostosis mandibulofacialis. Z Kinderheilkd 73:532, 1953 154. Nucci P, Brancato R, Carone F et al: Mandibulofacial dysostosis and corneal guttata. Am J Ophthalmol 108:204, 1989 155. Poswillo D: The pathogenesis of the Treacher-Collins syndrome. Br J Oral Surg 13:1, 1975 156. Keerl G: Zurformalen und causalen Genese der Dysostosis mandibulofacialis. Ophthalmologica 143:5, 1962 157. McKenzie J, Craig J: Mandibulofacial dysostosis (Treacher-Collins syndrome). Arch Dis Child 30:391, 1955 158. Sulik KH, Johnston MC, Smiley SJ et al: Mandibulofacial dysostosis (Treacher-Collins syndrome): A new proposal
for its pathogenesis. Am J Med Genet 27:359, 1987 159. Goldenhar M: Associations malformations de l'oeil et de l'oreille, en particular le
syndrome dermoide epibulbaire-appendices. Auricularies-fistula congenita
et ses relations avec la dysostose mandibulo-faciale. J Genet Hum 1:243, 1952 160. Pruzandky S, Miller MT: Ocular defects in craniofacial syndrome. In Goldberg
MF (ed): Genetic and Metabolic Eye Disease, p 245. 2nd ed. Boston: Little, Brown, 1986 161. Baum JL, Feingold M: Ocular aspects of Goldenhar's syndrome. Am J Ophthalmol 75:250, 1973 162. Mansour AM, Wang F, Henkind P et al: Ocular findings in facioauriculovertibral sequence (Goldenhar-Gorlin syndrome). Am J Ophthalmol 100:555, 1985 163. Feingold M, Gellis SS: Ocular abnormalities associated with first and second arch syndromes. Surv Ophthalmol 14:30, 1968 164. Rollnick BR, Kaye CI, Nagatoshi K et al: Oculoauriculovertebral dysplasia and variants: Phenotypic characteristics
of 294 patients. Am J Med Genet 26:361, 1987 165. Wilson GN: Cranial defects in the Goldenhar syndrome. Am J Med Genet 14:435, 1983 166. Buick V: Genetic aspects of hemifacial microsomia. Hum Genet 64:291, 1983 167. Cohen MM: Variability versus “incidental findings” in the first and second
branchial arch syndrome: Unilateral variants with anophthalmia. Birth Defects 7:103, 1971 168. Feingold M, Baum J: Goldenhar's syndrome. Am J Dis Child 132:136, 1978 169. Setzer ES, Ruiz-Castaneda N, Severn C et al: Etiologic heterogeneity in the oculoauriculovertebral syndrome. J Pediatr 98:88, 1981 170. Preston KL, McDonald M, Sebris SL et al: Validation of the acuity card procedure for assessment of infants with
ocular disorder. Ophthalmology 94:644, 1987 171. Raulo Y, Tessier P: Mandibulofacial dysostosis. Analysis: principles of surgery. Scand J Plast Reconstr Surg 15:251, 1981 172. Rogers BO: Embryologic development of the head and face. In Converse JM
et al (eds): Symposium on Diagnosis and Treatment of Craniofacial Anomalies, p 26. St
Louis: CV Mosby, 1979 173. Rogers BO: Embryologic development of the head and face. In Converse JM
et al (eds): Symposium on Diagnosis and Treatment of Craniofacial Anomalies, p 22. St
Louis: CV Mosby, 1979 174. Grabb WC: The first and second branchial arch syndrome. Plast Reconstr Surg 36:485, 1965 175. Krause VH: The syndrome of Goldenhar affecting two siblings. Acta Ophthalmol Copenh 48:494, 1970 176. Summitt R. Familial Goldenhar Syndrome. Birth Defects 5:106, 1969 177. Gorlin R, Pindborg JJ. Syndromes of the head and neck. New York: McGraw-Hill, 1964. 178. Hertle RW, Quinn GE, Katowitz JA: Ocular and adenexal findings in patients with facial microsomias. Ophthalmology 99:144, 1992 179. Kaye CI, Rollnick BR, Pruzansky S: Malformations of the auricle, isolated and in syndromes. IV. Cumulative
