DISORDERS OF PROTEIN AND AMINO ACID METABOLISM
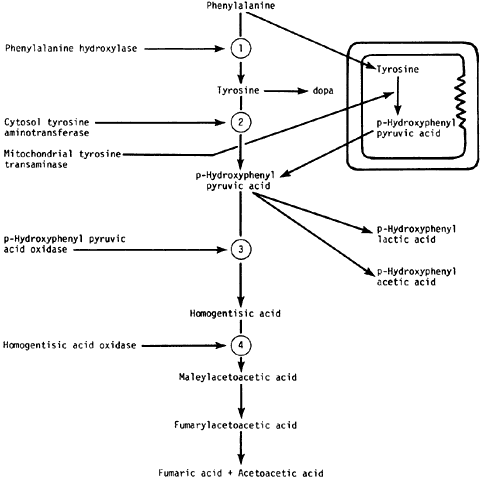
In the majority of diseases affecting the cornea that are caused by inborn errors of amino acid metabolism, the corneal change is unique and characteristic enough to form the basis for diagnosis. This section describes metabolic diseases related to enzyme blocks in the oxidation of phenylalanine and tyrosine (Fig. 1). Other disorders, such as Wilson's disease and the paraproteinemias, also are discussed.
|
Alkaptonuria (Ochronosis)
Alkaptonuria is a rare, autosomal-recessive metabolic disease in which the enzyme homogentisic acid oxidase, normally present in the liver and kidney, is missing. As a result of this defect, homogentisic acid, which is normally produced during the metabolism of phenylalanine and tyrosine, cannot be further metabolized and therefore accumulates and is excreted in the urine. The gene for this enzyme is present on chromosome 3q2.1 Urine containing homogentisic acid, if left to stand, gradually turns dark as the acid is oxidized to a polymerized melanin-like product. The rate of this reaction is accelerated by alkali.2 Homogentisic acid also reacts with Benedict's reagent and can produce a false positive urine test for glucose, resulting in an erroneous diagnosis of diabetes mellitus.
The cardinal features of alkaptonuria are the presence of homogentisic acid in the urine, pigmentation of cartilage and other connective tissues, sclerosis of cardiac valves and premature arteriosclerosis, and in later years, degenerative arthritis of the larger peripheral joints.
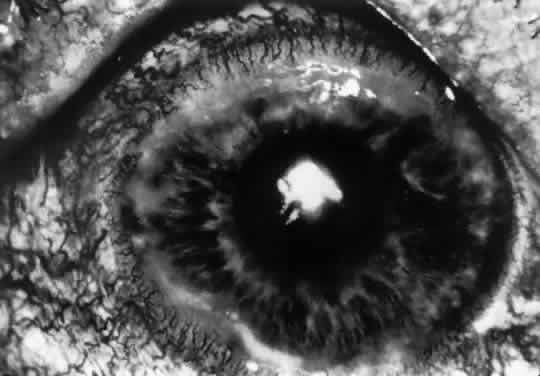
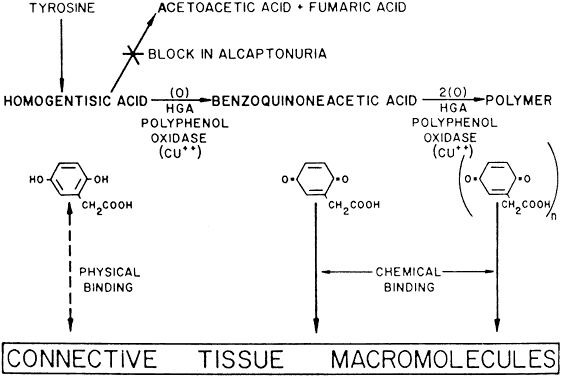
Tissue pigmentation (ochronosis) is not caused by the presence of homogentisic acid, which is bound physically to the connective tissue macromolecules, but rather by the chemical binding in the tissues of alternative pathway by-products resulting from the failure to remove homogentisic acid in a normal manner. These by-products are benzoquinoacetic acid and polymers of this compound. The production of both benzoquinoacetic acid and its polymers depends on the presence of the homogentisic acid polyphenol oxidase, which is present in the skin and cartilage of mammals. Once deposited, these polymers are indistinguishable from melanin (Fig. 2).3
|
Pigmentation of the cartilages of the ear, trachea, nose, tendons, heart valves, and prostate develops with age. Systemic complications are related to the joints, the genitourinary system, and the heart, where there is a higher than normal incidence of arteriosclerosis and valvular calcification.
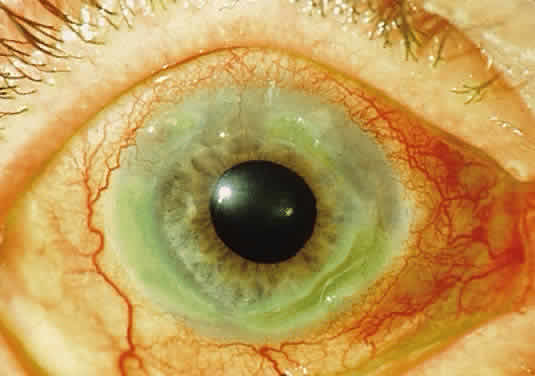
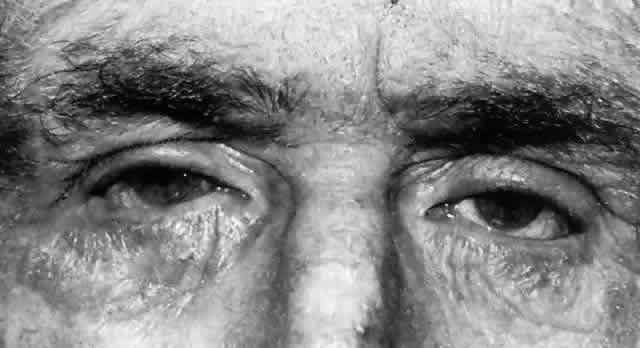
OCULAR FINDINGS. Ocular involvement is seen in 79% of patients with ochronosis (tissue pigmentation).4 In these patients, the ochronotic pigment is found in the sclera, conjunctiva, and limbic cornea. Vision usually is not impaired. The ocular abnormalities listed below are asymptomatic and related to pigmentary change5,6:
- Triangular patches of brown scleral pigmentation are seen anterior to the
insertions of the rectus muscles, primarily the horizontal recti. The
base of the triangle is near the limbus. The limbus often shows an
increased translucency, a sign of age-related degeneration in otherwise
normal eyes7; thus, the heavy pigmentation in that area supports the hypothesis that
pre-existing degeneration of connective tissue is a prerequisite for
ochronotic pigment deposition.
- Subepithelial brown particles may be seen in the superficial stroma near
the limbus.
- Pigmented pinguecula-like masses can be seen in the episclera, usually
between the limbus and the insertion of the muscles.
- Pigmentation of the tarsal plates and eyelids also has been described.
- Light microscopy reveals the amber globules or fiber-like structures in
association with degenerated collagen in the cornea, conjunctiva, episclera, and
sclera.8 Ultrastructurally, most of the pigment granules are extracellular, partly
altering the collagen fibers and fibrocytes. The ultrastructure of
the ochronotic pigment is similar to that of melanin, but histochemically
its behavior resembles that of elastin. Four patterns of deposition
have been observed with electron microscopy; these may represent biochemical
stages in the development of ochronotic deposits.9
DIAGNOSTIC EXAMINATION OF THE URINE. The urinary findings in this disease range from the nonspecific to the specific and are presented in that order here2,6:
Positive tests
Alkalization
Benedict's sugar reagent
Brigg's test (reduction of molybdate)
Reduction of silver in photographic film
Paper chromatography
Enzymatic spectrophotometric method
Negative tests
Glucose; enzymatic
Yeast fermentation
Fluorescence under ultraviolet light
DIFFERENTIAL DIAGNOSIS. Malignant melanoma must not be confused with the dark mass in the scleral region in a patient with alkaptonuria. Other etiologic agents of ochronosis are quinacrine and hydroquinone.
MANAGEMENT. The ideal form of therapy might be replacement of the missing enzyme. This is currently impractical, as are other measures such as dietary restriction of tyrosine and phenylalanine. Use of vitamin C has been advocated, but there are only a few reports of symptomatic relief.10 Most frequently ascorbic acid merely slows the rate of the urinary color change. Other treatments are purely symptomatic and do not halt the progression of the disease.
Tyrosinemia Type II (Richner-Hanhart Syndrome)
Tyrosinemia is classified as type I, type II, and neonatal.11 Tyrosinemia I is a rare, autosomal-recessive, metabolic defect characterized by hepatosplenomegaly, cirrhosis, fever, vomiting, renal glycosuria, generalized aminoaciduria, phosphaturia, and renal rickets. No skin or eye lesions are strictly characteristic of tyrosinemia type I.
Neonatal tyrosinemia is a transient disease. It primarily affects premature infants and is associated with lethargy. It also is without skin or eye lesions.
The clinical triad of skin lesions, ocular lesions, and mental retardation characterizes tyrosinemia type II. Painful skin lesions, which are limited to the palms and soles, may range from erosive to hyperkeratotic disturbances and may fade in a few days.12 Dermatologic involvement is usually coexistent with the eye lesions but may occur after the ocular symptoms, resulting in a delayed diagnosis.13 Mental retardation is a variable feature, with intelligence ranging from normal to severely retarded. The defect is caused by a deficiency of the enzyme soluble tyrosine aminotransferase, or a lack of parahydroxy phenylpyruvate hydroxylase, which leads to high serum tyrosine levels and increased urinary excretion of tyrosine and parahydroxy phenylpyruvate.14
OCULAR FINDINGS. Corneal lesions usually appear early in life, but later onset also occurs.12,15,16 Tearing, photophobia, and painful eyes are common. Pseudodendritic corneal erosions may involve the epithelium alone or may include Bowman's layer and anterior stroma. Central corneal ulcers with bases containing debris described as purulent and as thick and cheesy have been mentioned. All cultures are negative. Multiple intraepithelial opacities may be present and may be linear or arranged in a stellate pattern in the central cornea.17
Other ocular findings include whitish conjunctiva, discrete conjunctival plaques, and papillary hypertrophy. Rats fed a diet containing excessive L-tyrosine develop a disease mimicking human corneal disease.18 The corneal lesions in tyrosine-fed rats appear to be caused by the formation and growth of needle-shaped crystals within epithelial cells.19
A current pathogenetic hypothesis involves crystal production in the cornea as a result of a supersaturated state. When crystal formation is initiated in the central cornea, the disruption of cells forms biomicroscopically evident snowflake-like lesions. The crystals exert a force powerful enough to pierce cell membranes and displace nuclei. Lysosomal enzyme release, polymorphonuclear migration, vascularization, and subsequent healing20 follow cell rupture.
DIAGNOSIS. The diagnosis must be suspected clinically and confirmed by amino acid analysis of the blood and urine. The combination of skin lesions and pseudodendritic corneal ulcers is almost pathognomonic of this disorder. A tyrosine load test with ingestion of a tyrosine load of 150 mg/kg may be helpful in the diagnosis; in patients with Richner-Hanhart syndrome, this results in intense pain in the eyes, hands, and feet and induces a rim of intense erythema around the skin lesions.21
DIFFERENTIAL DIAGNOSIS. The eye lesions may suggest herpes simplex or other types of epithelial infections, developmental anomalies, vitamin deficiencies, or metabolic or storage diseases. The skin lesions are reminiscent of epidermolysis bullosa, dyskeratosis congenita, or keratosis palmaris et plantaris. However, the combination of eye and skin lesions in this pediatric age group is diagnostic. In the past, patients have been treated extensively for herpes simplex keratitis before a proper diagnosis was established. Clinical features helpful in distinguishing tyrosinemia from herpes simplex keratitis include bilaterally stellate, plaquelike lesions that lack club-shaped edges; minimal staining with rose bengal and fluorescein; and a lack of response to topical antiviral therapy.15,16,22
A low-tyrosine, low-phenylalanine diet is essential in the management of these patients. It usually results in a decrease of the serum and urine tyrosine levels and in relief of the discomfort in the eyes and skin within 24 hours.23 The photophobia and keratitis usually subside within 2 to 4 weeks.
The need for a metabolic examination (including assessment of serum tyrosine levels) in a young child with photophobia and bilateral pseudodendritic keratitis, even in the absence of cutaneous or developmental abnormalities, cannot be overemphasized. If the diagnosis is made early, dietary restrictions of phenylalanine and tyrosine lead to the resolution of ocular and cutaneous changes, and mental retardation may be prevented.10 Penetrating keratoplasty can be performed for end-stage corneal scarring and vascularization. Systemic steroids should be avoided after keratoplasty because dendritic lesions may recur on the new graft.24
Cystinosis
Cystinosis is a rare metabolic disorder characterized biochemically by an abnormally high intracellular content of free cystine; this, in turn, results in cystine crystal deposition in the eye, bone marrow, lymph nodes, leukocytes, and internal organs, including the kidneys.25
The primary defect involves impaired efflux of cystine from lysosomes, an active ATP-dependent process.26 The mutant gene for the infantile and juvenile forms has been mapped to chromosome 17p.27
Three cystinotic phenotypes have been reported and described: the infantile form, the benign adult form, and the intermediate adolescent form.28
INFANTILE FORM. Synonyms for this form are infantile nephropathic cystinosis, Fanconi's syndrome, and de Toni-Fanconi-Lignac syndrome.29,30 Symptoms, beginning in infancy, include polyuria, growth retardation, rickets, and progressive renal failure. In the past, renal failure has usually led to death before puberty, but dialysis and kidney transplantation now allow some patients to reach adulthood. The mode of inheritance is autosomal recessive. The ocular manifestations include photophobia, cystine deposition in the cornea and conjunctiva, and a peripheral retinopathy.
ADULT FORM. The benign adult form is asymptomatic, with the possible exception of photophobia, and is usually diagnosed during a routine slit lamp examination.31,32 Ages range from the teens through the mid-50s. Renal function is normal and patients have a normal life expectancy. The mode of inheritance is uncertain, but it may be autosomal recessive.
ADOLESCENT FORM. The adolescent form26,28 is characterized by a less severe nephropathy than is seen in the infantile form. Symptoms, which appear in the second decade, may or may not include rickets, renal failure, or the characteristic corneal and conjunctival deposits. Retinopathy is absent. Life expectancy is decreased because of renal dysfunction. The mode of inheritance is autosomal recessive.
