Gangliosides are important constituents of gray matter. They are glycosphingolipids that contain sialic acid in the oligosaccharide chain. At least ten different gangliosides have been identified, four of which constitute over 90% of the total: GM1, GDla, GD1b, and GT. Gangliosides GM2 and GM3 together constitute less than 2% of the total brain ganglioside content. The metabolism of ganglioside involves the removal of the terminal galactose to convert GM1-ganglioside to GM2-ganglioside. GM2-ganglioside is then hydrolyzed to GM3-ganglioside by the removal of N-acetylgalactosamine (Fig. 1).
|
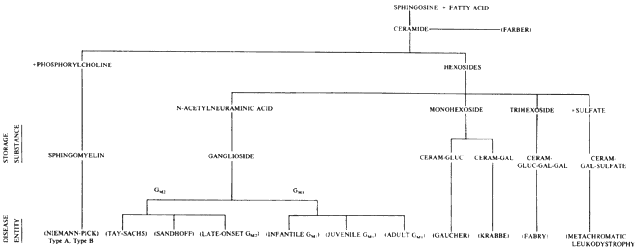
Ten ganglioside storage diseases are now recognized (Table 1). Seven of the disorders involve storage of GM2-ganglioside, and three involve the accumulation of GM1-ganglioside.
| Presentation | Disease | Age at Onset | Signs and Symptoms | Eye Findings | Defect | Stored Ganglioside |
| Infantile | Infantile Tay- Sachs | 2–3 mo | Increased startle, hypotonia, developmental and motor regression, megalencephaly, seizures | Cherry-red spots | Deficient hexosaminidase A, α-locus mutation(chromosome 15) | GM2- ganglioside |
| G m2-gangliosidosis AB variant and B1variant | 2–3 mo | Increased startle, hypotonia, developmental and motor regression, megalencephaly, seizures | Cherry-red spots | Normal hexosaminidase A and B with either activator deficiency(chromosom e 5) or in B1variant mutation in the active site for the α-locus(chromosome 15) | GM2- ganglioside | |
| Sandhoff's disease | 2–3 mo | Hypotonia, developmental and motor regression, megalencephaly, occasional organomegaly, seizures | Cherry-red spots | Absent hexosaminidase A and B, ß-locus mutation(chromosome 5) | Gm2- ganglioside GA2- ganglioside Globoside | |
| G M1-gangliosidosis type I | Birth-3 mo | Increased startle, hypotonia, developmental and motor regression, megalencephaly, organomegaly, coarse facial features, seizures | Cherry-red spots (50% of cases), mild corneal clouding | Deficient ß-galsctosidase (chromosome 3) | GM1- ganglioside | |
| Juvenile | Juvenile GM2-gangliosidosis | 2–6 yr | Dysarthria spasticity, ataxia, progressive dementia, seizures | Atypical cherry red spot, optic atrophy, pigmentary retinopathy | Deficient hexosaminidase A, α-locus mutation(chromosome 15) | GM2- ganglioside |
| Juvenile G M1-gangliosidosis | 2–4 yr | Cerebellar ataxia, dystonia, myoclonus, dementia, seizures | Cherry-red spots | ß-Galactosidase(chromosome 3) | GM1- ganglioside | |
| Juvenile Sandhoff's disease | 2–6 yr | Progressive cerebellar ataxia, psychomotor retardation, spasticity | Deficient hexosaminidase A and B, ß-locus mutation(chromosome 5) | GM2- ganglioside GA2- ganglioside Globoside | ||
| Chronic Gm2- gangliosidosis | First and second decade | Spinocerebellar degeneration, disabling dysarthria, tremor, late-appearing proximal muscle weakness and atrophy | 10% to 15% residual hexosaminidase A activity, α-locus mutation(ch romosome 15) | GM2- ganglioside | ||
| Adult | Adult G M1-gangliosidosis | Second decade | Ataxia, slurred speech, intellectual decline, mild vertebral changes, angiokeratoma | Deficient ß-galactosidase (chromosome 3) | GM1- ganglioside | |
| Adult GM2gangliosidosis | Second to fourth decade | Minimal spinocerebellar signs, prominent anterior horn cell disease with proximal muscle weakness and fasciculations, presence of manic-depressive illness in proband or family history of manic-depression in 40% of cases | 5% to 10% residual hexosaminidase A activity, ß-locus mutation(chromosome 15) | GM2- ganglioside |
THE GM2-GANGLIOSIDOSES
The GM2-gangliosidoses are a class of disorders resulting from the deficiency of ß-hexosaminidase A (Hex A) or ß-hexosaminidase B (Hex B) and Hex A. Hex A is an essential enzyme that normally cleaves the terminal sugar from the Tay-Sachs ganglioside and is identified by its action on artificial substrates.
Expression of Hex A activity requires three separate gene products: an α-subunit, a ß-subunit, and an activator protein, all encoded by genes located on two different chromosomes. Various forms of GM2-gangliosidosis have been described (Tay-Sachs disease, Sandhoff's disease, and variants) that can be traced to mutations in one or another of these three loci. The effect is usually a deficiency of Hex A activity or a concomitant Hex B and Hex A deficiency, permitting a pathologic accumulation of GM2-ganglioside in neurons.
