Most patients have no associated systemic disease and, therefore, are considered cases of nonsyndromic retinitis pigmentosa. A few cases have associated nonocular disease (i.e., syndromic retinitis pigmentosa). The most common syndromic form is Usher syndrome. About 15% of all cases of retinitis pigmentosa have an associated partial hearing loss (Usher syndrome, type II) and another 2% to 6% have associated profound congenital deafness and vestibular ataxia (Usher syndrome, type I).
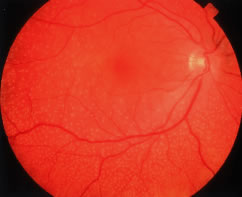
Patients with retinitis pigmentosa characteristically report difficulty with adaptation and night blindness in adolescence. They lose midperipheral and then far peripheral visual field in adulthood. As their condition progresses, they develop tunnel vision and a tendency to blue blindness. Most have reductions in central vision between ages 50 and 80. Signs on ophthalmoscopic examination include attenuated retinal arterioles, waxy pallor of the optic discs, and intraretinal bone spicule pigment around the midperiphery8–20 (Fig. 1). The bone spicule pigment is typically distributed 20 degrees to 40 degrees eccentric to the foveola in the zone where rods are normally in maximum concentration. Most show vitreous cells and a posterior vitreous detachment.14 Most patients develop posterior subcapsular cataracts in adulthood.21 An estimated 10% develop cystoid macular edema.22,23 Refractive errors such as astigmatism and myopia are common.24,25 Histologic studies of autopsy eyes have shown that loss of vision is due to degeneration of both rod and cone photoreceptor cells.26–34
|
ELECTRORETINOGRAMS
In 1945 Karpe35 reported that patients with advanced retinitis pigmentosa have very small or nondetectable (less than 10 μV) electroretinograms (ERGs). Subsequently it has been shown that patients with early retinitis pigmentosa can have reduced but easily detectable ERG responses.36–39 Responses are not only reduced in size but are also delayed with respect to the time interval between stimulus flash onset and corresponding response peaks (i.e., b-wave implicit time).38–42 These ERG changes can be detected in some instances many years before diagnostic abnormalities are visible on fundus examination.41
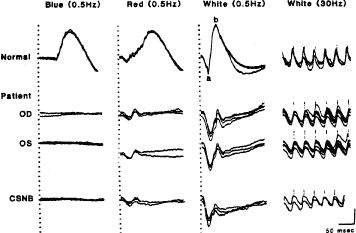
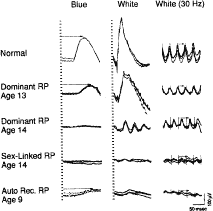
Figure 2 illustrates representative full-field ERGs from a normal subject and four children with early retinitis pigmentosa. Rod responses to single 0.5-Hz flashes of blue light (left column) are reduced and, when detectable, are delayed in b-wave implicit times. Cone responses to 30-Hz white flickering light (right column) are normal or reduced in amplitude and normal or delayed in b-wave implicit times. In most cases, cone b-wave implicit times are so delayed that a phase shift occurs between the stimulus flash onset (designated by the vertical lines) and the corresponding response peaks; each stimulus flash elicits the next-plus-one response in contrast to the normal. In mixed cone-rod responses to single 0.5-Hz flashes of white light (middle column), the cornea-negative a-wave generated by the photoreceptors is reduced in amplitude in all genetic types; this reflects the early involvement of the photoreceptors in this group of diseases.41
|
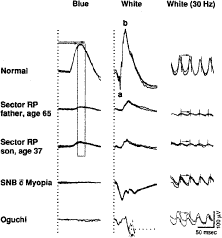
The reduced and delayed ERGs recorded from patients with widespread progressive forms of retinitis pigmentosa contrast with the reduced ERGs with normal b-wave implicit times observed in patients with self-limited sector retinitis pigmentosa. Figure 3 shows full-field ERGs from a father and son with dominantly inherited sector retinitis pigmentosa separated in age by almost 30 years; they have comparably reduced ERGs with normal b-wave implicit times. Patients with sector retinitis pigmentosa usually have patches of intraretinal pigment that are confined to one or two quadrants of the periphery of each fundus, with loss of peripheral rods and cones and consequent reductions in both rod and cone full-field ERG amplitudes. ERGs recorded from patients with sector retinitis pigmentosa are comparable to those recorded from patients who have large chorioretinal scars in the periphery.41