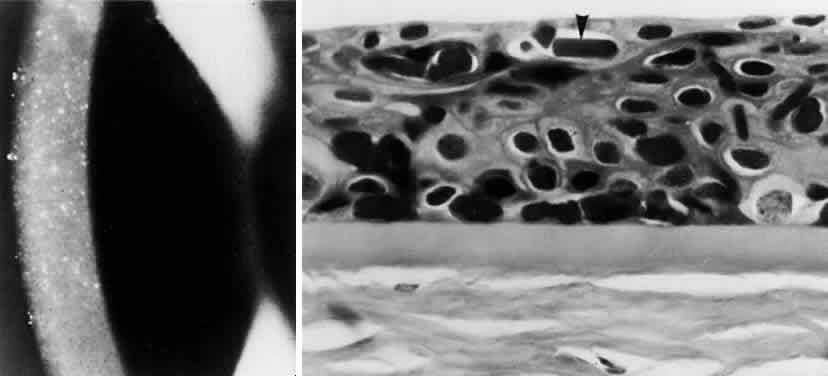
OCULAR FINDINGS. The outstanding clinical feature common to all three phenotypes is the corneal and conjunctival cystine crystal deposition (Figs. 3 and 4). Photophobia is often the only presenting visual symptom; this may be incapacitating and associated with blepharospasm.
|
|
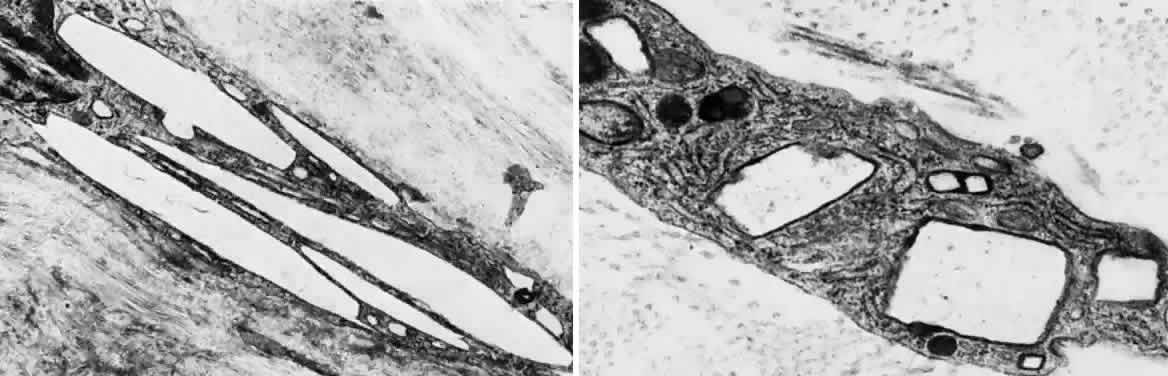
Corneal deposits appear as a layer of homogeneously distributed, fusiform or needle-shaped, iridescent crystals situated in the stroma beneath the epithelium. In the infantile form, anterior crystal deposition begins early in life (between 6 and 15 months of age) and proceeds posteriorly as the patient ages; deposition advances more rapidly in the periphery. The anterior location of the crystals may be associated with recurrent erosions.33 The depth of the stromal deposition and the density of crystals is always greater peripherally than centrally. More and larger crystals occur in the superficial stroma. No visual impairment occurs at this early stage. By the age of 7 years, most patients have crystals, either within or on the endothelial surface34,35; markedly decreased corneal sensitivity is also present.36 The spherical contrast sensitivity function is significantly lower in infantile cystinosis than in age-matched controls.37
The conjunctiva has a ground-glass appearance. Polychromatic, polymorphic, rectangular, or rhomboidal crystals can be seen with the biomicroscope.
The uvea contains an abundance of polymorphous crystals. Clinically, these can be seen as glistening dots on the surface of the iris. Thickened iris stroma and posterior synechiae may occur; pupillary block glaucoma38 also has been reported. The entire uvea has polymorphic crystal deposition, most heavily in the choroid. The sclera also has crystal deposition.
The retinal abnormality consists of a generalized depigmentation that may assume a patchy pattern. At first the pigmentary disturbance tends to be peripheral, but it progresses with age. Macular abnormalities have been observed.39 Intracellular crystals also have been seen in the retinal pigment epithelial cells during electron microscopy.
DIAGNOSIS. The ocular findings of cystinosis are sufficiently unique and characteristic to form the basis for a diagnosis of this disease. A specific diagnosis of cystinosis can be made by assaying for cystine in biopsied conjunctiva.40 Conjunctival biopsy is a simple and benign technique that may provide the diagnosis for this and other inborn errors of metabolism.
DIFFERENTIAL DIAGNOSES. Polychromatic corneal crystals similar to those in cystinosis also may be seen in multiple myeloma,41 Schnyder's crystalline dystrophy, Bietti's crystalline dystrophy, gout, and chrysiasis.
MANAGEMENT. The results of therapy with penicillamine and a cystine-free diet for infantile nephropathy have been disappointing.29 Oral cysteamine is the only available treatment for the nephropathy of infantile cystinosis. The cystine depletion that occurs through the formation of cystine-cysteamine-mixed disulfide42,43 apparently helps stabilize glomerular function and also improves growth and development.44 Cysteamine treatment decreases the deposition of additional cystine in the kidneys; but it does not reverse existing renal tubular and glomerular damage or gradual loss of kidney function.45 There is no evidence that corneal cystine deposition, posterior synechiae, or rod and cone dysfunction are reversible with cysteamine treatment; it may only prevent further ocular damage. Renal transplantation is an alternative therapy for patients with advanced renal disease. As a result of renal transplantation, children are now surviving to the second and third decades with normal renal function. Although cystine is not deposited in a grafted kidney, it appears to accumulate relentlessly in other organs and tissues, especially ocular tissue, and progressive visual impairment has been documented.46 Continued therapy with oral cysteamine after renal transplantation currently must be considered. Reversal of corneal crystal deposition by topically administered cysteamine has been reported.47,48 Although penetrating keratoplasty can be performed for advanced corneal cystinosis with endothelial decompensation, cystinosis can recur in the transplanted graft.33
Wilson's Disease (Hepatolenticular Degeneration) and Hypercupremia
Wilson's disease is an autosomal-recessive inborn error of metabolism in which excess copper deposition occurs, primarily in the liver (leading to cirrhosis), the kidneys (leading to renal tubular damage), the brain (leading to widespread failure of motor function while sparing sensory function), and Descemet's membrane of the cornea.49 The ATP7B gene on chromosome 13 that is mutated in this disease has been cloned and sequenced, and the resulting protein product is a copper-transporting ATPase. Because more than forty mutant forms of this gene have been identified, molecular diagnosis is only practical in family members of patients in whom the genetic defect has already been identified.50
Clinical manifestations rarely occur before 6 years of age and may be delayed until the fifth decade. Approximately 40% of patients present with symptoms of hepatic disease, and 40% develop symptoms referable to the nervous system.51
This copper storage disease is probably caused by an intrahepatic defect in copper metabolism that leads to impaired excretion of the metal in the bowel and, concurrently, to decreased ceruloplasmin (α2-globulin) synthesis. The result of this defect is that, for the first few years of life, increasing amounts of copper are stored in the liver. At some point, necrosis of the liver cells occurs, and the copper is released into the blood and deposited in other tissues.52
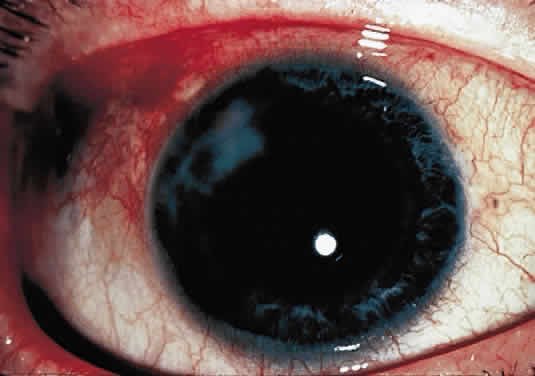
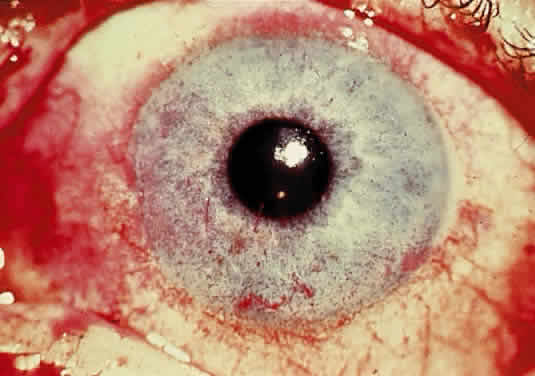
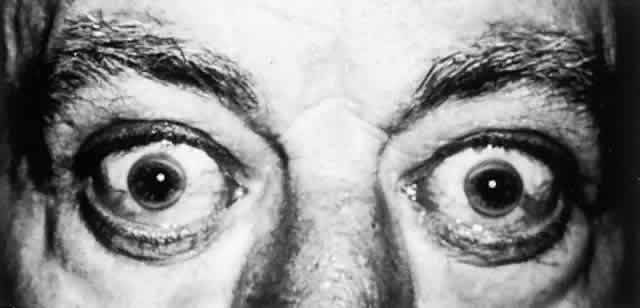
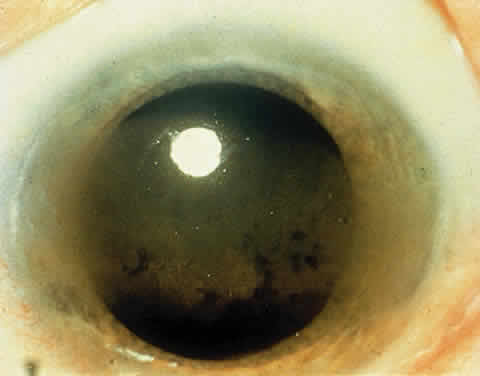
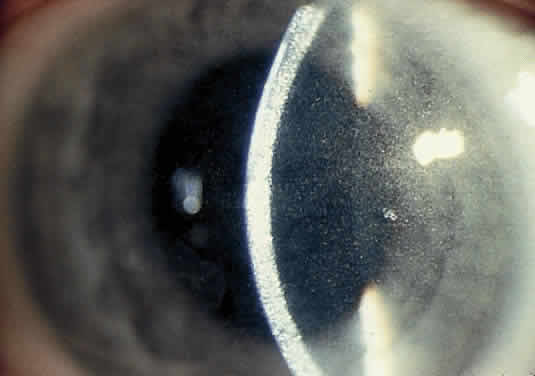
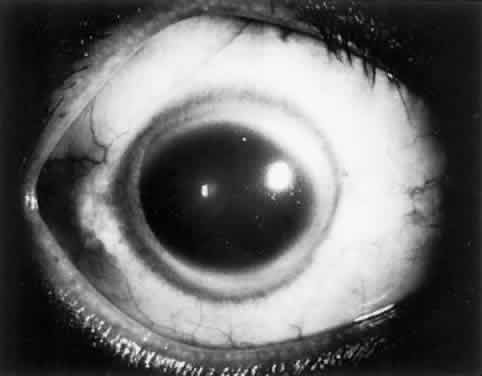
OCULAR FINDINGS. One of the few truly diagnostic and pathognomonic physical signs in clinical medicine is a Kayser-Fleischer corneal pigment ring found in patients with Wilson's disease (Fig. 5).This ring is recognized as a golden-brown, ruby-red, or green band of 1.0 to 3.0 mm, starting at the limbus but at the level of Descemet's membrane.53 The color of the ring is presumably caused by scattering and reflection of incident light and by photointerference effects created by the layers of copper granules. Such variables as size, shape, and unit density of the granules may account for the different appearances of the Kayser-Fleischer ring. The course of the Kayser-Fleischer ring has been well documented.54,55 The site of earliest pigment deposition is an arc in the superior periphery of the cornea from the 10- to 2-o'clock meridian. The arc spreads slowly toward the horizontal plane and gradually broadens. Later in the progression of the ring formation, a band appears inferiorly as a crescent stretching from the 5- to 7-o'clock positions. In time, the two arcs meet. With treatment, the sequence of events is reversed, and after the copper has reabsorbed, a pitted or beaten silver pattern may become apparent at the previous site of the ring. This is an indication that treatment has produced a negative copper balance.49 Decreased visual acuity is not a problem. Rarely, copper is deposited in the crystalline lens, giving an anterior subcapsular “sunflower” cataract.
|
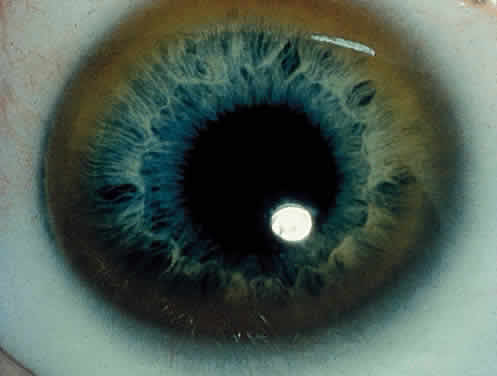
DIAGNOSIS. This syndrome should be suspected clinically. Serum copper levels, as well as ceruloplasmin levels, are low. More precise studies requiring measurement of the turnover of injected radioactive copper may be performed, or copper levels can be determined in liver biopsy specimens.
DIFFERENTIAL DIAGNOSES. Pigmented corneal rings clinically identical to Kayser-Fleischer rings have been reported in non-Wilsonian liver diseases, including primary biliary cirrhosis, progressive intrahepatic cholestasis of childhood, and chronic active hepatitis. Although these diseases cause an elevated level of copper in the blood, urine, and liver, only in Wilson's disease are subnormal levels of ceruloplasmin present.56 Pigmented corneal rings have also been associated with multiple myeloma57; and a patient with a poorly differentiated adenocarcinoma of the lung, associated with IgG monoclonal gammopathy and hypercupremia, presented with copper deposition on the cornea and lens.58
PATHOLOGY. Electron microscopic, x-ray energy spectroscopic, and atomic absorption spectroscopic studies of the Kayser-Fleischer rings have revealed electron-dense granules rich in copper and sulfur in both the peripheral and the central region of the cornea, but more abundantly at the periphery. The association of copper with sulfur suggests that a sulfur-containing moiety functions in binding copper.59
MANAGEMENT. D-penicillamine (Cuprimine) is an extremely effective chelating agent because of its ability to mobilize copper from the tissues and increase its excretion in urine.52,60 Treatment of Wilson's disease with penicillamine is effective when started before there is active liver disease. After prolonged treatment with penicillamine, Kayser-Fleischer rings may fade, neurologic signs may clear, and liver function abnormalities may disappear. However, Kayser-Fleischer ring regression does not correlate with neuropsychiatric improvement.61 With D-penicillamine or after liver transplantation,62 Kayser-Fleischer rings disappear in reverse order to their formation. In most patients, the neurologic and hepatic lesions remit; hence the importance of recognizing the significant corneal pigment rings.