Hex A is made up of two ß-subunits and two α-subunits, the latter coded for on the long arm of chromosome 15, The activator protein locus is located on chromosome 5. Hex B is a tetramer made up of four identical ß-subunits also coded for on chromosome 5 (Fig. 2).
|
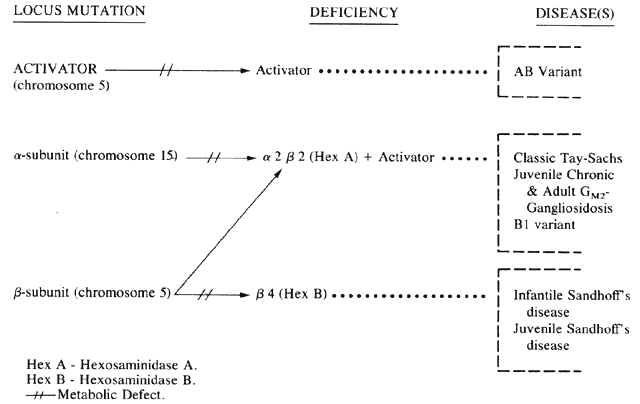
The extent of the deficiency of hexosaminidase determines the rate of ganglioside accumulation and hence the time of onset and the clinical severity of the disease. The most severe form is classic infantile Tay-Sachs disease characterized by an almost complete absence of Hex A activity with preservation of Hex B; hence it is called the B variant. Less severe forms have been described that are characterized by the presence of residual but insufficient levels of Hex A activity. These milder variants present as juvenile, chronic, or adult forms of GM2-gangliosidosis (see Table 1).
Tay-Sachs Disease (Infantile GM2-Gangliosidosis Type I)
Infantile GM2-gangliosidosis type I is the most common example of a sphingolipid storage disease. It is inherited as an autosomal recessive disorder because of a mutation in the α-locus on chromosome 15, resulting in a deficiency of Hex A. The carrier frequency for this gene is very high among those of Ashkenazi Jewish or French Canadian descent (1/27 and 1/25 individuals, respectively). Tay-Sachs screening programs primarily target these two populations.
CLINICAL MANIFESTATIONS. Children with Tay-Sachs disease show neurologic signs at birth. Observant parents notice an increased startle reaction to sound and hypotonia at 2 to 3 months of age. The startle response is a reflex myoclonic jerk consisting of tonic extension, adduction and elevation of the arms, clenching of the hands, flexion or extension of the legs, a startled facial expression, and a sharp cry. A brief metallic bang elicits it more reliably than a hand clap, and although it has been called hyperacusis, the sign does not denote increased auditory sensitivity. Occasionally the response is produced by light flashes and tactile stimuli. The increased startle reflex is characteristic of the early stages of Tay-Sachs disease and is without overt electroencephalogram (EEG) correlate. At 12 to 18 months of age it is superseded by segmental and diffuse myoclonus and by prolonged tonic seizures, both spontaneous and stimulus driven. The spikes and sharp waves of the EEG in the second year are replaced by a flattened, featureless, slow EEG by the third year.
The exaggerated startle response is not restricted to Tay-Sachs and Sandhoff's diseases. It occurs in other conditions, such as GM1-gangliosidosis, although not as early or as persistently. Apart from this sign and mild hypotonia the child appears to develop normally until the fourth to sixth month of life when signs of mental retardation and loss of motor skills become manifest. The infant fails to sit up or walk, marked axial hypotonia in combination with pyramidal signs develop, and there is variable spasticity of the limbs. After 18 months of age, progressive deafness, blindness, and megalencephaly occur as a result of storage of GM2-ganglioside and cerebral gliosis. By the end of the third year the child is demented, decerebrate, and blind. Progressive cachexia and aspiration pneumonia usually lead to death before age 10.