|
Full-field ERG testing can be used not only to identify which patients have widespread progressive forms of retinitis pigmentosa but also to determine which relatives are normal. Relatives of patients with retinitis pigmentosa, age 6 or older, with normal rod and cone ERG amplitudes and implicit times have not been observed to develop widespread progressive forms of retinitis pigmentosa at a later time.41–43
CARRIERS OF X-LINKED RETINITIS PIGMENTOSA
Females with the carrier state of X-linked retinitis pigmentosa can present with an area of bone spicule pigmentation in the periphery or an abnormal tapetal reflex in the macula. Less than 50% of carriers of child-bearing age show diagnostic findings on ophthalmoscopic examination, whereas more than 90% have abnormal ERGs.44 Abnormal ERGs in obligate carriers are either reduced in amplitude or delayed in cone b-wave implicit time, or both, in one or both eyes44,45 (Fig. 4). Daughters of obligate carriers can have either normal ERGs or abnormal ERGs similar to those recorded from obligate carriers.44
|
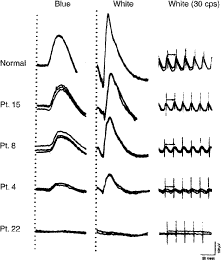
Female carriers of X-linked retinitis pigmentosa can have a slowly progressive retinal degeneration, although the natural course remains to be defined. Some female carriers have had considerable loss of visual field and substantial reductions in ERG amplitudes by age 70.44
The abnormal ERGs of female carriers of X-linked retinitis pigmentosa contrast with the normal full-field ERG amplitudes and normal fundi observed in obligate female carriers of autosomal recessive disease.44 Carriers of X-linked retinitis pigmentosa have a 50% chance of having an affected son and a 50% chance of having a carrier daughter with each childbirth.
NATURAL COURSE
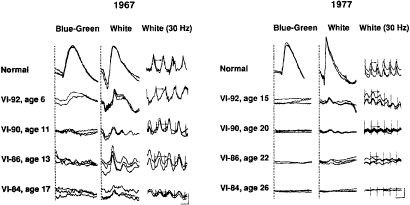
The ERG provides a quantitative measure of remaining retinal function in patients with retinitis pigmentosa. Figure 5 illustrates ERGs from affected patients followed over a 10-year period; responses become smaller as the condition progresses. Responses less than 10 μV are nondetectable with conventional recording techniques (i.e., without signal averaging). Patients with nondetectable conventional ERGs can still retain considerable visual function because most are not legally blind until ERG amplitudes decline to 0.05 μV or less.
|
Signal averaging with a bipolar artifact reject buffer and narrow-bandpass
filtering have extended the range of detectability of responses from
affected patients 100-fold or more so that responses can be quantitated
throughout almost the entire course of the disease. ERGs can now
be recorded that are as low as 1 μV to 0.5-Hz flashes of white light (normal 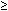 350 μV) with signal averaging alone and, with bandpass filtering and
signal averaging, as low as 0.05 μV to 30-Hz white flicker (normal
equal to or greater than 50 μV). Full-field ERG function could
be detected with at least one test criterion in 90% of an outpatient
population with retinitis pigmentosa and a visual field diameter greater
than 8 degrees. In an adult male with X-linked retinitis pigmentosa (whose
ERGs could not be quantitated without signal averaging) computer-averaged
ERGs show changes in ERG function over a 2-year period46,47 (Fig. 6).
350 μV) with signal averaging alone and, with bandpass filtering and
signal averaging, as low as 0.05 μV to 30-Hz white flicker (normal
equal to or greater than 50 μV). Full-field ERG function could
be detected with at least one test criterion in 90% of an outpatient
population with retinitis pigmentosa and a visual field diameter greater
than 8 degrees. In an adult male with X-linked retinitis pigmentosa (whose
ERGs could not be quantitated without signal averaging) computer-averaged
ERGs show changes in ERG function over a 2-year period46,47 (Fig. 6).