Lowe's Oculocerebrorenal Syndrome
This is an X-linked recessive metabolic disorder of unknown etiology. First recognized as a clinical entity by Lowe and co-workers,63 it occurs only in males and is characterized by mental and growth retardation, hypotonia, aminoaciduria, reduced ammonia production by the kidneys, rickets, and eye signs. The syndrome is listed here because aminoaciduria is one of the symptoms.
OCULAR FINDINGS. The most prominent ocular findings are congenital cataracts (over 96%) and congenital glaucoma (over 50%). A miotic pupil is common, but the retina is usually normal. A mild corneal haze has been reported, but this is probably the result of glaucomatous ocular hypertension.
DIAGNOSIS. This disorder should be considered in any male infant with both congenital cataracts and congenital glaucoma. In carrier females, the lenticular opacities are punctate, white to gray in hue, and vary in size from microns to several millimeters. Unlike congenital nuclear opacities, lenticular opacities are present in all layers of the cortex, indicating that they are well formed in adult life. Subcapsular, plaque-like cataracts are seen predominantly in older obligate carriers. Because female carriers have an identifiably higher number of lens opacities than do age-matched controls, genetic counseling may be indicated because 50% of their female offspring are expected to be carriers.64 Biochemical findings are typical and include metabolic acidosis and renal tubular acidosis. The basic enzyme deficiency is unknown, but may be related to biochemical abnormalities in glycosaminoglycan metabolism. The urinary findings include proteinuria, generalized aminoaciduria, organic aciduria, and on occasion, glycosuria.65
DIFFERENTIAL DIAGNOSIS. Rubella can cause both congenital cataracts and glaucoma but usually produces only one or the other.
MANAGEMENT. A lensectomy-vitrectomy is indicated for visual rehabilitation in patients with dense cataracts. Although all Lowe's syndrome patients are retarded, the degree of retardation is not necessarily severe. When these patients are visually rehabilitated by surgery and subsequent refractive correction, they function better than do those patients with visual stimuli deprivation.66
Porphyria
CONGENITAL ERYTHROPOIETIC PORPHYRIA.
This disease, also known as Gunther's disease, is an autosomal-recessive disorder. It is caused by a deficiency of uroporphyrinogen III synthase activity, resulting in an overproduction of porphyrins (type I) for which the body has no use. Different molecular defects have been detected.67 The clinical signs include hydroa aestivate (a vesicular or bullous eruption of the face, backs of the hands, and other exposed areas of the body), erythrodontia (brownish discoloration of the teeth), hypertrichosis, and splenomegaly.68 Neurologic symptoms, hypertension, and abdominal colic are not found.
Ocular Findings. Ocular manifestations of this disorder include cicatricial ectropion, loss of eyelashes and eyebrows, bilateral exophthalmos, optic atrophy, retinal hemorrhages, and chorioretinitis.69–71 Conjunctivitis may result in marked scarring and adhesion of the conjunctiva to the sclera. Decreased corneal sensitivity, corneal scarring and vascularization, corneal perforation at the limbus, and scleromalacia may occur. Histopathology of tissue from a patient who underwent a penetrating keratoplasty and conjunctival resection revealed a thickened basement membrane of the vessels within the conjunctival and corneal stroma. Microfibrillar material was seen in the extracellular spaces of the conjunctival stroma. Inflammatory cells were noted in the corneal stroma, and there was a loss of keratocytes. Descemet's membrane lacked the normal fetal and postnatal banding and the endothelium was severely damaged.72 The changes in the vessels and conjunctival stroma were similar to those described in the skin of porphyria patients exposed to ultraviolet light.
Laboratory Investigations. Normochromic anemia and excessive excretion of uroporphyrin I and coproporphyrin I in urine and feces are found. The diagnosis should be confirmed by detection of decreased uroporphyrinogen III synthase activity.
VARIEGATE PORPHYRIA. This hepatic porphyria is characterized by acute attacks involving abdominal and neurologic manifestations, as well as by chronic skin lesions that are caused by photosensitivity.
Ocular Findings. In addition to the previously mentioned ocular signs, optic neuritis, optic atrophy, and exudates similar to those in acute intermittent porphyria are seen in affected homozygotes.70
Laboratory Investigations. Analysis of urine and stools reveals increased levels of protoporphyrin, coproporphyrin, uroporphyrin, aminolevulinic acid, and porphobilinogen, the latter being present only in acute attacks.68,70,71
Management. Intravenous heme leads to a rapid recovery of acute attacks. No specific therapy for ocular lesions is available. Scleral patch grafts have been used successfully to treat the scleromalacia perforans that develops in some of these patients.73
Amyloidosis
Amyloid deposition produces dysfunction in any organ by replacing its functioning mesenchymal or parenchymal cells. All amyloid proteins are insoluble fibrinous proteins that share a unique fibrillar ultrastructure. Depending on the origin of the amyloid protein, the amyloid fibrils deposit either in certain organs or systemically. Amyloid deposition does not incite any inflammatory response; however, a foreign body giant cell reaction may be seen occasionally around deposits in the ocular adnexa.74
Amyloidosis can be classified as primary (localized and systemic), secondary (localized and systemic), and heredofamilial according to the presence or absence of underlying disease (primary or secondary amyloidosis) and genetic associations (heredofamilial).
More recently, amyloidosis has been classified according to the protein precursor. The nomenclature for this classification includes A (for amyloidosis) followed by the initial of the protein precursor (e.g., AL for amyloidosis with light chain immunoglobulins as precursors).75 The most common type of systemic amyloidosis currently seen is the light chain amyloidosis (AL), which is associated with primary amyloidosis and sometimes with multiple myeloma. Fifteen to 20% percent of patients with myeloma have amyloidosis; fewer than 20% of patients with amyloidosis have myeloma.76 In secondary systemic amyloidosis the major amyloid deposit is protein AA, a degradation product of a serum acute phase reactant, serum amyloid A (apoSAA).
Histologic examination of amyloid deposition has shown a pericollagenous distribution associated with the primary and myeloma-related types (pattern type I) and perireticulin deposition in the secondary form of amyloidosis, associated with parenchymatous organ invasion (pattern type II).
PRIMARY AMYLOIDOSIS (SYSTEMIC AND NONSYSTEMIC TYPES). Primary amyloidosis is the form associated with clinically important depositions of amyloid in the eyes or ocular adnexa.74,77
Ocular Findings
CORNEA
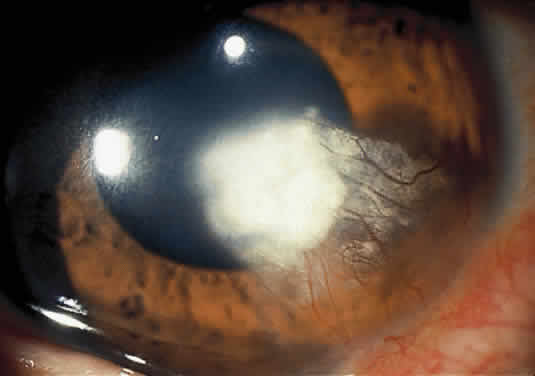
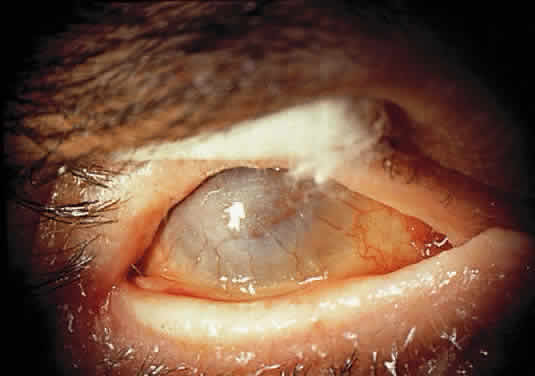
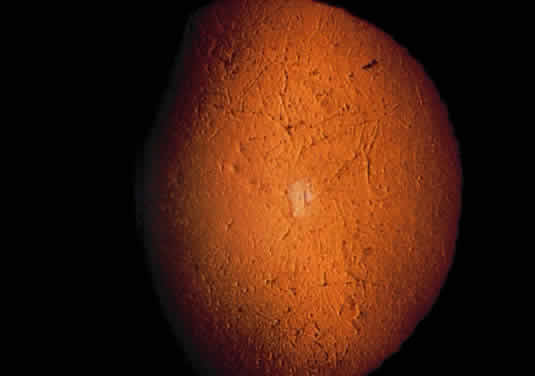
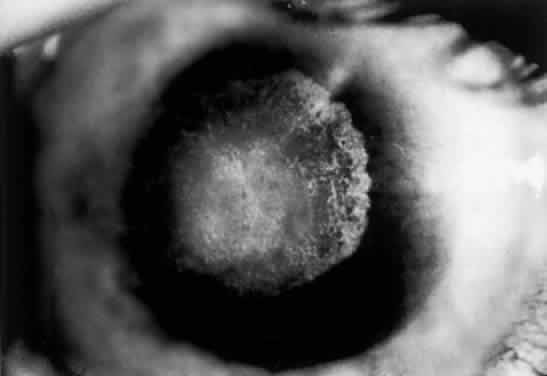
Nonsystemic Familial Type. Lattice corneal dystrophy (Biber-Haab-Dimmer) is usually considered to be a localized amyloidosis of the cornea and could be classified as a special type of primary localized autosomal dominant amyloidosis (see corneal dystrophies section) (Fig. 6).78 Rarely, a form of primary familial amyloidosis of the cornea may occur. The lesions appear as centrally located, raised, gelatinous masses with a mulberry-like surface (Fig. 7).79
|
|
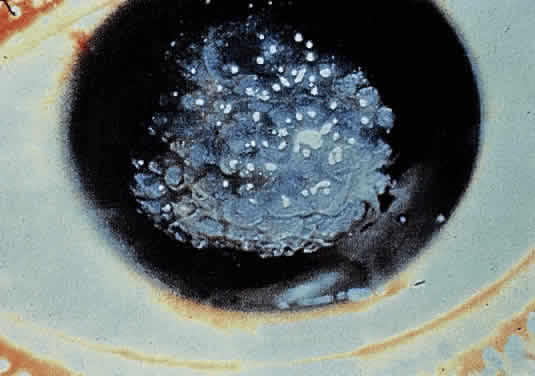
Nonfamilial Type. Accumulations of amyloid in the cornea without apparent systemic involvement or a hereditary pattern have been described. Most often this occurs as the result of pre-existing ocular disease.80
Systemic Familial Type. Rarely, patients with lattice corneal dystrophy may have a familial primary systemic amyloidosis known as Meretoja syndrome. These patients develop lattice dystrophy in young adulthood and, in later years, skin changes, cranial nerve palsies, peripheral neuropathies, and visceral complaints. The lattice dystrophy that develops with systemic involvement is milder with regard to visual loss but more extensive in that it reaches to the corneal periphery without the peripheral lucid interval seen with localized lattice.81
CONJUNCTIVA. Clinically detectable conjunctival involvement is not a feature of systemic amyloidosis. However, nonfamilial amyloidosis confined to the conjunctiva has been reported as an example of primary amyloidosis. Conjunctival amyloidosis is often asymptomatic and may be present for years before the patient seeks medical attention. Typically, there is a discrete, nonulcerative, yellow, waxy, firm, nontender subconjunctival swelling (Fig. 8). This may be located in the palpebral fornix or bulbar conjunctiva, including the limbal area. The conjunctival area is usually smooth but may be friable and may show recurrent bleeding. However, antecedent local diseases have been incriminated in this amyloid deposition.77
|
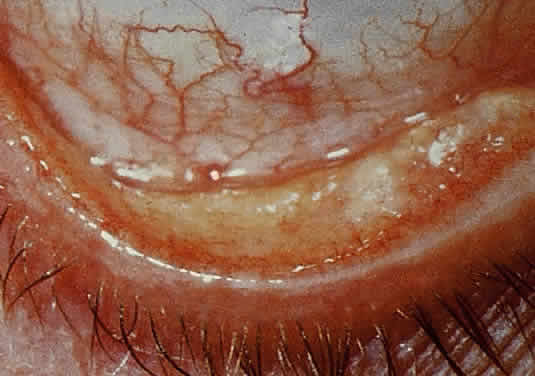
EYELID. The skin of the eyelid is the site of predilection for the characteristic cutaneous eruption of primary systemic amyloidosis.77 Typically, the lesions are symmetric, bilateral, small, smooth, and discrete or confluent papules. These lesions may be yellow and waxy or purple and hemorrhagic. A positive family history is often present. Primary localized amyloidosis does not affect the skin of the eyelids.7
VITREOUS. Vitreous opacities in amyloidosis may be uniocular or they may be asymmetric if both eyes are involved; these opacities may be the presenting feature of systemic amyloidosis. Vitreous involvement is much more common in primary familial systemic amyloidosis (more than 20 cases reported) than in nonfamilial amyloidosis (only 5 cases reported).82
ORBIT. Orbital involvement does occur in primary nonsystemic amyloidosis; proptosis is a prominent feature. Orbital amyloidosis can present as an acquired isolated sixth nerve palsy in a young adult.83 Amyloid also occurs in the orbits of some patients with primary familial systemic amyloidosis.
NEURO-OPHTHALMIC MANIFESTATIONS. Pupillary abnormalities are the most common neuro-ophthalmic sign found in primary familial systemic amyloidosis. Pupils have been described as dilated and unequal with absent or sluggish reactions to light, which at times may be accompanied by poor accommodative responses.74 Amyloid may be deposited in ciliary nerves. External ophthalmoplegia, diplopia, and optic neuropathy have been reported in some cases of familial amyloidosis.
SECONDARY SYSTEMIC AMYLOIDOSIS. Secondary systemic amyloidosis is associated with a number of underlying afflictions that have a significant chronic inflammatory component, such as tuberculosis, osteomyelitis, and leprosy. Small, clinically insignificant deposits of amyloid in the eye have been described rarely, and ophthalmologic signs attributable to the presence of amyloid are virtually unknown.77
SECONDARY LOCALIZED AMYLOIDOSIS. This form of amyloidosis has been reported to occur in association with trichiasis,84 granular corneal dystrophy,85 and keratoconus,86 or after chronic keratitis.
Diagnosis. Amyloidosis must be suspected on clinical grounds. Definitive diagnosis depends on histochemical reactions, including metachromasia with crystal violet, fluorescence with thioflavin T, birefringence and dichroism with Congo red, and positive staining with direct cotton dyes such as Sirius red. The mere recognition of amyloid in an ocular structure can no longer be considered a complete diagnosis. In primary systemic amyloidosis, the diagnosis is readily made by pathologic examination of the vitreous aspirate. The possibility of amyloidosis in extraocular sites, underlying or associated ocular disorders, familial aspects, and myelomatosis and other immunoglobulin abnormalities must be considered.