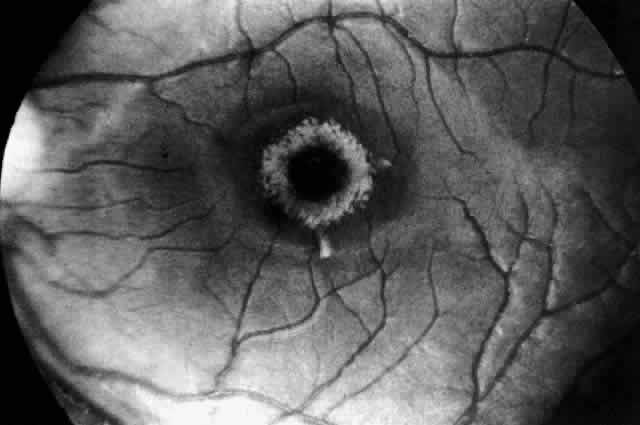
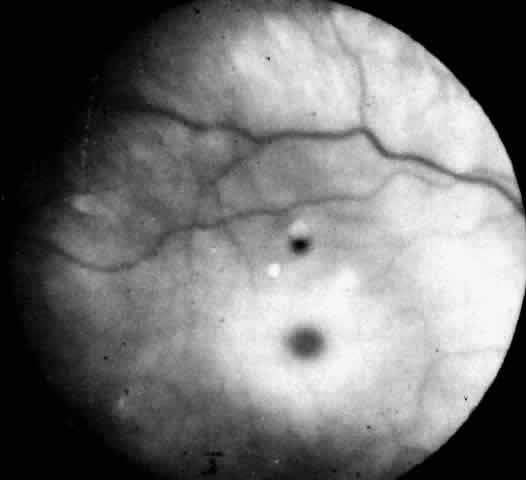
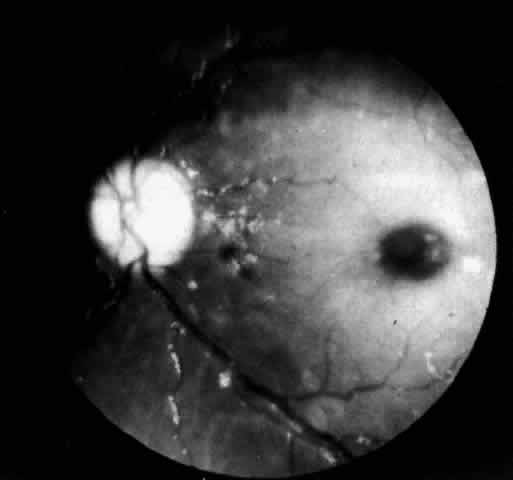
OCULAR MANIFESTATIONS. Tay, a British ophthalmologist, was the first to recognize the macular cherry-red spot in 1881 .1 In 1896, Sachs, an American neurologist, emphasized the association of this ocular manifestation with signs of progressive involvement of the central nervous system (CNS) characterized by dementia, blindness, convulsions, and early death.2
Recognition of the cherry-red spot at the macula is a major diagnostic criterion of Tay-Sachs disease (Fig. 3). It is caused by the accumulation of intracytoplasmic membranous bodies in retinal ganglion cells.
|
The circular appearance of the fundoscopic lesion reflects the anatomy of the macula. No ganglion cells are present at the very center of the macular region, the foveola, and the central red spot simply represents the normal choroidal background color. The ganglion cell layer surrounding the foveola is several cells thick, and loading of these neurons by storage products results in loss of retinal transparency and a white parafoveal halo. Peripheral to the macular region the ganglion cell layer is only one cell thick, and lipid accumulation in these cells is, therefore, less conspicuous.
Tay-Sachs disease is also the most common storage disease causing macular cherry-red spots (Table 2).3 The variation in the shade of the red spot reflects racial fundus pigmentation. The halo is opaque, slightly elevated, and 1.5 disc diameters in width. The outer border is less sharp than the inner border.
TABLE 2. Storage Diseases with Macular Cherry-Red Spots
| Disease | Enzyme | Diagnostic Sample(s) |
| GM2-gangliosidosis | ||
| Tay-Sachs disease | Hex A deficient | Leukocyte serum |
| Sandhoff's disease | Hex A and B deficient | Leukocyte serum |
| AB variant | Hex A, B normal | GM2loading studies in fibroblasts |
| B1variant | Hex A deficient with sulfated substrate | Leukocyte fibroblasts |
| Juvenile GM2-gangliosidosis | Partial Hex A deficiency | Leukocyte serum |
| Infantile GM1-gangliosidosis | ß-Galactosidase deficient | Leukocyte fibroblasts |
| Niemann-Pick disease | ||
| Type A, infantile | Sphingomyelinase deficient | Leukocyte fibroblasts |
| Type B | Sphingomyelinase deficient | Leukocyte fibroblasts |
| Farber's lipogranulomatosis | Ceramidase | Leukocyte fibroblasts |
| Sialidoses | ||
| Type 1 | Sialidase deficient | Fresh fibroblasts |
| Type 2 | Sialidase and ß-galactosidase deficient | Fresh fibroblasts |
Hex, hexosaminidase.