pedigree data. Birth Defects 16:163, 1979 180. Smahel Z: Craniofacial changes in hemifacial microsomia. J Craniofac Genet Dev Biol 6:151, 1986 181. Converse JM, Wood-Smith D, McCarthy JG et al: Bilateral facial microsomia: diagnosis, classification and treatment. Plast Reconstr Surg 54:413, 1974 182. Bennon RD, Mulliken JB, Kaban LB et al: Microtia: A microform of hemifacial microsomia. Plast Reconstr Surg 76:859, 1985 183. Rollnick BR, Faye CI: Hemifacial microsomia and variants: Pedigree data. Am J Med Genet 15:233, 1983 184. Vento AR, LaBrie RA, Mulliken JB: The OMENS classification of hemifacial microsomia. Cleft Palate Craniofac J 28:68, 1991 185. Converse JM, McCarthy JG, Coccaro J et al: Clinical aspects of craniofacial
microsomia. In Converse JM, McCarthy JG, Wood-Smith D (eds): Symposium
on Diagnosis and Treatment of Craniofacial Anomalies, p 461. St
Louis: CV Mosby, 1979 186. Lauritzen C, Monro IR, Ross RB: Classification and treatment of hemifacial
microsomia, Scand J Plast Reconstr Surg 19:33, 1985 187. Ross RB: Lateral facial dysplasia (first and second branchial arch syndrome, hemifacial
microsomia). Birth Defects 11:51, 1975 188. Seeds JW, Cefalo RC, Herbert WHP: Amniotic band syndrome. Am J Obstet Gynecol 144:243, 1982 189. Lubinsky M, Sujansky E, Sanger W et al: Familial amniotic bands. Am J Med Genet 14:81, 1983 190. Page EW, Villee CA, Villee DB: Human Reproduction: the Core Content of
Obstetrics, Gynecology and Perinatal Medicine, pp 169–199, 230–368. Philadelphia: WB
Saunders, 1976 191. Higginbottom MC, Jones KL, Hall BD et al: The amniotic band disruption complex: Timing of amniotic rupture and variable
spectra of consequent defects. J Pediatr 95:544, 1979 192. Van der Meulen JCH: Oblique facial clefts: Pathology, etiology and reconstruction. Plast Reconstr Surg 76: 212, 1985 193. Dumaman G, Heckler FR: Constriction ring syndrome. In Bentz MJ (ed): Pediatric
Plastic Surgery, pp 1009–1016. Stamford, CT: Appleton & Lange, 1998 194. Braude LS, Miller M, Cuttone J: Ocular abnormalities in the amniogenic band syndrome. Br J Ophthalmol 65:299, 1981 195. Hollsten DA, Katowitz JA: The ophthalmic manifestations and treatment of the amniotic band syndrome. Ophthalmol Plast Reconstr Surg 6:1, 1910 196. Miller MT, Deutsch TA, Cronin C et al: Amniotic bands as a cause of ocular abnormalities. Am J Ophthalmol 104:270, 1987 197. Hertle RW et al: Visual loss in patients with craniofacial synostosis. J Pediatr Ophthalmol Strabismus 28:344, 1991 198. Marchac D, Renieir D, Broumand S: Timing of treatment for craniosynostosis and faciocraniosynostosis: A 20-year
experience. Br J Plast Surg 47:211, 1994 199. Barden RC, Ford ME et al: Effects of craniofacial deformity in infancy on the quality of mother-infant
interactions. Child Development 60:819, 1989 200. Jackson IT: Reconstruction of the lower eyelid defect in Treacher-Collins syndrome. Plast Reconstr Surg 67:365, 1981 201. Van der Meulen JCH, Hauben DJ, Vaandrager JM et al: The use of a temporal osteoperiosteal flap for the reconstruction of malar
hypoplasia in Treacher-Collins syndrome. Plast Reconstr Surg 74:687, 1984 202. Hertle RW, Quinn GE, Katowitz JA et al: Visual abnormalities and strabismus
in a craniofacial clinic. Ophthalmology (Suppl) 150, 1990 203. Diamond GR, Katowitz JA, Whittaker LA et al: Variations in extraocular muscle and structure in craniofacial dysostosis. Am J Ophthalmol 90: 416, 1980 |