Management. Medical treatment may be specific for the various amyloidoses. Liver transplantation has been successful in some familial cases. Treatment of the underlying inflammatory condition is of questionable benefit in secondary amyloidosis; however, in primary amyloidosis, where a presumed monoclonal plasma cell secretes excess light chain, alkylating agents have been used with modest impact on survival. Recently, the combination of melphalan and prednisone in this setting was shown to be superior to colchicine, leading to prolonged survival.87 When surgery is indicated, the treatment of choice for vitreous involvement of amyloidosis is vitrectomy. Pars plana vitrectomy can be effective in restoring visual acuity, although recurrences are common. Reopacification of the retrolental vitreous is the most common reason for vitrectomy revision, which is required in 24% of patients.88 Penetrating keratoplasty may be required for advanced corneal disease although the deposition frequently recurs in the graft. Phototherapeutic keratectomy with excimer laser has been used for superficial depositions in the cornea.89,90
Gout and Urate Keratopathy
Gout is a clinical disorder of purine metabolism manifested by hyperuricemia, recurrent attacks of acute arthritis that are usually responsive to colchicine, and on occasion, tophaceous deposition of monosodium urate. Nephropathy is a frequent complication.91 Primary gout is usually attributed to an inborn error of metabolism. A deficiency of hypoxanthine-guanine phosphoribosyl transferase has been found in familial gout and may represent the enzymatic deficiency in at least one variety of the disease.92 The largest subgroup consists of patients in whom the biochemical defect is as yet undefined. Other enzymes may be involved.
Secondary gout or hyperuricemia may occur in toxemia of pregnancy and chronic renal insufficiency, as well as in other conditions that may be associated with breakdown of cell nuclei, such as malignant lymphomas, chronic myelogenous leukemia, hemolytic anemia, and primary polycythemia.
Increased endogenous urate production, as well as impaired renal urate excretion, has been cited as the basis for the hyperuricemia. The characteristic pathologic feature of gout is the deposition of urate crystals in articular and periarticular structures. In the eye, precipitation of urate crystals has been described in the lens, sclera, tarsal plates, tendons of extraocular muscles, and cornea.93 The ocular deposition of urate crystals supports the clinical finding that urate deposits develop in avascular rather than vascular tissue.
Genetic factors may include both cumulative gene action and single-gene effects.
OCULAR FINDINGS. Clinical manifestations of ocular gout include chronic hyperemic conjunctivitis, scleritis, episcleritis (Fig. 9), and tenonitis. In the past, uveitis has been associated with gout; but in a review of 69 patients with severe gout, none had any evidence of uveitis, either past or present.94 Monosodium urate crystals may be deposited in the corneal epithelium.95 The urate crystal deposition may offer one of two possible appearances. Slit lamp examination shows a fine, golden-yellow, scintillating crystal formation, diffusely distributed in the cornea, more heavily in the interpalpebral fissure and extending to the limbus. These crystals appear epithelial and subepithelial; they are best visualized by retroillumination.
|
Crystals interpreted as urates also have been described in band keratopathy occurring in a patient with gout.96 This keratopathy cannot be differentiated from calcium deposits solely by slit lamp examination. The site of urate crystals in the corneal epithelium is intranuclear.95 Urate-like crystals also have been seen in the corneal stroma in a disease termed keratitis urica. This is probably a localized dystrophic corneal disease and not associated with gout.96
DIAGNOSIS. A high index of suspicion is necessary to establish gout as a contributing factor to corneal crystals, band keratopathy, conjunctivitis, and iritis. Accordingly, the ophthalmologist should consider gout in the differential diagnosis of every patient with bilateral chronic conjunctivitis. High levels of uric acid in the urine and blood are helpful in establishing the diagnosis. Diuretic therapy is currently one of the major causes of secondary hyperuremia.97 Other causes of corneal crystals include cystinosis and monoclonal gammopathy. (See Table 1 for the differential diagnoses of crystals and band keratopathy.)
MANAGEMENT. Allopurinol is the most common antihyperuricemic agent used to treat gout. Long-term use of allopurinol has been associated with lens changes, primarily in the anterior and posterior subcapsular area. Recent evidence, however, suggests that long-term allopurinol therapy has a cataractogenic action only in patients in whom the drug has become photobound within the lens, possibly as a result of exposure to ultraviolet radiation.98
Paraproteinemias: Multiple Myeloma
Multiple myeloma is characterized by multiple plasma cell tumors, commonly manifesting themselves as osteolytic bone lesions. The neoplastic cells secrete monoclonal immunoglobulins, which may deposit as amyloid protein in different tissues, and may be excreted in the urine as immunoglobulin light chains (Bence Jones proteinuria). The disease results in bone pain, depletion of blood elements, hypercalcemia, renal insufficiency, amyloidosis, and immune disturbances. The incidence of multiple myeloma increases with age and is around 3 per 100,000. This incidence is equal in men and women, but blacks are affected more commonly than whites.99 Recently, a Herpes virus (HHV-8) has been implicated in the development of the disease.100
OCULAR FINDINGS. The ocular manifestations of multiple myeloma may be divided into two groups: those attributable to plasmacytoma growth in and about the eye, and those attributable to hematologic and serum protein abnormalities.101 The ocular manifestations include intraorbital or intraocular plasmacytomas,102,103 ciliary body cysts,104,105 retinal vascular tortuosity, hemorrhages and exudates,106 and the ophthalmic manifestations of intracranial plasmacytomas (papilledema).106 Crystals may be seen in the lens, and the cornea may show a band keratopathy resulting from the hypercalcemia. Clusters of myeloma cells may occur on the corneal endothelium. Conjunctival involvement in patients with multiple myeloma includes sludging of red cells in conjunctival vessels; occasionally malignant plasmacytomas of the conjunctiva arise anew or in association with advanced multiple myeloma.107
The presence of numerous delicate, scintillating crystals in the cornea has been recognized as a rare manifestation of hypergammaglobulinemia.41,108–110 In some cases, the corneal crystals are interspersed throughout the corneal stroma; in others they are most evident in the anterior cornea,103 including the epithelium.110 Posterior stromal corneal deposits are uncommon. The deposits usually are not vascularized, and corneal sensation is normal. Crystals may be detectable clinically in the bulbar conjunctiva.41,108,109 Aronson and Shaw described corneal crystals as probably consisting of cholesterol41; however, these crystals probably consist of partial or complete immunoglobulins110 (Fig. 10). Cysts of the ciliary body have been reported in 33% to 50% of myeloma patients, and retinal vascular lesions have been reported in up to 66% of these patients.111 Although corneal and orbital involvement are less common, orbital involvement is the first manifestation of systemic disease in about 75% of cases. Primary plasmacytomas may arise in the soft tissues or in the surrounding bones with secondary orbital invasion. The orbital roof and frontal bones often are affected, with resultant proptosis and downward displacement of the globe. The most common clinical signs are eye pain, diplopia, and visual impairment associated with a history of slowly progressive proptosis for weeks to months. Choroidal folds, venous engorgement of the disc, ptosis, and pupillary involvement may occur.
|
Polychromatic, dustlike deposition of copper in Descemet's membrane of the central cornea (with peripheral sparing) and in the anterior and posterior lens capsule occurs when the myeloma protein has strong copper-bonding properties.57
Corneal crystalline deposits also may occur in benign monoclonal gammopathy.110 Corneal opacities also have been reported in a patient with cryoglobulinemia and reticulohistiocytosis.112
DIAGNOSIS. Serum protein electrophoresis, examination of the urine for the presence of Bence Jones protein, and bone marrow aspiration are required. The differential diagnoses must include other causes of corneal crystals (see Table 1).
MANAGEMENT. Lamellar keratoplasty has been performed for visual rehabilitation.110 However, the treatment is basically directed toward the systemic disease. Chemotherapy and radiotherapy are effective in reducing the tumor mass in the orbit. Clinical improvement occurs in more than half of patients treated with alkylating agents such as melphalan. Radiotherapy is effective in treating orbital plasmacytomas and localized lytic bone lesions. Hypercalcemia is treated with hydration and intravenous furosemide (to promote urinary calcium excretion), as well as with steroids. The new bisphosphonate agents, such as pamidronate, are effective in correcting the hypercalcemia of multiple myeloma, and have recently been approved by the FDA for prevention of bone fractures in this disease.113
Ciliary body epithelial cysts are usually not diagnosed in life and do not require treatment. Retinopathy may improve with systemic treatment. Both penetrating and lamellar keratoplasties have been performed for advanced corneal disease, but recurrence after lamellar keratoplasty has been reported.110 Marked clearing of the corneal crystals with corresponding improvement of visual acuity has been reported to occur after chemotherapy.114
DISORDERS OF LIPOPROTEIN AND LIPID METABOLISM
The plasma lipoproteins are complex water-soluble macromolecules involved in the transport of lipid to and from tissues. These macromolecules contain triglyceride, cholesterol ester, and phospholipid in combination with specific apoproteins. The lipoproteins can be separated by electrophoresis into four main groups; in order of increasing mobility on paper electrophoresis, these are the chylomicrons, β-lipoproteins, prebetalipoproteins, and α-lipoproteins (Table 2). A further characterization based on lipoprotein size and density is achieved by ultracentrifugal analysis. The normal range of plasma lipid concentrations is noted in Table 3.
TABLE 15-2. Designation of the Plasma Lipoproteins
| Name | Synonym | Predominant Lipid |
| Chylomicrons | Chylomicron | Triglyceride |
| β-Lipoproteins | Low-density lipoprotein | Cholesterol |
| Prebetalipoproteins | Very-low-density lipoprotein | Triglyceride |
| α-Lipoproteins | High-density lipoprotein | Phospholipid |
TABLE 15-3. Normal Range of Plasma Lipid Concentrations
| Lipid | Concentration |
| Total cholesterol | 120–250 mg/100 ml |
| % as ester | 66–72 |
| Triglyceride | 40–125 mg/100 ml |
| Total phospholipid | 160–310 mg/100 ml |
A number of genetically determined disturbances of plasma lipoproteins have been identified. Although the role of the lipoproteins in the metabolism of the eye has not yet been precisely determined, changes in the eye may reflect more widespread disease.115
Hyperlipoproteinemia
Five basic phenotypes (Table 4) of hyperlipoproteinemia have been defined116,117; these five phenotypes are distinguished by the class of lipoprotein that is elevated in each.
TABLE 15-4. Hyperlipoproteinemias
| Elevation in Plasma | ||||||
| Type | Name | Lipoprotein | Lipid | Xanthelasma | Corneal Arcus | Lipemia Retinalis |
| I | Familial lipoprotein lipase deficiency | Chylomicrons | Triglycerides | + | - | + |
| Familial apoprotein CII deficiency | ||||||
| IIa | Familial hypercholesterolemia | LDL | Cholesterol | + | + | - |
| Multiple lipoprotein-type hyperlipidemia | ||||||
| Polygenic/exogenous hypercholesterolemia | ||||||
| IIb | Multiple lipoprotein-type hyperlipidemia | LDL and VLDL | Cholesterol and triglycerides | - | + | - |
| Familial hypercholesterolemia | ||||||
| III | Familial dysbetalipoprotenemia | VLDL remnants | Triglycerides and cholesterol | + | + | + |
| IV | Familial hypertriglyceridemia (mild form) | VLDL | Triglycerides | + | - | + |
| Multiple lipoprotein-type hyperlipidemia | ||||||
| Sporadic hypertriglyceridemia | ||||||
| Tangier disease | ||||||
| V | Familial hypertriglyceridemia (severe form) | VLDL and chylomicrons | Triglycerides and cholesterol | - | - | + |
| Familial apoprotein CII deficiency | ||||||
| Multiple lipoprotein-type hyperlipidemia | ||||||
LDL, low-density lipoprotein; VLDL, very low-density lipoprotein.