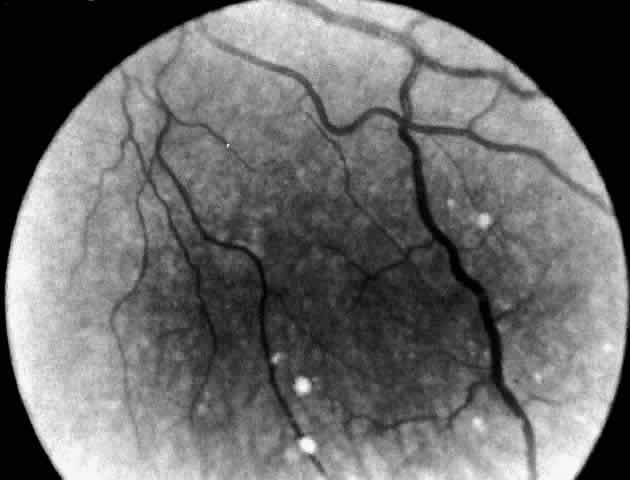
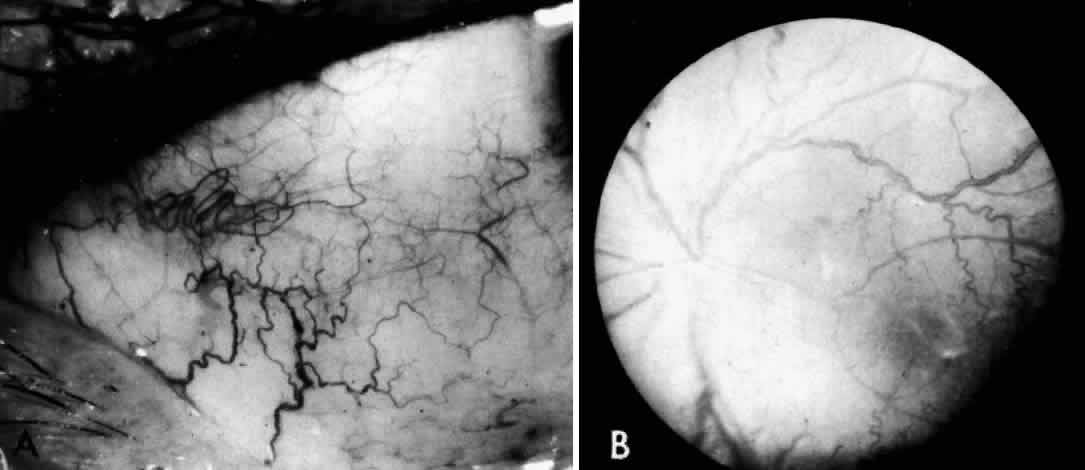
A more widespread opacification of the retina can occur due to involvement of the ganglion cells in the posterior pole. This was observed by Wray4 in a 3½-year-old child with Tay-Sachs disease. The child had black hair and brown eyes, and the cherry-red spot at the macula was brown. The patient was blind, with marked optic atrophy (Fig. 4).
|
A dynamic process of development of the macular cherry-red spot occurs paralleling the infant's progressive neurologic disorder. The cherry-red spot can be observed as early as 2 months of age and is conspicuous at age 4 to 6 months. Loss of visual acuity may occur without noticeable change in the circular halo. But, in time, the ganglion cells atrophy and optic atrophy and loss of the nerve fiber layer occurs. At this stage blindness coincides.
The frequency of the disappearance of the cherry-red spot is unknown,3 and Cogan has actually never seen it disappear. Nevertheless, the physician should include biochemical testing in the evaluation of a neurologically impaired child presenting with only optic atrophy.
A number of patients reported to have classic Tay-Sachs disease have presented without the expected cherry-red spots. Most of these cases were documented between 1908 and 1964 before biochemical confirmations were available. This suggests that the patients had another storage disease or, alternatively, the diagnosis of Tay-Sachs disease may have been correct but the eye examination was performed after the cherry-red spots had disappeared.
In the final prognosis, infants with Tay-Sachs disease become blind, usually by 18 months of age. The pupils are areflexic to light. In the rare patient noted to have brisk pupillary responses despite blindness, the visual loss may be cortical in origin. At this stage, the visual evoked response (VER) is no longer elicited although the flash electroretinogram (ERG) remains intact.
Infants with Tay-Sachs disease also show a degradation of eye movements relative to the stage of the illness.5 In the early stage there is loss of exploratory and voluntary eye movements, followed by loss of pursuit and optically elicited and vestibular movements. In the late stage, conjugate vertical eye movements elicited by the doll's head maneuver are lost. Terminally, the eyes may become fixed in a downward deviated position.
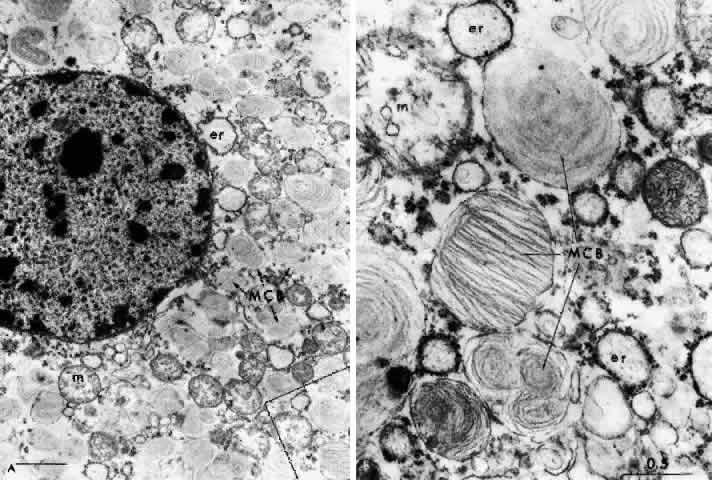
NEUROPATHOLOGY. The pathology in Tay-Sachs disease is diffuse. Intraneuronal storage of GM2-ganglioside occurs in neurons throughout the cortex, brain stem, cerebellum, spinal cord, autonomic ganglia, rectal submucosa, and retina. The affected cells are distended by intralysosomal inclusion bodies composed of closely packed, eccentrically arranged, electron-dense lamellae—the membranous cytoplasmic bodies. The membranous cytoplasmic bodies are characteristic of all the gangliosidoses and represent aggregates of gangliosides and their derivatives with cholesterol and phospholipid, which have oriented to form membranes.