|
Among 94 patients, ages 6 to 49 years, with the common forms of retinitis pigmentosa, full-field ERGs declined significantly over a 3-year interval in 66 of 86 patients (77%), with detectable responses at baseline. Patients lost on average 16% of remaining full-field ERG amplitude per year to single flashes of white light (95% confidence limits, 13.1% to 18.6%) and 18.5% of remaining amplitude per year to 30-Hz white flicker (95% confidence limits, 15.1% to 21.5%). Patients lost on average 5.2% of remaining foveal cone ERG amplitude per year, indicating that loss of retinal function was primarily extrafoveal in these patients. They lost on average 4.6% of remaining visual field area per year in the Goldmann perimeter with a V-4e white test light, whereas visual acuity and dark-adaptation thresholds remained relatively stable. Bone spicule pigment increased in 54% for whom comparisons could be make over a 3-year interval, suggesting that observation of increased pigmentation as a means of following this condition is not as sensitive as full-field ERG testing.46
Caution must be exercised in applying these population ERG results to predict longitudinal patterns in individual patients, because standard deviations derived from standard errors have revealed considerable variation around the mean for these patients.46 However, these results, describing the natural course on a quantitative basis, provide a frame of reference for planning interventions in similar populations to stabilize or slow the course of retinitis pigmentosa, particularly if monitored with full-field ERG testing.
MOLECULAR GENETIC STUDIES
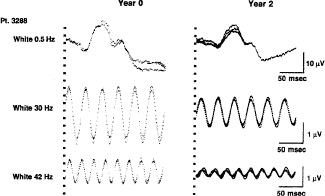
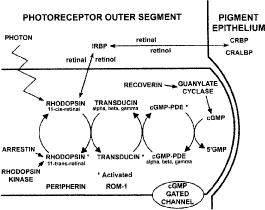
Substantial genetic heterogeneity exists among patients with retinitis pigmentosa with abnormalities in over 25 genes identified as of 2001; an updated list is maintained on the RetNet site (http://www.sph.uth.tmc.edu/Retnet/sum-dis.htm). Genes so far identified as causes of nonsyndromic retinitis pigmentosa, as well as Usher syndrome, are given in Table 2.48–87 Molecular genetic analyses have revealed defects in genes encoding proteins involved in the phototransduction cascade (Fig. 7), the retinoid cycle (Fig. 8), and structural components of photoreceptors, as well as in photoreceptor and retinal pigment epithelial proteins with unknown functions.
TABLE 2. Estimated Proportions of Cases of Retinitis Pigmentosa (RP) Caused
by Identified Genes
| Inheritance Pattern/Gene | Percent of all RP (including Usher Syndrome) | |
| Autosomal Dominant (ADRP) (-40% of all cases of retinitis pigmentosa) | ||
| 1. rhodopsin (3q) | 10% (90/363 ADRP cases) | 48–51 |
| 2. peripherin/RDS (6p) | 3–4% (based on a survey of 227) | 52 |
| 3. RP1 (8q) | 2% (based on a survey of 187) | 53 |
| 4. NRL (14q) | 1% | 54 |
| 5. FSCN2 (17q) | 1.3% (4/120 ADRP cases in Japan) | 55 |
| 6. PRPC8 (17p) | -1% (7/332 ADRP cases in UK) | 56 |
| 7. PRPF31 (19q) | -3.2% (4/50 ADRP cases in UK) | 57 |
| Autosomal Recessive (ARRP) (-50% of all cases of retinitis pigmentosa, including isolates) | ||
| 1. rhodopsin (3q) | 0.4% (1/126 ARRP cases) | 58 |
| 8. rod PDEβ (4p) | 2% (4/92 ARRP cases) | 59–61 |
| 9. rod PDEα (5q) | 1% (3/173 ARRP cases) | 62,63 |
| 10. rod channel α (4p) | 1% (3/173 ARRP cases) | 64 |
| 11. myosin VIIa (11q) | -2% (Usher IB) | 65 |
| 12. RPE65 (1p) | 0.7% (2/147 ARRP cases) | 66–68 |
| 13. CRALBP (15q) | 0.5% (3/324 ARRP cases plus isolates) | 69 |
| 14. TULP1 (6q) | 0.3% (1/162 ARRP cases) | 70 |
| 15. USH2A (1q) | >10% | 71 |
| 16. ABCA4 (i.e., ABCR) (1p) | <1% (a few cases have been reported) | 72,73 |
| 17. arrestin (2q) | 0.7% (3/120 ARRP cases in Japan; 0/85 in the U.S.) | 74,75 |
| 18. RGR (10q) | <1% | 76 |
| 19. CRB1 (1q) | ? (10 families with ARRP) | 77 |
| 20.NR2E3 (15q) | ? 1% | 78 |
| 21. MERTCK (2q) | ? 1% | 79 |
| 22. LRAT (4q) | ~1% (3/267) | 80 |
| 23. harmonin (11p) | ? 1% (Usher IC) | 81 |
| 24. CDH23 (10q) | ? 1 % (Usher ID) | 92,83 |
| 25. USH3 (3q) | ? 1 % (Usher III) | 84 |
| X-linked (XLRP) (~10% of all cases of retinitis pigmentosa) | ||