Type I (Hyperchylomicronemia). A deficiency of lipoprotein-lipase and Apo CII result in an elevation of chylomicrons, which are normally absent from fasting serum.118 Most patients are asymptomatic; however, attacks of abdominal pain, and sometimes pancreatitis, do occur. There may be eruptive xanthomas of the skin or mucous membrane. No cardiovascular complication, tendinous tuberous xanthoma, or corneal arcus is seen. Occasionally, lipemia retinalis is present. This disorder is transmitted as an autosomal-recessive trait.115–116
Type II (Hyperbetalipoproteinemia and Prebetalipoproteinemia). In these disorders there are elevated levels of low-density lipoproteins (LDLs; type IIa) or elevated levels of both LDLs and very-low-density lipoproteins (VLDLs; type IIb).118 The most serious systemic manifestation is cardiovascular disease, including peripheral vascular disease and myocardial infarction. The serum is clear, and there is an elevated cholesterol level. Corneal arcus, xanthelasma, or conjunctival xanthoma typically are present. This disorder is transmitted as an autosomal-dominant trait. Type II is expressed most severely in the homozygous state. Death before the fourth decade is common. 115–116
Type III (Dysbetalipoproteinemia). VLDL triglyceride and VLDL cholesterol are both elevated in this rare disorder.118 The serum is creamy. A variety of xanthomas are seen, including palmar, tuberous, and subperiosteal varieties and the more characteristic tuberoeruptive variety. Corneal arcus and lipemia retinalis are present early in the disease. Cardiovascular diseases, including peripheral vascular disease and myocardial infarction, are the most serious systemic manifestations. Type III disease is inherited as an autosomal-recessive trait.115–116
Type IV (Hyperprebetalipoproteinemia). Serum cholesterol in this disease may be normal or slightly elevated; however, the triglycerides are elevated and overproduction of VLDLs has been reported.118 There is no apparent increased risk of coronary heart disease in this type.118 Corneal arcus and xanthelasma are not seen; but lipemia retinalis may be evident. Type IV is transmitted as an autosomal-dominant trait.115–116
Type V (Hyperprebetalipoproteinemia and/or Hyperchylomicronemia). This disease involves familial lipoprotein lipase deficiency with resulting hyperchylomicronemia, and familial apoprotein CII deficiency, in which either chylomicrons or VLDL (VLDLa prebetalipoprotein) or both are high.118 Triglyceride is elevated in these diseases. Attacks of abdominal pain and, less commonly, of pancreatitis may be seen. Eruptive xanthomas do occur. Lipemia retinalis may be seen, but there is no xanthelasma or corneal arcus. The genetic factors involved are uncertain.115,116
OCULAR FINDINGS. As a generalization, the ocular features of hyperlipoproteinemia may be correlated with the presence of elevated levels of cholesterol or triglycerides (Table 5). Higher than normal levels of plasma cholesterol may be associated with xanthelasma and with presenile arcus of the cornea. However, both of these conditions also may be present when plasma cholesterol levels are normal. The incidence of xanthelasma and corneal arcus increases with age; it is highest in persons with type II hyperlipoproteinemia and usually low in those with type IV disease. Xanthelasma and corneal arcus are highly associated with each other and with increased levels of plasma cholesterol and LDL cholesterol (LDL-C), especially in young people.119
TABLE 15-5. Genetically Determined Hyperlipoproteinemia and Typical Ocular
Signs
| Primary Type | Lipoprotein Abnormality | Increased Lipid Fraction | Lipemia Retinalis | Corneal Arcus | Xanthelasma |
| I | Chylomicrons | Triglyceride | + | - | - |
| II | β-Lipoprotein | Cholesterol | - | + | + |
| III | Broad-beta band | Cholesterol | + | + | + |
| (β-mobility, pre-beta density) | Triglyceride | ||||
| IV | Prebetalipoprotein | Triglyceride | + | - | - |
| V | Chylomicrons | ||||
| Prebetalipoprotein | Triglyceride | + | - | - |
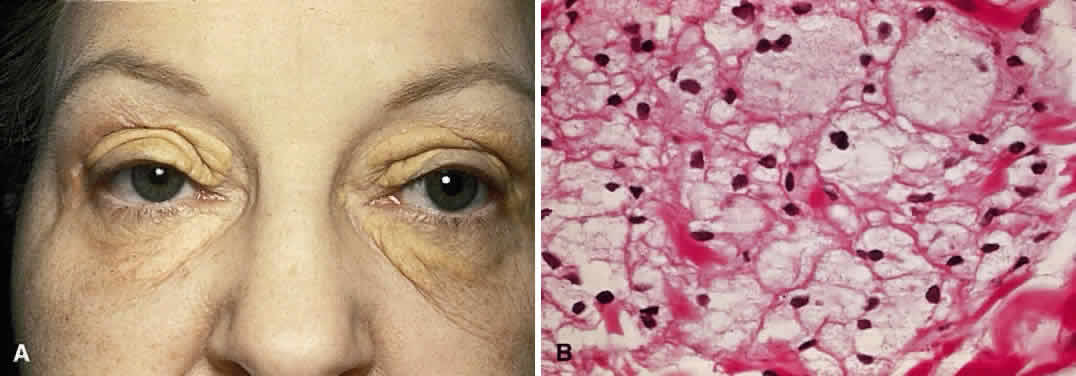
Xanthelasma is a planar xanthoma that can affect both lids. The patches are yellow-orange and slightly elevated (Fig. 11A). In children, xanthelasmas are likely to be associated with increased levels of plasma cholesterol. There is a less clear-cut relationship in adults.120 These lesions are caused by subcuticular lipid-filled macrophages that contain esterified cholesterol as the predominant lipid (see Fig. 11B).121
|
Arcus lipoides is a circular lipid infiltration of the peripheral stroma of the cornea, separated from the limbus anteriorly by a distinct, clear zone. It is formed by the junction of two arcs commencing first above and then inferiorly. The lesion is granular and is made up of two wedge-shaped opacities based on Descemet's and Bowman's membranes and extending into the stroma. The outer edge is better defined than the inner edge.115
Arcus senilis is an opacity of the peripheral cornea occurring in middle-aged or older persons (Fig. 12). Pathologically, it is characterized by a hyaline sudanophilia of Descemet's and Bowman's membranes and a granular sudanophilia of the corneal stroma.122 The chemical composition of senile arcus appears to be chiefly cholesterol ester with some cholesterol and a neutral glyceride, with the density of the opacity related to the concentration of sterol ester deposited.123 Arcus in elderly persons is generally not related to disturbed metabolism.
|
Arcus juveniles corneae may be associated with extensive lipochemical alteration in the serum of young persons.124 The presence of arcus in a young patient requires investigation into the possibility of a lipoprotein or lipid metabolism disorder. A higher prevalence of arcus has been reported in the black population, especially in young blacks, in the absence of elevated cholesterol.125
Other ocular lesions associated with hypercholesterolemia include tuberous xanthomas of the lids and conjunctival, retinal, and choroidal deposits.126 Turgidity of the plasma is associated with hypertriglyceridemia, which may be noted in the plasma or serum. This lactescence is identified in the retinal vessels as lipemia retinalis.115 Lipemia of the limbal vessels also has been seen. With longstanding hypertriglyceridemia, all arteries and veins, including those at the disc, are cream-colored and show a broadened light reflex in the fundus.126 Twenty-six patients with hyperlipidemia (serum cholesterol or triglyceride > 95th percentile for age) were compared with normal individuals for the presence of arteriovenous nicking, “sclerosis,” narrowing, and tortuosity. No differences were found in the retinal arterioles of patients with hyperlipidemia compared with the control group.127 Thus, examining the retinal arterioles is not helpful in detecting hyperlipidemia. Eruptive xanthomas may be seen on the lids and, less commonly, on the iris and in the retina.126
MANAGEMENT. Treatment includes prevention and management of complications through lifestyle alterations, such as dieting and exercising, and through drug therapy. There are three types of cholesterol-lowering drugs: bile acid-binding resins, niacin, and HMG-CoA reductase inhibitors. For hypertrygliceridemia, niacin and gemfibrozil are available. When a combination of these therapies is inadequate, apheresis can be performed every 7 to 14 days.128 Whether a decrease in serum lipid levels causes a regression of xanthomas is controversial.
Familial Lecithin:Cholesterol Acyltransferase (LCAT) Deficiency
In this rare metabolic disorder, absence of the enzyme that transforms fatty acid from lecithin to cholesterol results in abnormally high serum levels of free cholesterol and lecithin.115,129–130 The plasma may be turbid or milky. Erythrocyte lipid composition is abnormal, with target cell formation, anemia, and reduced erythrocyte life span. Phagocytosis of excess lipid results in sea-blue histiocytes in the bone marrow and spleen.131 Premature atherosclerosis may develop atherosclerosis, as well as renal failure secondary to lipid deposition in glomeruli.
The disorder is inherited as an autosomal-recessive trait.130 The LCAT gene is situated on chromosome 16 in close proximity to the α-haptoglobulin locus.132 The gene defect is not geographically restricted to Scandinavia, as has been previously reported.133–134 Corneal opacification, the single obligatory clinical sign of LCAT deficiency, has been noted in all homozygous cases reported in the literature.135 In contrast, anemia was present at the time of diagnosis in 92% of the reviewed cases, and proteinuria was present in 76% of cases.
OCULAR FINDINGS. Corneal opacities, which are present in all patients from early childhood, are diagnostic. Numerous small, fine gray dots are distributed throughout the stroma, causing diffuse haze without affecting the visual acuity. The epithelium, endothelium, and Descemet's membrane are spared. Near the limbus, the dots become more numerous, forming a gray, circular band resembling arcus senilis. The lipoid arcus is nearly always present. A relatively clear zone separates the circular band from the limbus in most patients. The outer border of the arcus is irregular because of differences in the number of stromal dots. In addition to the pathognomonic diffuse cloudiness in the corneal stroma, and the peripheral arcus lipoides, anterior and posterior crocodile shagreen have been described.136 Whether the two conditions are related is not known; however, the deposition of material in LCAT deficiency may alter the arrangement of collagen lamellae.
DIAGNOSIS. LCAT activity can be measured in plasma. Total cholesterol is variable: there is increased unesterified cholesterol and markedly decreased esterified cholesterol. VLDL is elevated. Because the general signs and symptoms of LCAT deficiency are nonspecific (anemia, proteinuria) and the biochemical changes cannot be detected by routine laboratory tests, the ophthalmologist is in a key position to recognize the disease based on findings from a routine slit lamp examination.
DIFFERENTIAL DIAGNOSIS. The ringlike condensation in the periphery differs from typical arcus lipoides in its indistinct, irregular, blurred margin and a limbal zone of mild opacities containing vacuoles. Both share a clear zone between the peripheral opacity and the limbus.
An arcus lipoides in young people is called anterior gerontoxon, or arcus juvenilis. It may accompany hypercholesterolemia or congenital anomalies such as blue sclera, megalocornea, or aniridia. However, in these disorders, the central part of the cornea is free from deposits. The fine, dustlike opacities seen in the posterior corneal stroma in cornea farinata, the vaguely defined “snowflake” opacities in the central cloudy dystrophy of François, and the diffuse opacity of Tangier disease may resemble the opacities found in the central part of the cornea in familial LCAT deficiency. In Fabry's disease, corneal opacities are quite different.130
MANAGEMENT. Fat-restricted diet and kidney transplantation in cases of renal failure remain the principal forms of therapy.118 Ocular therapy is usually not needed because visual acuity and visual fields are normal early in the disease.
Hypolipoproteinemia
TANGIER DISEASE. Tangier disease is a rare disorder involving a genetically determined inability to synthesize the polypeptide necessary for elaboration of HDL.137 HDLs are almost completely absent. Plasma, cholesterol, and phospholipids are reduced, and triglycerides are normal or elevated. The clinical picture is characterized by hepatosplenomegaly, lymph node enlargement, peripheral neuropathy, and corneal clouding. The two pathognomonic features are the presence of hypocholesterolemia and enlarged orange tonsils.137 The enlarged orange tonsils reflect the generalized storage of cholesterol ester in the reticuloendothelial system, which affects the lymph nodes, liver, and spleen. The neuropathy includes sensory loss, loss of tendon reflexes, and weakness and wasting of muscles.115 The early symptoms of note are mildly fatty stools and fatigue. This disorder is autosomal recessive and is expressed in the homozygous state.
Ocular Findings. Corneal manifestations are not invariably present in this disease. The corneal stroma is diffusely affected with a generalized haze, made up of numerous equidistant dots.115 Corneal arcus is not seen; however, a peripheral density in the 3- and 9-o'clock positions has been reported.117 Vision is unaffected.
Diagnosis. The diagnosis should be suggested by the clinical findings. Exact diagnosis requires determination of lipoprotein fractions, most specifically by immunoelectrophoresis. The corneal findings do not include the sparkling multicolored iridescence of the crystals found in cystinosis or multiple myeloma.
Management. No ocular therapy is required.
Schnyder's Hereditary Crystalline Dystrophy
This is a rare autosomal-dominant disorder characterized by the accumulation of cholesterol crystals beneath the central corneal epithelium. When the progression of this disease was described in affected and unaffected family members in one study during a nine-year period, no correlation was observed between Schnyder's corneal dystrophy and serum lipid levels.138
OCULAR FINDINGS. The corneal dystrophy is bilateral, with onset usually occurring early in life. Although the corneal changes do not typically progress after childhood, there are reports of progression later in life.139 The chief feature is a round or oval, discoid or ring-shaped central opacity, predominantly made up of fine, needle-shaped polychromatic crystals140 (Fig. 13). The opacity occupies the anterior portion of the stroma, including Bowman's layer, but ultrastructural changes in the posterior stroma and in the endothelium also have been described.141 The epithelium is normal, and the stroma outside the crystalline lesion is clear. In most cases, vision is only mildly disturbed. Corneal sensation may be abnormal. Both Vogt's limbus girdle and arcus lipoides are frequently seen in this disorder. Patients more than 40 years of age have a diffuse progressive stromal haze.142 Neovascularization of the cornea does not occur.
|
A raised cholesterol level has been found in a number of these patients. Because of the systemic implications of undiagnosed hyperlipidemia, fasting levels of cholesterol and triglycerides and lipoprotein electrophoresis should be checked in patients with Schnyder's dystrophy. Histologic analysis of the corneal crystals that occur with this dystrophy has shown that they contain neutral lipids and both esterified and unesterified cholesterol.143
It is possible that the basic mechanism in Schnyder's dystrophy is a local defect in the handling of lipid by stromal keratocytes.
DIFFERENTIAL DIAGNOSES. Gout, cystinosis, Bietti's corneal dystrophy, and dysproteinemias such as multiple myeloma, Waldenström's macroglobulinemia, cryoglobulinemia, Hodgkin's lymphoma, and benign monoclonal gammopathy are included.
MANAGEMENT. Reduction of cholesterol intake is usually not beneficial in preventing progression of the dystrophy. Lamellar or penetrating keratoplasty may be indicated when the dystrophy is advanced and visually disabling; but recurrence of crystalline deposits in grafted corneas appears to be quite common. Recurrence of crystalline deposits may happen early and the amount of the deposit may be greater in lamellar than in full-thickness grafts.144
Lipoid Proteinosis (Urbach-Wiethe Disease; Hyalinasis cutis et mucosae)
This is a rare autosomal-recessive disorder associated with deposition of hyalinized material in the skin, mucous membranes, and brain. The lesions appear as clusters of yellow to white papules on the elbows, axillae, knees, perineum, scrotum, tongue, larynx, and eyelids.52 The clinical presentation is characterized by hoarseness beginning in infancy and by multiple lesions of both the mucous membrane and skin; some patients also have presented with psychomotor seizures. Skin papules may become atrophic or hyperkeratotic; these lesions can ulcerate, leaving a pitted scar.145–146 Lipid is not part of the storage material, and thus lipoid proteinosis is a misnomer.
OCULAR FINDINGS. Ocular manifestations include retinal drusen and waxy papules along the lid margins. Hyaline material sometimes infiltrates the lid margins. Nodular, beadlike yellow-white excrescences appear on the lid margin and may cause madarosis. A chronic blepharitis may arise concurrently.146–147 Bilateral corectopia with upwardly and nasally displaced oval pupils was reported in one 32-year-old patient.148
DIAGNOSIS. Serum lipids may be elevated but are usually normal.145,147 The enzyme defect is unknown. Skin lesion biopsies show lower than normal lipid content149 and characteristic, diffuse dermal deposits of amorphous hyalinized material, in part composed of neutral mucopolysaccharides, occurring around blood vessels.150 Lid involvement is highly diagnostic.
MANAGEMENT AND PROGNOSIS. Lid nodules can be excised. The life span is normal.
Secondary Lipid Keratopathy
There are many factors that may predispose to lipid deposition in the cornea, but the most common causes are herpes simplex and herpes zoster disciform keratitis. Lipids also may be deposited in the corneal tissues. In the presence of inflammation, vascularization, injury, and corneal edema, the limbal vessels are regarded as the chief source of entry of lipid into the cornea (Fig. 14).115 The deposits appear as dense, yellow-white infiltrates. Cholesterol has been found to be the lipid of note in the case of lipid keratopathy without high blood lipids.151
|
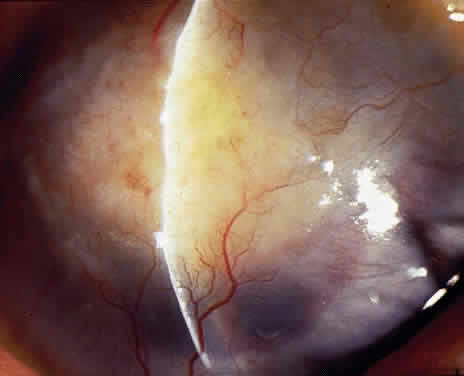
MANAGEMENT. Initial treatment involves controlling the primary inflammatory disease. Argon laser treatment to occlude feeder vessels that have been identified by fluorescein angiography has reportedly been successful in reducing the extent of lipid keratopathy in 62% of cases and its density in 49%.152 Visual acuity was improved in 48% of cases. The use of an Abraham laser lens is important to prevent extensive iris atrophy.153 Complications of corneal argon laser photocoagulation in the treatment of corneal stromal vascularization and secondary lipid keratopathy include iris atrophy, intracorneal hemorrhage, and crystalline deposits.154 Currently, argon laser is seldom used to treat keratopathies.