The first histologic study of the eye in Tay-Sachs disease was performed by Collins in 1892.6 Emphasis has since been placed on the staining properties of the storage material, and loss of ganglion cells and extravasation of storage material occurs.7 In Tay-Sachs disease, unlike most other retinal storage diseases, the ganglion cells die early and all of them are involved.
The retinal ganglion cells in the inner portion of the bipolar cell layer and in the inner reticular layer store GM2-ganglioside. As they become affected, the nucleus in the swollen cell locates eccentrically and membranous cytoplasmic bodies fill the cytoplasm; when the cells burst the material is deposited extracellularly without significant phagocytosis or gliosis.
These deposits are strongly birefringent, and striking macular birefringence may be readily visualized in sections of the retina (Fig. 5) even in the unstained state as well as when lightly stained with cresyl violet or sudanophilic dyes.8 Loss of the ganglion cells and atrophy of the optic nerve, which is also evident, are accompanied by a thinning of the nerve fiber layer.
|
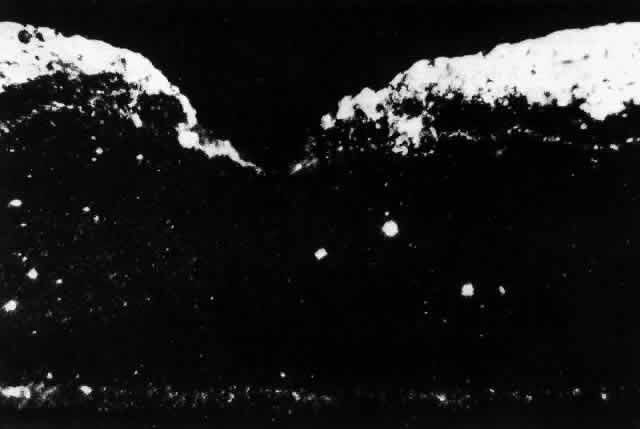
Histochemical stains have characterized the retinal lipid as a glycolipid,7 and lipid chromatography of retinal cells shows a prominent spot of GM2-ganglioside.9
Although ultrastructural studies of the eye have focused primarily on the ganglion cell,8–10 the amacrine cells in the inner nuclear layer of the retina are equally loaded with GM2-ganglioside yet the horizontal cells, bipolar cells, and photoreceptor cells are unaffected.9
DIAGNOSTIC TESTS. The diagnostic test in Tay-Sachs disease is the assay for Hex A in serum and/or leukocytes, cultured skin fibroblasts, or cultured amniotic fluid cells obtained by amniocentesis at 16 weeks' gestation. In the homozygous Tay-Sachs infant or fetus, Hex A is markedly deficient and the test confirms the diagnosis.
Early prenatal diagnosis, between the 8th and 11th weeks of gestation, is now possible by aspiration of tissue from the fetal chorionic villus through a transcervical catheter. The enzyme assay is performed on fresh and cultured villus cells. This technique reduces the waiting period for anxious parents, and earlier termination of pregnancy can be carried out if the fetus is affected.
Heterozygous carriers of the gene for Tay-Sachs disease can also be detected by serum and leukocyte assay. Heterozygotes have reduced levels of Hex A. Genetic counseling of carriers permits reduction in the incidence of the disease through the use of early prenatal monitoring of a pregnancy.
Infantile GM2-Gangliosidosis: The AB and B1 Variants
The AB and B1 variants of infantile GM2-gangliosidosis are rare. These variants are distinguished biochemically from classic Tay-Sachs disease and Sandhoff's disease by the presence of normal Hex A and Hex B levels in the serum. The defect in the AB variant is a deficiency in activator protein due to a mutation on chromosome 5.11 The defect in the B1 variant is secondary to a mutation involving the active site for the a-locus on chromosome 15 (see Fig. 2). The end result is that GM2-ganglioside accumulates in neurons.12
CLINICAL AND OCULAR MANIFESTATIONS. The story of the index case of the AB variant of Tay-Sachs disease is not widely known. The patient was a male infant of American Indian and English descent. He was said to have developed normally during the first few months of life but failed to sit at the appropriate period and mental retardation was questioned. Cogan consulted on the case in 1965 at the Osteopathic Hospital in Portland. Examination at the age of 21 months showed a typical cherry-red spot, but the nerve-heads were pink and the patient responded to appropriate visual stimuli. The pupils reacted vigorously to light. Neurologically the child had moderate spastic quadriparesis and extensor plantar responses. The case was reported in the literature by Harrell as Tay-Sachs disease in a non-Jewish male.13 The patient died at age 32 months.
NEUROPATHOLOGY. Cogan considered the case reported by Harrell so atypical for Tay-Sachs disease that he asked to be notified when the patient died. Kuwabara removed the eyes. Histopathologic study of the eyes showed the deposition of a substance that was weakly sudanophilic, strongly periodic acid-Schiff (PAS) positive, cresyl violet positive, and strongly birefringent in intact ganglion cells of the retina. The staining characteristics were similar to those of classic Tay-Sachs disease but did not show customary destruction of the ganglion cells nor the usual optic atrophy. By electron microscopy the laminated bodies were abundant in the ganglion cells, presenting both as intact membrane-bound inclusions and free unbound bodies in the cytoplasm.