| 26. RPGR (Xp21.1) | ~ 8% | 85,86 |
| 27. RP2 (Xp11.3) | 1% | |
| Digenic(only a few families described to date) | ||
| 28. ROM1 (11q) peripherin/RDS (6p) | (these two genes account for <1% of all cases) | 7,52 |
| Mitochondrial (only one family, with Usher type III, described to date) | ||
| 29. MTTS2 | <1% | 87 |
Note: Excluding syndromic RP except for Usher syndrome; genes currently identified account for 50% to 60% of cases of RP. Percentages are approximate and are based on the breakdown of RP cases according to genetic type reported by Fishman (Arch Ophthalmol 1978;96:822.), Macrae (Birth Defects: Original Article Series 1982;18:175), and Bunker et al. (Am J Ophthalmol 1984;97:357) and on the assumptions that all isolate cases are autosomal recessive and that Usher syndrome type I accounts for about 6% of all cases of RP. The values are calculated based on frequency of cases in a published series multiplied by the proportion of RP with that inheritance pattern (e.g., dominant rhodopsin mutations were found in 90/363 cases of ADRP; ADRP accounts for about 40% of all cases of RP; accordingly, the percentage of cases caused by dominant rhodopsin mutations is 90/363 = 24.8% × 0.4 = 9.92 ≈ 10%). A current listing of genes causing retinitis pigmentosa and allied hereditary retinal diseases can be accessed on the World Wide Web at: http://www.sph.uth.tmc.edu/Retnet/sum-dis.htm; Table prepared 9/15/01.
|
|
Of the more than 25 genes so far found to be causes of retinitis pigmentosa, some are known to play a role in the phototransduction cascade in rod photoreceptors (e.g., rhodopsin, the α- and β-subunits of rod cyclic guanosine monophosphate-phosphodiesterase (cGMP-PDE), the α-subunit of the rod cGMP gated channel, and arrestin). Other genes are thought to encode structural proteins in the outer segments (peripherin/RDS and ROM1). Others are involved in the recycling of vitamin A (CRALBP, RPE65, LRAT, and ABCA4). Some genes encode transcription factors (NRL and NR2E3) or proteins involved in the escort of opsin from the inner to the outer segment (RPGR and TULP1). The more than 25 genes (see Table 2) account for about 50% to 60% of cases of retinitis pigmentosa in the United States.
It should be noted that defects in the same gene can result in different phenotypes; for example, most rhodopsin mutations lead to retinitis pigmentosa, but some cause stationary night blindness. Defects in the ABCA4 gene can result in juvenile macular degeneration or a generalized cone-rod degeneration. Some mutations in the USH2A gene lead to retinitis pigmentosa with partial hearing loss, whereas another mutation in the same gene results in only retinitis pigmentosa. Variable clinical expression can exist among patients with the same gene defect, suggesting that factors other than the gene defect itself (e.g., diet, environment, modifier genes) affect the course of the disease with possible implications for therapy.
TREATMENT TRIALS
Many treatments have been attempted for the common forms of retinitis pigmentosa including various vitamins and minerals, vasodilators, tissue therapy with placental extract, cortisone, cervical sympathectomy, injections of a hydrolysate of yeast ribonucleic acid (RNA), ultrasound, transfer factor, dimethyl sulfoxide (DMSO), ozone, muscle transplants, and subretinal injections of fetal retinal cells.88–95 None of these has been shown to have proven therapeutic benefit. A study of patients evaluated before and after receiving electric stimulation, autotransfused ozonated blood, and ocular surgery in Cuba showed that this intervention provided no benefit and raised the possibility that this intervention was aggravating the course of the disease.95 None of these attempts at treatment was conducted with a randomized, controlled, double-masked protocol, which is necessary to avoid possible patient or examiner biases. Most of these studies were performed without electroretinographic data as an end point for evaluating efficacy, so that the amount of remaining retinal function could not be quantitated in an objective manner.