Fabry's Disease
Fabry's disease is a lysosomal storage disorder caused by the defective activity of the lysosomal hydrolase α-galactosidase A,155 an enzyme that is encoded on Xq22 (in the long arm of chromosome X).156 This defect leads to the progressive and systematic accumulation of glycosphingolipids, particularly globotriaosylceramide, in vascular endothelial cells and smooth-muscle cells.155–157
Most of these patients have angiokeratomas (telangiectatic lesions of the skin) that appear as purplish-red maculopapular cutaneous nodules in the inguinal, scrotal, and umbilical regions.158 Patients with Fabry's disease may experience severe tingling and burning pains in the hands and feet around the time of puberty. Pain is worse in hot weather and with exercise and may last a number of years. With aging, the onset of atherosclerosis occurs, with a propensity for early myocardial infarction and cerebrovascular problems. However, most patients live until their late 40s, at which time they succumb to renal failure because of lipid in the glomeruli of the kidneys. Female heterozygotes may have some of the manifestations of this disease, but the disease is usually milder in females than in affected males.
Fabry's disease is transmitted as an X-linked recessive trait. Of the carrier's sons, 50% are affected, and half of her daughters are carriers.
OCULAR FINDINGS. The ocular deposition of this glycosphingolipid results in unique and diagnostic eye findings in both the severely affected homozygous male and the more minimally affected heterozygous carrier female. The ocular findings, which are recognized as one of the distinctive hallmarks of this disease, are among its earliest clinical manifestations.159,160
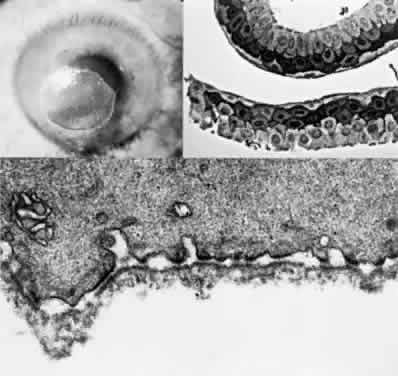
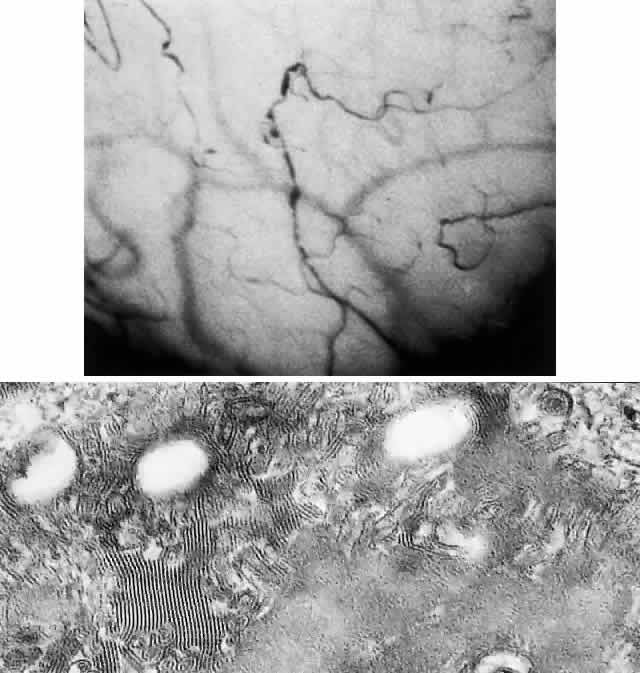
Cornea. Whorl-like corneal opacities (Fig. 15) are seen in almost all homozygotes and heterozygotes, with more severe involvement in heterozygotes.159 In female carriers, these corneal changes are usually the only manifestations of the disorder. They occur primarily in the subepithelial layers of the cornea, at or near the level of Bowman's layer. These linelike opacities are made up of a multitude of small dots, usually in the cornea. They are mostly cream-colored but range from white to golden brown. The corneal dystrophy (cornea verticillata) appears in a number of patterns. The more fully developed vortex patterns also involve the upper cornea. Lesions appear as a diffuse haze in younger patients and may be seen as early as 6 months of age.161 Corneal dystrophy does not impair visual acuity.159,160 Conjunctival inclusion tortuosities, isolated dilation or saccular aneurysms, as well as sheathing, may be present (Fig. 16).
|

|
Lens. Two specific types of lenticular changes may be seen: a granular, anterior capsular cataract with a radial pattern and an unusual posterior subcapsular cataract with whitish, translucent spokelike deposits of granular material radiating from the central part of the posterior cortex.
Retina. Fundus lesions may be either primary or secondary to systemic hypertension or renal disease. The first alterations occur in the veins, which assume a tortuous appearance; later, in the second decade of life, they may show segmentation and dilatation. The arteries also become tortuous and show sheathing and generalized or localized dilatations.
Other findings include papilledema, optic atrophy, lid edema, and angiokeratoma of the lids. The neurologic sequelae are protean and are the result of cerebrovascular involvement.
DIAGNOSIS. The enzyme deficiency can be diagnosed in tears,162 serum, urinary sediment, leukocytes, or cultured fibroblasts.163 Conjunctival biopsy may be of value and should be strongly considered in these patients.161 It is important to diagnose Fabry's disease in carriers for the purpose of genetic counseling; such diagnoses must be based on family history and examination of family members. Where a high index of suspicion is present, conjunctival biopsy and examination of the urine for excess glycolipids are indicated.
DIFFERENTIAL DIAGNOSES. A number of drugs can produce vortex patterns in the superficial cornea: quinacrine, chloroquine, hydroxychloroquine sulfate, amiodarone, indomethacin, and chlorpromazine. In chloroquine keratopathy, the radiating lines are more pronounced in the lower half of the cornea. Striate melanokeratosis may have a vortex pattern but is usually found in more heavily or darkly pigmented eyes,164 and is associated with uveitis and keratitis.
Corneal verticillata, a familial whorl-like corneal opacity, is actually the expression of Fabry's disease in otherwise asymptomatic heterozygotes.159 The ocular pathology of a hemizygous male with Fabry's disease, who died after renal transplantation, revealed reduplication of the corneal basement membrane as well as osmophilic inclusion bodies in both epithelial and endothelial cells.165 It is thus plausible that the diffuse haziness of the cornea seen on slit lamp examination is caused by epithelial deposits of lipid, and that reduplication of the basement membrane is responsible for whorl-like dystrophy.
MANAGEMENT. Neither the corneal lesion nor the cataract affects visual acuity. The frequency and intensity of the pain crisis can be decreased with chronic use of phenytoin and carbamazepine. Hemodialysis and renal transplant have been successful in saving lives.166 The enzyme is biochemically available, but replacement and specific therapeutic methods for the treatment of the disease are still in experimental stages.156,167
Gaucher's Disease
This autosomal-recessive sphingolipidosis is caused by a defect in the enzyme acid α-glucosidase,168 a lysosomal hydrolase encoded by a gene on chromosome 1 (q21 to q31),156,168 which results in the accumulation of glucosylceramide in reticuloendothelial cells. Gaucher's disease is characterized by organomegaly, hematologic problems, and skeletal complications. There are three recognized clinical phenotypes.52
Type I is a chronic, nonneuropathic, adult-onset disorder which accounts for 90% of cases of Gaucher's disease. Splenomegaly, anemia, thrombocytopenia, pathologic bone fractures, episodes of bleeding, and a yellow skin pigmentation are all features of this disease. There is no cerebral involvement.
In type II (infantile, acute neuropathic form), afflicted infants fail to thrive and have progressive hepatomegaly, splenomegaly, and dysphagia early in life. Later, persistent retroflexion of the head and signs of pseudobulbar palsy develop.
Type III is a subacute, neuropathic form with onset in the teenage years. The neurologic features follow a milder course.
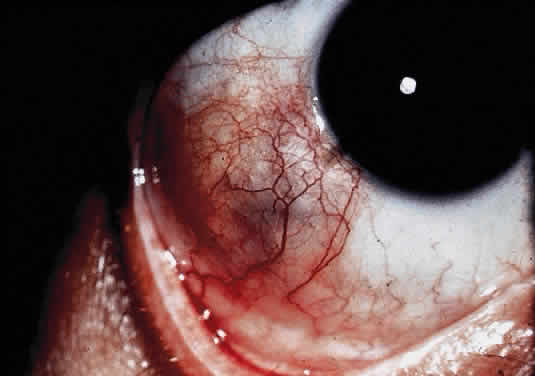
OCULAR FINDINGS. A pingueculum-like lesion containing typical Gaucher's cells is the only significant ocular feature169 (Fig. 17). The pingueculum-like mass or thickening of the bulbar conjunctiva in the horizontal meridian, with its base at the limbus, eventually enlarges and assumes a yellow color.170 The nasal and temporal bulbar conjunctiva are involved with equal frequency but only in one fourth of all patients with the chronic form of the disease.
|
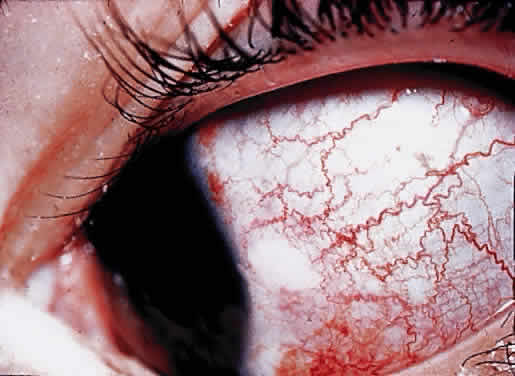
Biopsy of the conjunctival lesions reveals typical Gaucher's cells. In the infantile form, pseudobulbar palsy may cause oculomotor disturbances.171
DIAGNOSIS. Tentative diagnosis is based on the finding of Gaucher's cells in bone marrow. The cytoplasm of these cells has a “wrinkled paper” appearance caused by the deposition of sphingolipid material. Tissue demonstration of decreased levels of glucocerebroside activity confirms the diagnosis. Because 1 of 18 Ashkenazi Jews carries the trait, screening should be carried out in this population.
MANAGEMENT. Bone marrow transplantation has been successful in curing some patients with Gaucher's, but the associated mortality and morbidity are too high.172 Enzyme replacement therapy using placental α-glucosidase has been successful. The efficacy of the recombinant enzyme is currently being studied.156
DISORDERS OF CARBOHYDRATE METABOLISM
This group of disorders includes primarily the various forms of mucopolysaccharidosis (MPS) and diabetes mellitus, which are discussed in a later section of this chapter.
Currently there are at least seven well-defined syndromes among the systemic disorders of acid mucopolysaccharide metabolism (Table 6). The MPSs are inherited as autosomal-recessive traits, with the exception of Hunter's syndrome, an X-linked disease. A deficiency of one of the enzymes involved in the metabolism of glycosaminoglycans (GAGs) is the hallmark. Glycosaminoglycans, which are long-chain, complex carbohydrates linked to proteins, are a major component of connective tissue. Examples of GAGs are keratan sulfate, chondroitin sulfate, hyaluronic acid, and heparan sulfate.