Pathologic examination of the brain showed a similar storage substance in the neurons as was present in the eyes and similarly a relatively good preservation of white matter. This later was especially evident in the cerebellum, which provided a striking contrast to the usual loss of white matter in Tay-Sachs disease.14
A sample of frozen liver tissue was sent to Sandhoff, who made the first demonstration of normal hexosaminidase levels, thus establishing the AB variant as a biochemical entity.
DIAGNOSTIC TESTS. TO establish the diagnosis it is necessary to evaluate GM2-ganglioside metabolism in cultured skin fibroblasts or amniotic fluid cells over a period of time. In both the AB and B1 variants there are normal levels of Hex A and Hex B but cultured cells from an affected infant or fetus are unable to metabolize 88% to 95% of radioactively labeled GM2-ganglioside.15 In the B1 variant Hex A is shown to be deficient if the enzyme assay is performed using a sulfated synthetic substrate.
Sandhoff's Disease (GM2-Gangliosidosis Type 2)
In 1968, Sandhoff and colleagues16 described a phenotypic variant of Tay-Sachs disease. The disorder is panethnic. It is characterized by the neuronal and visceral deposition of GM2-ganglioside, its asialo derivative, and GA2-globoside. Storage of GA2-globoside is particularly prominent in the kidney. The hexosaminidase deficiency is marked by the absence of Hex A and Hex B; hence Sandhoff called this the O variant. Others have called it Tay-Sachs type 2. The hexosaminidase deficiency is due to a defect in the ß-locus on chromosome 5 that codes for the ß-subunit, an essential gene product for both enzymes (see Fig. 2).
CLINICAL MANIFESTATIONS. Clinical features of Sandhoff's disease are almost indistinguishable from classic Tay-Sachs disease except for, in some cases, the presence of moderate hepatosplenomegaly and mild skeletal dysostosis. The infants have no signs of renal disease. Death occurs before 10 years of age, generally from pneumonia.
OCULAR MANIFESTATIONS. Cherry-red spots in the macula were present in four infants with Sandhoff's disease known to O'Brien.17 Additional cases have now been observed. The halo is opaque white and funduscopically identical to Tay-Sachs cherry-red spots. Blindness and optic atrophy occur.
The cornea in infants with Sandhoff's disease is usually clear clinically, but Tremblay and Szots18 reported a unique patient with slight corneal clouding. The combination of macular cherry-red spots and corneal clouding found in Sandhoff's disease also occurs in GM1-gangliosidosis type I, Niemann-Pick disease, and sialodosis type 1 with ß-galactosidase deficiency.
NEUROPATHOLOGY. In Sandhoff's disease there is an accumulation of GM2-ganglioside as in Tay-Sachs disease, as well as of asialo-GM2 and GA2-globoside. Ultrastructurally, the metabolic products are stored in almost every tissue as cytoplasmic bodies or cytosomes. These vary in their concentration and configuration, resembling typical membranous cytoplasmic bodies or pleomorphic cytoplasmic inclusions, zebra bodies, vesicles, or granules. Histologic evidence of lipid storage in the viscera is present. Lipid-laden histiocytes appear in the liver and spleen, and there are droplets of fat in the cytoplasm of renal tubular epithelial cells. The accumulation of asialo-GM2-ganglioside is much more marked in CNS tissue and in the liver. Characteristically there is storage also of another glycolipid (a kidney globoside) in the kidney, liver, and spleen as well as in the brain.
There are few reports in the literature describing the ocular pathology of Sandhoff's disease,18–20 but the findings hardly differ from that of Tay-Sachs disease. The major distinctions are the character of the membranous cytoplasmic bodies within the ganglion cells of the retina and the involvement of the cornea. In an electron microscopic study, Brownstein and co-workers2° found abundant pleomorphic storage cytosomes in all neurons of the retina, including the inner segments of the photoreceptor cells, and in astrocytes in the optic nerve.
The ultrastructure of the cornea was also studied even when the corneas were normal on clinical examination. Storage cytosomes in keratocytes were found resembling those noted in the mucopolysaccharidoses (MPS) and mucolipidoses, in which the storage material has been identified as acid mucopolysaccharide. An excess accumulation of acid mucopolysaccharide was not evident histochemically however, implying that the storage material in the cornea was a different substance although one that leaves an ultrastructural residue similar to that observed in the MPS. Brownstein and co-workers explained the normal clinical appearance of the cornea as due to the relatively low concentration of storage material in the keratocytes.
DIAGNOSTIC TESTS. Enzyme assay for Hex A and Hex B levels in the serum or leukocytes confirms the diagnosis of Sandhoff's disease. This test shows homozygous patients with Sandhoff's disease to have absent Hex A and Hex B and clearly distinguishes infants with Sandhoff's disease from Tay-Sachs cases. Prenatal diagnosis of Sandhoff's disease has been successfully performed.21 An additional test in Sandhoff's disease is thin layer chromatography of the urine for oligosaccharides. This procedure detects a pattern of complex carbohydrate excretion typical for Sandhoff's disease.