Claims of success with one or another treatment for patients with retinitis pigmentosa that are based solely on subjective reporting of improved visual function are to be interpreted with caution. Spontaneous fluctuations in visual acuity and visual field are well known in this condition. Given the slow course of retinitis pigmentosa without treatment, it will usually require several years to assess whether or not any proposed treatment has an effect on stabilizing or slowing the course of the disease. The problem of assessing treatments may be further complicated by the genetic heterogeneity of this condition and the stage of disease at which treatment is initiated.
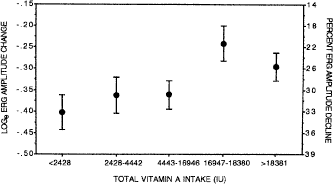
The common forms of retinitis pigmentosa have now yielded to treatment. In a randomized, controlled, double-masked trial among 601 patients ages 18 to 49, the course of retinal degeneration as monitored by the ERG was slower on average among patients taking a daily supplement of vitamin A palmitate, 15,000 IU/day, than among those not on this dose. Furthermore, the course appeared to be faster on average among patients taking a daily supplement of 400 IU/day of vitamin E than those not on this dose.96 Among a subset of 125 patients who could perform visual field testing with great precision, those on vitamin A showed a slower rate of decline of visual field than those in the control group.97 The optimal total intake of vitamin A palmitate in this study population appeared to be approximately 18,000 IU/day (i.e., 3000 IU of vitamin A in a regular diet and 15,000 IU/day of vitamin A as a supplement); higher intake did not provide greater benefit (Fig. 9). These findings have led to the recommendation that most adult patients with the common forms of retinitis pigmentosa should take a daily supplement of 15,000 IU of vitamin A in the palmitate form under the supervision of their ophthalmologist and avoid high-dose supplementation with vitamin E. For the average patient in the trial, it has been estimated that treatment with vitamin A could add 7 additional years of useful vision96; that is, the average patient with 1.3 μV of cone function who starts vitamin A at age 32 would retain useful vision until age 70 rather than age 63 if left untreated. For some patients with larger pretreatment ERGs, vitamin A supplementation could make the difference whereby these patients retain some vision for their entire lives.
|
Vitamin A may provide its benefit through the rescue of remaining cones, thereby, explaining how one supplement can help patients with many different rod-specific gene defects. The daily intake of a vitamin A supplement may provide protection against possible transient decreases in serum retinol concentrations that may adversely affect photoreceptor function. With respect to vitamin E, it has been hypothesized that a daily dose of 400 IU may adversely affect the course of retinitis pigmentosa at least in part by inhibiting the absorption or transport of vitamin A, because it was observed that the patients receiving vitamin E had slight but significant decreases in serum retinol concentration compared with those not receiving vitamin E.96
Because doses greater than 25,000 IU/day of preformed vitamin A can be associated with liver disease when taken over the long term, patients should have a fasting serum vitamin A and liver function profile before starting this treatment and annually thereafter while on this treatment. Beta-carotene is not predictably converted to preformed vitamin A and, therefore, should not be considered as a substitute for treatment with vitamin A palmitate. Because of the risk of birth defects associated with high-dose vitamin A supplementation, women who are pregnant or planning to become pregnant should not take this supplement. Because patients younger than age 18 were not included in this study, no formal recommendation can be made for patients with retinitis pigmentosa younger than this age.96,97 No toxic side effects have been observed in adults with retinitis pigmentosa in good general health on this dose of vitamin A who have been followed for up to 12 years.98
RETINITIS PIGMENTOSA ASSOCIATED WITH HEREDITARY ABETALIPOPROTEINEMIA
In 1950 Bassen and Kornzweig described an 18-year-old girl, born of first cousins, who had a malabsorption syndrome, a generalized retinal degeneration, a diffuse neuromuscular disease similar to Friedreich's ataxia, and a peculiar crenation of the red blood cells, now called acanthocytosis.99–101 In 1958 low serum cholesterol (less than 50 mg per dl) was observed.102 Soon thereafter, an absence of low-density plasma lipoproteins or so-called β-lipoproteins was found, and the term abetalipoproteinemia was assigned to this recessively inherited disorder.103–105 Other classes of lipoproteins have also been found to be abnormal.106
Patients with hereditary abetalipoproteinemia can assimilate fat into the intestinal mucosa, but a defect exists in its removal from this site because of the lack of chylomicra. Intestinal biopsies have revealed normal-sized villi filled with lipid droplets that are essentially triglycerides. Mutations in the gene encoding a microsomal triglyceride transfer protein have been found in patients with this condition.107 It appears that the liver and then the retina become depleted of vitamin A. Abnormal ERGs have been reported in a 15-month-old child108 and a 6-year-old patient109 in whom the fundi were still normal. The original case described by Bassen and Kornzweig showed multiple white dots in the early stages, but by age 31, the patient developed multiple areas of pigment epithelial cell atrophy. In other cases, the typical intraretinal pigment associated with retinitis pigmentosa has been noted in the retinal periphery.