TABLE 15-6. Differential Features of Systemic Mucopolysaccharidoses and
Mucolipidoses
| Excess Mucopolysacchariduria | ||||||
| Classification | Genetics | Enzyme Defect | Heparan Sulfate | Dermatan Sulfate | Keratan Sulfate | |
| MPS I-H (Hurler) | Allelic AR | α-L-Iduronidase | + | + | - | |
| MPS 1-S (Scheie) (formerly MPS V) | Allelic AR | α-L-Iduronidase | + | + | - | |
| MSP I-H/S (Hurler-Scheie compound) | Allelic AR | α-L-Iduronidase | + | + | - | |
| MPS II (Hunter) | ||||||
| A (severe) | Allelic XR | Iduronosylsulfate sulfatase | + | + | - | |
| B (mild) | + | + | - | |||
| MPS III (Sanfilippo) | ||||||
| A | AR | Heparan-N-Sulfate sulfatase | + | - | - | |
| B | N-acetyl-x-D-glucosaminidase | + | - | - | ||
| C | Acetyl-transferase | - | - | + | ||
| D | N-acetylglucosamine-6-sulfate sulfatase | |||||
| MPS IV (Morquio) | ||||||
| A | AR | Galactosamine-6-sulfatase | ||||
| B | β-Galactosidase | |||||
| MPS V (Vacant) | ||||||
| MPS VI (Maroteaux-Lamy) | N-Acetylgalacto-samine-4-sulfatase | |||||
| A (severe) | Allelic AR | (Arylsulfatase B) | - | + | - | |
| B (mild) | - | + | - | |||
| MPS VII (Sly) | β-Glucuronidase | |||||
| (β-Glucuronidase deficiency) | AR | chondroitin sulfate | ||||
| GM1 gangliosidosis | AR | |||||
| Type I | β-Galactosidase A, B, C | - | - | + | ||
| Type II | AR | β-Galactosidase B, C | - | - | - | |
| ML I (lipomucopoly-saccharidosis) | AR | α-N-Acetyl neuraminidase | - | - | - | |
| ML II (I-cell disease) | AR | Multiple | - | - | - | |
| ML III (pseudo-Hurler polydystrophy) | AR | Multiple | - | - | - | |
| ML IV | AR | ? | - | - | - | |
| Systemic Features | ||||||
| Classification | Genetics | Enzyme Defect | Psycho-motor Retardation | Skeletal Dysplasia | Facial Dysplasia | Hepato-spleno-megaly |
| MPS I-H (Hurler) | Allelic AR | α-L-Iduronidase | + | + | + | + |
| MPS 1-S (Scheie) (formerly MPS V) | Allelic AR | α-L-Iduronidase | ± | + | + | ± |
| MSP I-H/S (Hurler-Scheie compound) | Allelic AR | α-L-Iduronidase | ± | + | + | + |
| MPS II (Hunter) | ||||||
| A (severe) | Allelic XR | Iduronosylsulfate sulfatase | + | + | + | + |
| B (mild) | ± | + | + | + | ||
| MPS III (Sanfilippo) | ||||||
| A | AR | Heparan-N-Sulfate sulfatase | + | ± | + | + |
| B | N-acetyl-x-D-glucosaminidase | + | ± | + | + | |
| C | Acetyl-transferase | ± | + | + | + | |
| D | N- acetyl-glucosamine -6-sulfate sulfatase | |||||
| MPS IV (Morquio) | ||||||
| A | AR | Galactosamine-6-sulfatase | ||||
| B | β-Galactosidase | |||||
| MPS V (Vacant) | ||||||
| MPS VI (Maroteaux-Lamy) | N-Acetylgalacto-samine-4-sulfatase | |||||
| A (severe) | Allelic AR | (Arylsulfatase B) | - | + | + | + |
| B (mild) | - | + | + | + | ||
| MPS VII (Sly) | β-Glucuronidase | + | + | ± | + | |
| (β-Glucuronidase deficiency) | AR | |||||
| GM1 gangliosidosis | AR | |||||
| Type I | β-Galactosidase A, B, C | + | + | + | + | |
| Type II | AR | β-Galactosidase B, C | + | - | - | - |
| ML I (lipomucopoly-saccharidosis) | AR | α-N-Acetyl neuraminidase | + | + | + | ± |
| ML II (I-cell disease) | AR | Multiple | + | + | + | + |
| ML III (pseudo-Hurler polydystrophy) | AR | Multiple | ± | + | + | - |
| ML IV | AR | ? | + | - | - | - |
| Ocular Features | ||||||
| Classification | Genetics | Enzyme Defect | Corneal Clouding | Optic Atrophy | Retinal Pigmentary Degeneration | |
| MPS I-H (Hurler) | Allelic AR | α-L-Iduronidase | + | + | + | |
| MPS 1-S (Scheie) (formerly MPS V) | Allelic AR | α-L-Iduronidase | + | + | + | |
| MSP I-H/S (Hurler-Scheie compound) | Allelic AR | α-L-Iduronidase | + | NR | + | |
| MPS II (Hunter) | ||||||
| A (severe) | Allelic XR | Iduronosylsulfate sulfatase | - | + | + | |
| B (mild) | + | + | + | |||
| MPS III (Sanfilippo) | ||||||
| A | AR | Heparan-N-Sulfate sulfatase | - | + | + | |
| B | N-acetyl-x-D-glucosaminidase | - | + | + | ||
| C | Acetyl-transferase | + | + | - | ||
| D | N-acetylglucosamine-6-sulfate sulfatase | |||||
| MPS IV (Morquio) | ||||||
| A | AR | Galactosamine-6-sulfatase | ||||
| B | β-Galactosidase | |||||
| MPS V (Vacant) | ||||||
| MPS VI (Maroteaux-Lamy) | N-Acetylgalacto-samine-4-sulfatase | |||||
| A (severe) | Allelic AR | (Arylsulfatase B) | + | + | - | |
| B (mild) | + | - | - | |||
| MPS VII (Sly) | β-Glucuronidase | ± | NR | NR | ||
| (β-Glucuronidase deficiency) | AR | |||||
| GM1 gangliosidosis | AR | |||||
| Type I | β-Galactosidase A, B, C | ± | + | NR | ||
| Type II | AR | β-Galactosidase B, C | - | + | ± | |
| ML I (lipomucopoly-saccharidosis) | AR | α-N-Acetyl neuraminidase | ± | - | NR | |
| ML II (I-cell disease) | AR | Multiple | ± | - | NR | |
| ML III (pseudo-Hurler polydystrophy) | AR | Multiple | + | - | NR | |
| ML IV | AR | ? | + | + | NR | |
MPS, mucopolysaccharidosis; ML, mucolipidosis; AR, autosomal recessive; XR, X-linked recessive; NR, not recorded; +, present; ±, variable; --, absent.
The characteristic clinical features of these disorders, including skeletal abnormalities, gargoyle facies, hepatosplenomegaly, cardiac disease, mental deficiency, deafness, and ocular involvement, occur with varying degrees of severity and overlap. The tissue-specific differences in the structure of mucopolysaccharides and the predominant types accumulated in various organs account for the variability in clinical manifestations. Ocular manifestations include progressive corneal clouding, optic atrophy, and retinal pigmentary degeneration. In these disorders, excess dermatan, chondroitin, and keratan sulfates accumulate in the cornea and in other body tissues, causing visceral manifestations, and heparan sulfate accumulates in the retina and in the central nervous system, causing mental deficiency. The accumulation of these abnormal substances results from the faulty catabolism of mucopolysaccharides in the lysosomes caused by deficiencies of lysosomal acid hydrolases (Figs. 18 and 19). In the cornea, this causes characteristic pathologic changes consisting of intracytoplasmic vacuolization of epithelium, subepithelial histiocytes, stromal keratocytes, and endothelium. In the conjunctiva, membrane-bound vacuoles found in the cytoplasm represent fine granular material in lysosomes. Large storage vacuoles form in histiocytes, which are called gargoyle cells.
|
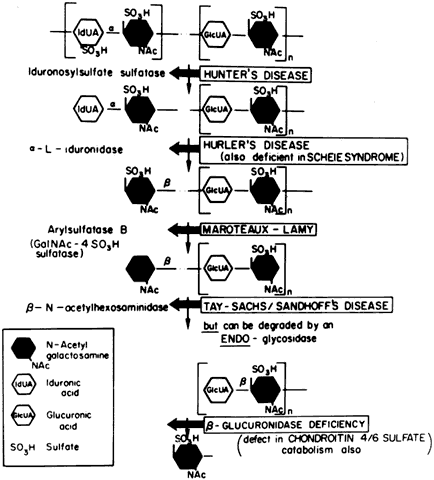
|
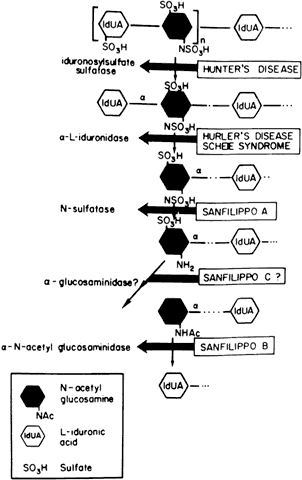
Night blindness associated with retinal pigmentary degeneration and bone-spicule appearance is seen only in MPS types that involve storage of heparan sulfate. Papilledema, a frequent finding, has been attributed to the hydrocephalus that occurs secondary to meningeal thickening with the storage material; narrowing of the scleral canal of the nerve head, as a result of posterior scleral thickening with mucopolysaccharide accumulation, may play a role in the optic nerve head swelling. Acute and chronic glaucoma may be seen in MPS I-H, I-S, and I H-S.
Conjunctival biopsy is an important diagnostic step in these disorders, owing primarily to its popularization by Kenyon and colleagues173 and by others.174 This simple procedure has become increasingly applicable as a reliable screening test for patients with suspected lysosomal diseases. A small fragment from the upper bulbar conjunctiva is easily removed, using topical anesthetic without sedation, and sent for ultrastructural evaluation (Fig. 20). The diagnosis of mucopolysaccharidosis is based primarily on the characteristic clinical findings; specific enzymatic assays can be performed for definitive diagnosis and further categorization of the different types and subtypes.
|

MPS 1-H (Hurler's Syndrome)
This autosomal-recessive syndrome, which becomes manifest in infancy, is one of the most severe mucopolysaccharidoses. The basic genetic defect is the absence of the lysosomal enzyme α-L-iduronidase, which renders the cells incapable of cleaving iduronic acid residue in polysaccharide chains. As a result of the enzyme defect, both heparan sulfate and dermatan sulfate are excreted in the urine.175
The infant may develop normally for a few months before mental and physical deterioration begins.176 The clinical features include dwarfism, grotesque facies, hepatomegaly, progressive mental retardation, skeletal abnormalities, hirsutism, and cardiovascular disease. Progressive corneal clouding occurs in all patients and may conceal the retinal degeneration that develops concurrently. The usual cause of death, which occurs before 10 years of age, is respiratory infection and cardiac failure. The change in body tissue is caused by an accumulation of mucopolysaccharides (Fig. 21).
|
OCULAR FINDINGS. The cornea is diffusely clouded by the progressive accumulation of intracellular and extracellular acid mucopolysaccharides. Initially, fine, punctate stromal opacities are present in the anterior stroma; these opacities progress to involve the entire stroma, resulting in visual loss (Fig. 22).177 The epithelium and endothelium are spared. Abnormal mucopolysaccharides accumulate in all cellular components of the cornea, with profound alteration of Bowman's layer and of the corneal lamellae.
|
Other ocular findings, such as buphthalmos, megalocornea, and glaucoma177have been reported. A picture similar to retinitis pigmentosa, indistinguishable from other forms of heredofamilial retinal pigmentary dystrophies, is a frequent ophthalmoscopic finding. Narrowing of the retinal vessels, optic atrophy, or optic nerve head swelling are commonly seen; the electroretinogram is diminished or extinguished in many cases.
DIAGNOSIS. Conjunctival biopsy demonstrates connective tissue cells containing abnormal vacuolization by single membrane-limited cytoplasmic bodies. These bodies contain predominantly fibrillogranular and occasional membranous lamellar inclusions. They are thought to represent lysosomes swollen with acid mucopolysaccharides and glycolipid.173 Excessive levels of dermatan sulfate and heparan sulfate are found in the urine. The definitive diagnosis is made by direct assay of α-L-iduronidase activity in white blood cells or cultured skin fibroblasts. A similar assay is performed on amniocytes for prenatal diagnosis.
MANAGEMENT. Bone marrow transplant is the most effective treatment available for Hurler's disease. However, this form of therapy is not widely available because of its high cost, its high mortality, and the lack of matched related donors. Corneal transplantation may be required for severe corneal disease; but in such cases vision remains poor because of optic atrophy or retinal degeneration. Use of gene therapy to treat Hurler's disease is under investigation.178
MPS I-S (Formerly MPS V, Scheie's Syndrome)
This autosomal-recessive syndrome, characterized by claw hands, stiff joints, aortic insufficiency, deafness, and hernia, is a variant of Hurler's syndrome.176,179 The facial features are coarse but not grotesque. The predominant ocular feature is a peripheral corneal clouding that progresses centrally with age. Pigmentary retinal degeneration occurs in the first decade, with night blindness and visual field constriction in the second and third decades. Acute glaucoma may be seen. The same enzyme, α-L-iduronidase, is deficient; as in Hurler's syndrome,175 and as with the SS and SC hemoglobinopathies, they are presumably allelic. Patients with MPS I-S usually have a normal stature and a relatively normal life span.
DIAGNOSIS. The MPS spot test is positive. Urine mucopolysaccharide values show both dermatan sulfate and heparan sulfate excreted in a 70:30 ratio, as in MPS 1-H. Conjunctival biopsy is of value. The Scheie fibroblasts show identity to the Hurler fibroblasts in the Neufeld mixed fibroblast culture.180 It has been demonstrated that residual α-L-iduronidase activity in Hurler fibroblasts is heat stable, whereas that in Scheie fibroblasts is heat labile; the enzyme activity in MPS I H-S is intermediate between the two. The differential diagnoses include MPS VI-B (mild phenotype).
MANAGEMENT AND PROGNOSIS. Penetrating keratoplasty may be considered if vision is markedly reduced, even though poor results have been reported.179 Life expectancy is well into middle age.52
MPS I H-S (Hurler-Scheie Compound)
A clinical disorder with severity midway between the Hurler and Scheie syndromes has prompted the suggestion of a genetic compound of the two alleles, H and S. This disorder is inherited as an autosomal-recessive trait. Most of these patients have severe bone involvement but only minor intellectual impairment. These patients have a longer life expectancy than do those with the Hurler syndrome.52 Micrognatia (receding chin) is a distinctive clinical feature. Arachnoid cysts with spinal rhinorrhea are characteristic. These cysts are also seen in Hurler's syndrome, where they may lead to sella turcica enlargement.
OCULAR FINDINGS. Ocular findings include corneal clouding, a chronically elevated optic disc, a diminished or extinguished electroretinogram, and pigmentary retinopathy. Progressive corneal clouding requiring corneal transplantation by the end of the first decade is generally the rule.
DIAGNOSIS. An MPS urine spot test is positive, with excessive excretion of dermatan sulfate and heparan sulfate. A conjunctival biopsy is helpful. Biochemical and enzymatic assays are identical to those seen in both MPS 1-H and MPS I-S.
MANAGEMENT. Penetrating keratoplasty is suggested for vision of less than 20/100 (6/30).* Even with an extinguished electroretinogram, vision may be improved.52
The authors acknowledge the contributions of George T. Frangieh, MD, and John S. Lee, MD, to the preparation of previous versions of this chapter.
MPS II (HUNTER'S SYNDROME)
There are two phenotypes of this X-linked recessive syndrome: mild (type A) and severe (type B). Both types are caused by mutations at the X-linked locus for the enzyme iduronate sulfatase, and are allelic.52,176 The deficiency of iduronate sulfatase results in accumulation of dermatan and heparan sulfates.
The severe form demonstrates many of the same features as Hurler's syndrome, including similar skeletal and mental changes, but with less severity than in Hurler's syndrome. Only males are affected. Death occurs before age 15.
In the mild phenotype, survival may extend past age 50. Intelligence is normal and skeletal involvement moderate. Characteristic pebbling of the skin may be found over the neck, scapula, or thigh.52
OCULAR FINDINGS. Corneal clouding is absent, although histologic examination reveals deposits of acid mucopolysaccharides in clinically clear corneas. Pigmentary retinopathy with an abnormal extinguished electroretinogram may lead to visual impairment of varying degrees. Elevated and blurred disc margins account for the diagnosis of chronic papilledema and may culminate in optic atrophy.175
DIAGNOSIS. An MPS spot test is necessary. Dermatan sulfate and heparan sulfate are present in the urine. A conjunctival biopsy is helpful.181 Definitive diagnosis is made by assaying for sulfoiduronate sulfatase in fibrocytes and, for prenatal diagnosis, in amniocytes. MPS II is clinically distinguished from MPS I-H by the lack of corneal clouding. Patients with Sanfilippo's syndrome, types A and B, are generally less dwarfed.
MANAGEMENT. Bone marrow transplant has caused stabilization of symptoms in this syndrome. More long-term follow-up studies are needed to confirm its potential benefit.178
MPS III (Sanfilippo's Syndrome, Types A, B, C, and D)
The four types of Sanfilippo's syndrome are caused by different nonallelic mutations. Type A is most severe, with an earlier onset and earlier death than types B, C, or D. Excessive heparan sulfate, but not dermatan sulfate, is present. In type A there is a deficiency of the enzyme heparan-N-sulfatase; in type B, of N-acetyl-α-D-glucosaminidase (NAG); in type C, of N-acetyltransferase; and in type D, of N-acetylglucosamine-6-sulfate sulfatase. This autosomal-recessive syndrome is characterized by severe mental retardation, minimal skeletal dysostosis, moderate hepatosplenomegaly, and moderate dwarfing.176,182 Cardiac abnormalities have not been associated with the syndrome. Radiographic findings include oval vertebral bodies, sclerotic mastoids, and thickened posterior calvaria.