Juvenile Sandhoff's Disease
Juvenile Sandhoff's disease is rare. The absence of Hex A and Hex B in this disorder is attributed to a mutation in the ß-locus on chromosome 5 allelic to the mutation in infantile Sandhoff's disease.22
CLINICAL MANIFESTATIONS. Onset of neurologic symptoms occurs at age 2 to 6 years. A 10-year-old boy with progressive cerebellar ataxia and psychomotor retardation was described by Wood and McDougall.23 Cherry-red spots have not been reported.
DIAGNOSTIC TESTS. Assay for hexosaminidase activity shows an absence of Hex A and Hex B.
Juvenile GM2-Gangliosidosis
Juvenile GM2-gangliosidosis differs from classic Tay-Sachs disease by its later onset and more protracted course.
CLINICAL MANIFESTATIONS. In juvenile GM2-gangliosidoses ataxia is the dominant feature. Four families of Puerto Rican descent and a family of Lebanese Canadian origin have been described by Andermann.24 The children present between 2 and 6 years of age with progressive dysarthria, spasticity, and cerebellar ataxia. Seizures occur later. Death occurs in the teen years.
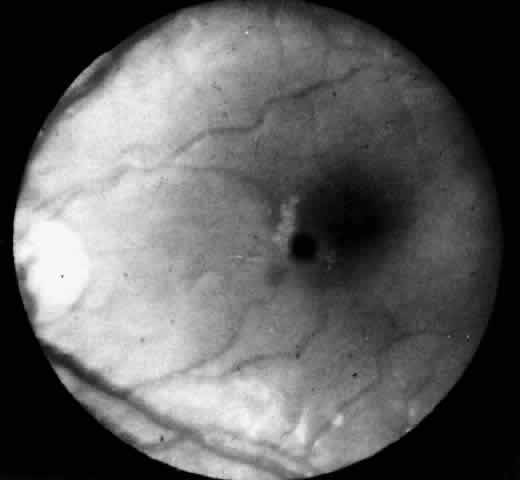
OCULAR MANIFESTATIONS. By 1984, 24 patients with late-onset GM2-gangliosidosis had been reported but only 4 had macular changes in the form of atypical cherry-red spots, unlike those seen in cases of Tay-Sachs disease. In two of the three cases (cases 4 and 5),25 symptoms began in the late-infantile period (the second year of life). The cherry-red spots were of an unusual nature and described by Brett and colleagues25 as an irregular, poorly defined area of pallor surrounding the fovea. The fovea itself was not clearly demonstrated and was not circular. A photograph of this maculopathy in a blind boy aged 5 years, 8 months (case 4) with onset of the disease at age 14 months is available for comparison with the typical cherry-red spot in classic Tay-Sachs disease.25 The child died at 6 years, 10 months of age. No autopsy examination was made.
The onset of symptoms in four additional cases of late-onset GM2-gangliosidosis was in the juvenile period (the fourth year or later). An atypical cherry-red spot was seen in only one child (case 6) with mild bilateral optic atrophy.25 Cherry-red spots at the macula are not mentioned in previously reported cases of juvenile GM2-gangliosidosis reviewed by Brett and colleagues25 or in the single case studies by Menkes and associates26 but one child showed the beginnings of retinitis pigmentosa 4 years from onset.
Johnson and associates27 found a boy with mild hand tremor since age 21/2 years to have macular cherry-red spots at age 4 years and juvenile cerebellar ataxia. Visual acuity was 20/100 (6/30)* with optic atrophy. The electroretinogram (ERG) was normal. The child had full eye movements and no nystagmus, but both eyes showed slowing of voluntary and optokinetic saccadic movements.
* Metric equivalent given in parentheses following Snellen notation.
DIAGNOSTIC TESTS. Assay of hexosaminidase shows a marked deficiency of Hex A. This is attributed to a mutation in the a-locus on chromosome 15 at a site allelic to the classic Tay-Sachs mutation (see Fig. 2).
Chronic GM2-Gangliosidosis
CLINICAL MANIFESTATIONS. Chronic GM2-gangliosidosis usually presents in the first or second decade with signs of spinocerebellar degeneration, dysarthria, and disabling tremor. Later in the course signs of anterior horn cell disease develop with muscle wasting and fasciculations. The affected teenager shows a deterioration in school performance as intellectual function declines.28
OCULAR MANIFESTATIONS. The fundi are normal. Macular cherry-red spots have not been observed. Episodic uncontrollable vertical eye movements, facial grimacing, and dystonic postures were observed in affected members of a family with atypical spinocerebellar degeneration.29 Oculogyric crisis may occur.
DIAGNOSTIC TESTS. Biochemical tests show a 10% to 15% residual Hex A activity.
Adult GM2-Gangliosidosis
All cases of adult GM2-gangliosidosis described have been in persons of Ashkenazi Jewish descent but unreported non-Jewish cases are known to us.
CLINICAL MANIFESTATIONS. Intellect is usually preserved, but probands may have a manic-depressive illness. Proximal muscle weakness manifests itself in the second to fourth decade. Fasciculations are prominent, and muscle biopsies confirm the presence of group atrophy. Computed tomography of the brain shows cerebellar atrophy.
DIAGNOSTIC TESTS. In adult GM2-gangliosidosis a 5% to 10% residual Hex A activity is present.