OCULAR FINDINGS. The corneas are clinically clear, but less severe intracytoplasmic vacuolation of corneal cells and the accumulation of granular material around keratocytes is present. Pigmentary retinopathy, which may be indistinguishable from retinitis pigmentosa, is frequently present. Optic atrophy may be seen.52 Sanfilippo found 1 patient with corneal opacity.183
DIAGNOSIS. Urine contains high levels of heparan sulfate. Electron microscopic evaluation of conjunctival biopsy confirms the diagnosis. Definitive diagnosis is based on specific enzyme assays.
DIFFERENTIAL DIAGNOSIS. This syndrome is differentiated from the other MPS syndromes, in which the corneas are clear, by an increased severity of behavioral problems and mental retardation, and by less skeletal involvement. The genetic pattern of inheritance also helps in making the diagnosis.
TREATMENT. Psychotropic agents may be needed for the behavioral disorders.
MPS IV (Morquio's Syndrome, Types A and B)
Morquio's syndrome is an autosomal-recessive disorder. Type A is caused by the absence of galactosamine-6-sulfatase, and type B is caused by 8-galactosidase deficiency. Keratan and chondroitin sulfate are deposited. In type A, the activity of the enzyme neuraminidase was also found to be markedly decreased.
These patients are notably dwarfed and have distinctive skeletal changes by the first 10 years of life. The joints are loose and hyperextensible, the wrists are enlarged, and the hands are misshapen.176 The facies are characteristic, with broad mouth, prominent maxilla, short nose, and widely spaced teeth. No abnormalities are apparent at birth; however, during the second year of life, awkward gait, retarded growth, knock knees, sternal bulging, and flaring of the rib cage become evident. Hypoplasia of the odontoid process may lead to atlantoaxial dislocation and spinal cord compression later in the course of the disease. Death occurs late in childhood as a result of respiratory paralysis secondary to spinal cord compression and recurrent pneumonia. Intelligence is normal or only mildly impaired.176
OCULAR FINDINGS. Corneal clouding is usually not present before the age of 10, but is the most common ocular feature. Corneal epithelium and Bowman's membrane appear normal under the slit lamp, but a homogeneous cloudiness of the corneal stroma develops.184 No retinal dystrophy is seen.
DIAGNOSIS. Diagnosis is made on the basis of typical clinical features and excessive excretion of keratan sulfate and chondroitin sulfate in the urine. Assay for the specific enzyme is definitive for the diagnosis. Differential diagnoses include other causes of spondyloepiphyseal dysplasia.
MANAGEMENT AND PROGNOSIS. Orthopedic surgery is the main approach. Patients may survive to the sixth decade.52 Corneal transplantation may be necessary in adolescents with normal intelligence.
MPS VI (Maroteaux-Lamy Syndrome, Polydystrophic Dwarfism, Types A and B)
The deficient enzyme in Maroteaux-Lamy type A syndrome is arylsulfatase-B.185 Urinary excretion of heparan sulfate (85% of patients) and dermatan sulfate (15%) is present.176 Both severe and mild phenotypes have been described. Type A, the classic form, is more severe than type B, the allelic form. The extent of corneal clouding is the same in both, but skeletal dysplasia is less pronounced in the latter.
MPS VI is an autosomal-recessive syndrome characterized by clinical features similar to those of MPS I-H, including dwarfism. It is distinguished from the other mucopolysaccharidoses, except MPS IV, by retention of normal intelligence, at least until the later stages of the disorder. Growth retardation is first noted at age 2 or 3. Restriction of articular movement soon follows. Serious cardiac abnormality may develop, and hepatosplenomegaly is a feature. Hypoplasia of the odontoid process may cause spinal cord compression and spastic paraplegia. Patients with type A generally die in their teens as a result of hydrocephalus secondary to meningeal involvement.
OCULAR FINDINGS. Diffuse corneal opacification throughout the entire stroma is a feature.186 The opacities develop fairly early, and the cornea increases in thickness—especially peripherally, where the clouding is most dense.176 Papilledema 176 and optic atrophy have been reported.187 Abnormalities have been noted on the electroretinogram.
DIAGNOSIS. Assays for the specific enzymes are available. A conjunctival biopsy confirms the diagnosis.
PROGNOSIS. Hydrocephalus and cardiac failure lead to death.
MPS VII (Sly Syndrome, β-Glucouronidase Deficiency)
This latest addition to the MPS group is caused by β-glucuronidase deficiency, an enzyme encoded on chromosome 7.188 In this autosomal-recessive disorder there may be two allelic forms leading to two clinical phenotypes, one being more severe.176,189
The severe form shows rapid progression to extensive mental, motor, and growth retardation; hepatosplenomegaly; massive ascites; inguinal hernias; thoracolumbar gibbus; corneal clouding; and skeletal radiographic changes similar to those of the mucopolysaccharidoses.
The milder phenotype is characterized by hepatosplenomegaly, skeletal abnormalities, and unusual facies. Mental retardation is not present at birth, but mental and physical development slows with age.190
OCULAR FINDINGS. Corneal clouding is variable and usually mild. A mild pigmentary retinopathy develops later in life.
DIAGNOSIS. Excess chondroitin sulfate is excreted in the urine. Alder-Reilly phenomenon is present, with metachromatic granules in leukocytes; there is an absence of β-glucuronidase demonstrated in cultured fibroblasts.
Glycogen Storage Disease Type I (von Gierke's Disease)
Von Gierke's disease is an autosomal-recessive disorder that is caused by absence of the enzyme glucose-6-phosphatase in the liver, kidney, and intestinal mucosa. It is the only glycogenosis with corneal involvement. Delayed growth, feeding difficulties, massive hepatomegaly, hypoglycemia, and upper respiratory infections are noted in infancy. As development continues, the child has short stature and poor muscle tone. Chemical evaluation demonstrates a low fasting blood sugar, high blood lipids, high uric acid, and an increased platelet level.191 Xanthoma is evident in 10% of these patients.
OCULAR FINDINGS. Faint, brown, cloudy infiltration of the corneal periphery has been described.140 Yellowish discrete perimacular lesions also have been noted.192 Increased bilateral subcutaneous fat in the lower eyelids and inverted eyelashes have been described in three adult Japanese patients with Von Gierke's disease.193
DIAGNOSIS. The most reliable method of diagnosis is direct measurement of liver glucose-6-phosphatase activity in biopsy samples.
COMBINED DISORDERS OF MUCOPOLYSACCHARIDE AND GLYCOLIPID METABOLISM
Mucolipidoses
This group of disorders has phenotypic and biochemical features overlapping those of the mucopolysaccharidoses and of the sphingolipidoses.
Ultrastructurally, the affected cells show cytoplasmic vacuoles filled with fine fibrillogranular material (similar to systemic MPS), and many also contain membranous lamellar inclusions (similar to systemic lipidoses).
MUCOLIPIDOSIS I. Also known as dysmorphic sialidosis, this rare storage disease is characterized by a mild Hurler-like manifestation with moderate progressive mental retardation, skeletal changes of dysostosis multiplex, no excess mucopolysacchariduria, and peculiar inclusions in cultured fibroblasts.176,194–195 Peripheral neuropathy, tremor, and cerebellar signs may develop. There is increased sialic acid and a deficiency of the enzyme α-N-acetyl neuraminidase in cultured mucolipidosis I fibroblasts.196 Most cases have been sporadic; however, autosomal-recessive inheritance is the most likely mode.
Ocular Findings. Corneal clouding secondary to fine epithelial and anterior stromal opacities, cataracts, cherry-red spot in the macula,52 and optic atrophy with constriction of the visual fields are noted.
Diagnosis. The basis for diagnosis is microscopy. Fibroblasts and conjunctival biopsy show small, membrane-bound vacuoles containing fibrogranular and membranous lamellar bodies. Differential diagnoses must include the syndromes causing a cherry-red spot in the macula.
Management and Prognosis. No treatment is known and the prognosis is uncertain.
MUCOLIPIDOSIS II (I-CELL DISEASE). This severe, autosomal-recessive mucolipidosis is a Hurler-like disorder with severe radiologic features and striking fibroblast inclusions (thus “I-cell” for inclusion cell). Mucolipidosis II results from the absence of N-acetylglucosamine phosphotransferase, the enzyme that attaches a recognition phosphate group onto a mannose residue in hydrolases. This deficency results in abnormal enzymes not recognized and taken up by fibroblasts.197
Early in development, hernia, thoracic deformities, congenital dislocation of the hips, and hyperplastic gums become evident. Radiographic studies show bony changes of dysostosis multiplex that are more severe than those seen in MPS 1. Retarded psychomotor development and restricted joint mobility are features of this disease.176,198 Patients develop coarse facial features over time.
Ocular Findings. Early in the course of the disease, the corneas are clear. Late corneal clouding that correlates positively with survival occurs in 40% of cases, and glaucoma occurs in 6% of cases.198 There is no cherry-red spot. The disease may be apparent during ultrastructural observation of the conjunctiva.198–199
Diagnosis. Conjunctival biopsy confirms the presence of a storage disorder after the clinical diagnosis has been made at birth. Neuraminidase activity has been shown to be deficient in this disorder. In no other storage disorder that resembles the Hurler syndrome do the symptoms occur as early as in I-cell disease.52
Prognosis. Death occurs in the first few years of life.
MUCOLIPIDOSIS III (PSEUDO-HURLER POLYDYSTROPHY). This is a relatively mild, autosomal-recessive disorder caused by the same enzyme deficiency as in mucolipidoses I and II.200 These cases have many features of the Hurler syndrome but a much slower clinical evolution and no mucopolysacchariduria. The children usually present at approximately 3 years of age with joint stiffness, coarse facial features, and short stature. Skeletal and joint diseases progress slowly. The arms and hands are most markedly involved and carpal tunnel syndrome is common, with wasting of the thenar eminence. Aortic valve disease takes place in a majority of cases. The cornea shows a fine, ground-glass clouding (Fig. 23).176
|
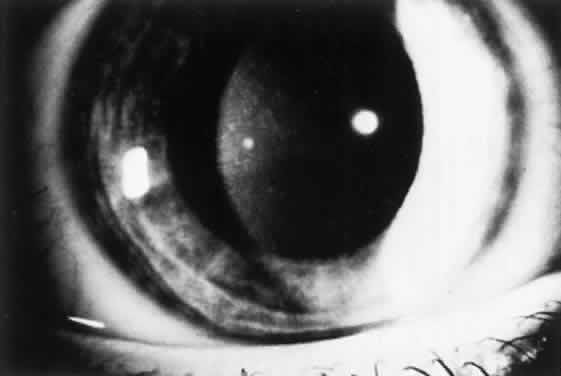
Ocular Findings. Corneal clouding and hyperopic astigmatism appear to be the constant ocular features. Visual acuity, however, remains unchanged. Epiretinal membranes, optic nerve edema, and retinal vascular tortuosity have been reported.201
Diagnosis. Excess intracellular storage of acid mucopolysaccharides and an excess of glycolipids are seen ultrastructurally in the conjunctival biopsy; these appear as fibrillogranular inclusion bodies and membranous lamellar vacuoles, respectively. Maroteaux-Lamy syndrome must be considered among the differential diagnoses.
Management and Prognosis. Correction of high hypermetropic refractive error is necessary. Progressive joint stiffness occurs.
MUCOLIPIDOSIS IV. This is a storage disease in which corneal clouding is an early sign,202 with no other evidence of systemic involvement until 1 year of age. At that time mild psychomotor retardation becomes evident and progresses, increasing in severity as the child develops.163,203 No other signs of storage disease are found.
The disorder is autosomal recessive and is probably caused by a deficiency of ganglioside neuraminidase.204 The cases described have been found in Ashkenazi Jews.
Ocular Findings. Diffuse, generalized corneal clouding, involving the epithelium more severely than the stroma, may be present at birth or within the first few months of life. In time, the corneal opacities may increase in density, causing poor fundus visualization.205 Epithelial edema has been noted.
The electroretinogram may be subnormal or extinguished.205–206 Retinal atrophy with a pale nerve may be seen.206 A progressive rod-cone impairment similar to tapetoretinal dystrophy has been suggested.207
Diagnosis. This disorder must be considered in a child who presents with corneal clouding during the first year of life. Conjunctival biopsy shows the intracellular inclusions characteristic of the mucolipidoses.208 Prenatal diagnosis is possible.203,209 Congenital glaucoma, congenital hereditary endothelial corneal dystrophy, Hurler's syndrome, and Scheie's syndrome should all be considered among the differential diagnoses.
Management. Removal of the corneal epithelium results in corneal clearing; however, the clouding recurs with re-epithelialization.205 Conjunctival transplantation with donor conjunctiva from an unaffected sibling may improve long-term corneal clarity.210
Gangliosidoses
GENERALIZED GANGLIOSIDOSIS (GM1, GANGLIOSIDOSIS I).
This autosomal-recessive syndrome is characterized by nystagmus, a cherry-red spot in the macula, hepatomegaly, and psychomotor retardation in an infant who fails to thrive. The clinical features resemble Hurler's syndrome and are present at birth. A prominent midface is caused by a protuberant maxilla. Other possible findings include telangiectatic skin lesions and heart disease.52 This syndrome is caused by the universal absence of all fractions of the enzyme α- galactosidase.211
There is an abnormal storage of two substances: a ganglioside that is stored in the brain and visceral organs, and a keratan sulfate-like mucopolysaccharide that accumulates in the abdominal organs.212–213
Ocular Findings. Nystagmus, decreased visual acuity, retinal hemorrhages, and optic atrophy may be noted. A cherry-red spot is present in at least 50% of patients.214 Mild, diffuse corneal clouding has been reported.215 Conjunctival vascular tortuosity may be a feature.216 GM1 gangliosides accumulate in the cytoplasm of corneal epithelium and keratocytes.215
Diagnosis. α-Galactosidase deficiency can be demonstrated in leukocytes.211 The urine has increased keratan sulfate levels. High levels of GM1 ganglioside are found in red blood cells, and foamy storage cells are present in the bone marrow.217
Differential Diagnoses. Hurler's syndrome has a later onset. Tay-Sachs and Niemann-Pick diseases do not include facial dysmorphism. The facial dysmorphism of I-cell disease is more severe.
Management and Prognosis. There is no therapy for ocular problems. This syndrome is fatal by the second year of life.