THE GM1-GANGLIOSIDOSES
Infantile GM1-Gangliosidosis (GM1-Gangliosidosis Type I)
The earliest well-documented report of GM1-gangliosidoses type I appeared in 1959,30,31 followed by seven cases in 1964.32 The disease is inherited as an autosomal recessive disorder caused by a deficiency of ß-galactosidase.
The absence of ß-galactosidase isoenzymes A, B, and C leads to the storage of a variety of macromolecules with a terminal ß-galactosyl residue. These are primarily GM1-ganglioside and complex carbohydrates. The accumulation of the latter compound explains the visceral and skeletal manifestations of this disease. The storage of GM1-ganglioside in the brain is responsible for the neurologic manifestations.33
CLINICAL MANIFESTATIONS. Dysmorphic facial features, macular cherry-red spots, and an enlarged liver delineate GM1-gangliosidosis. Unlike the GM2-gangliosidoses, symptoms of infantile GM1-gangliosidosis may be present at birth with edema of the extremities, hypotonia, and failure to thrive. Psychomotor development halts during the third to sixth months of life. Spasticity, tonic spasm, and hyperreflexia then develop. Head size may be normal, or megalencephaly and seizures occur. The infant becomes decerebrate and blind. Frontal bossing, depressed nasal bridge, hypertelorism, epicanthal folds, large low-set ears, and macroglossia are variable features. Progressive hepatomegaly is noted early with or without splenomegaly. Mild changes of dysostosis multiplex are often present. The peripheral blood smear contains vacuolated lymphocytes, and the bone marrow contains foamy histiocytes. Death from pneumonia usually occurs by 2 years of age.
OCULAR MANIFESTATIONS. A systematic ocular examination is most important in infantile GM1-gangliosidosis. Blindness develops early, and cherry-red spots have been found in half the patients. The macular cherry-red spot resembles that seen in Tay-Sachs disease. Nystagmus and optic atrophy have been reported34 as well as somewhat tortuous retinal vessels and flame-shaped retinal hemorrhages.35 Mild clouding of the cornea may also occur, but slit lamp examination of the cornea has not been done routinely in these infants to determine the frequency of small corneal opacities. Strabismus, lid edema, and ptosis are also described.
NEUROPATHOLOGY. Membranous cytoplasmic bodies are present in central and autonomic nervous system neurons. Vacuoles in cells of the liver, spleen, and cerebral blood vessels are indicative of widespread accumulation of intracellular complex carbohydrates. Renal glomerular epithelial cells also store complex carbohydrates.
The first pathologic report of the eye in infantile GM1-gangliosidosis was made by Emery and associates.35 The infant had cherry-red spots of the macula and mild diffuse corneal clouding. Light and electron microscopy showed lipid storage in the ganglion cells of the retina. The cytoplasm of these cells, particularly of those in the macular region, appeared foamy and filled with membranous cytoplasmic bodies with a morphologic appearance resembling the intralysosomal inclusions present in cerebral neurons.34
DIAGNOSTIC TESTS. In early infantile GM1-gangliosidosis type I, ß-galactosidase activity is deficient. The assay can be performed on leukocytes, cultured skin fibroblasts, amniotic fluid cells, and chorionic villus samples.
Urine thin-layer chromatography for oligosaccharides is abnormal, showing a pattern of complex carbohydrate excretion typical for this disease.
Juvenile GM1-Gangliosidosis (GM1-Gangliosidosis Type II)
Infantile GM1-gangliosidosis type I and juvenile GM1-gangliosidosis type II are recognized to be allelic mutations at the structural gene locus for acid-ß-galactosidase on chromosome 3.
CLINICAL MANIFESTATIONS. The disease begins in the first or second year of life with motor and mental regression, quadriplegia, dystonia, pseudobulbar palsy, cerebellar ataxia, dementia, and occasionally seizures. Foamy histiocytes are present in the bone marrow. There is no hepatosplenomegaly or significant facial or skeletal deformity. Because the clinical features are quite uncharacteristic, biochemical studies are easily overlooked. Survival until 10 years of age is possible.36
OCULAR MANIFESTATIONS. The eye does not provide a clue to the diagnosis. Vision is normal, and changes in the retina and cornea are absent. Nonparalytic squints are frequent. Late in the course optic atrophy may occur.
DIAGNOSTIC TESTS. Marked deficiency of ß-galactosidase activity in leukocytes and cultured fibroblasts confirms the diagnosis.
Adult GM1-Gangliosidosis (GM1-Gangliosidosis Type III)
CLINICAL MANIFESTATIONS. Adult GM1-gangliosidosis type III presents in the second decade with progressive cerebellar ataxia, dysarthria, and spasticity. The patient maintains normal intellect until quite late in the course. Seizures are rare. Vertebral changes, if present, are mild, and organomegaly is not usually a feature. Occasionally skin angiokeratomas develop.37
OCULAR MANIFESTATIONS. The eye may not be affected although some patients have had mild optic atrophy. Occasional mild corneal clouding occurs.38
DIAGNOSTIC TESTS. Markedly deficient ß-galactosidase activity in leukocytes or cultured fibroblasts confirms the diagnosis.