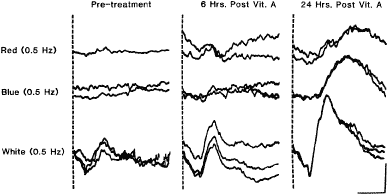
Patients with this condition are treated with a low-fat diet and supplements of the fat-soluble vitamins A, E, and K. Vitamin A supplementation has been shown to restore elevated dark-adaptation thresholds and reduced ERG responses to normal in patients with the early stages110,111 (Fig. 10). More advanced cases have not responded, but in one such case in which the retina was examined after the death of the patient, widespread loss of photoreceptor cells was observed.112 Vitamin A therapy may not maintain retinal function over the long term, because patients have been reported in whom vitamin A levels have been restored to normal and yet the retinal degeneration has appeared to progress.113,114 Because these patients have low serum vitamin E levels, supplementation with vitamin E in addition to vitamin A has been advocated with reported stabilization of retinal function.115-19
|
RETINITIS PIGMENTOSA ASSOCIATED WITH REFSUM DISEASE
Refsum disease is a recessively inherited condition in which the patient accumulates exogenous phytanic acid.120,121 Findings include a peripheral neuropathy, ataxia, an increase in cerebrospinal fluid protein with a normal cell count, and retinitis pigmentosa. All have elevated serum phytanic acid. Some cases have anosmia, neurogenic impairment of hearing, electrocardiogram (EKG) abnormalities, and skin changes resembling ichthyosis. Patients show a granular fundus with areas of depigmentation around the periphery and have a subnormal ERG in the early stages or show more typical retinitis pigmentosa with a nondetectable ERG in more advances stages.122
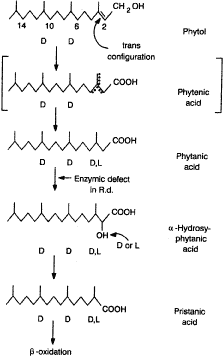
A defect exists in the conversion of phytanic acid to alpha-hydroxy phytanic acid, specifically in the introduction of a hydroxyl group on the alpha carbon of phytanic acid123 (Fig. 11). The pathogenesis appears to involve accumulation of phytanic acid in a variety of tissues including the retinal pigment epithelium (RPE).124
|
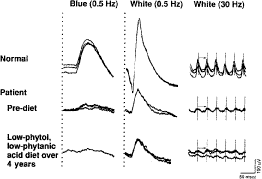
Treatment consists of restricting not only animal fats and milk products (i.e., foods that contain phytanic acid) but also green leafy vegetables containing phytol.125 Success of treatment depends on the patient maintaining his or her body weight; if body weight becomes reduced, phytanic acid is released from tissue stores, resulting in an increase of phytanic acid in serum and exacerbation of symptoms. Refsum126 has reported two patients whose serum phytanic acid levels were lowered to normal and who showed improvement in motor nerve conduction velocity, some relief of ataxia, and return of the cerebrospinal fluid protein to normal. Moreover, the retinitis pigmentosa and hearing impairment did not progress; one of these patients was followed for 10 years and the other for many years. One young adult with a mild form of this disorder has been followed on a low-phytol, low-phytanic acid diet for 4 years; full-field ERGs, reduced about 75% below normal before commencement of this diet, have remained about the same over this period42 (Fig. 12). Long-term effects of this diet on retinal function continue to be studied.
|
FREIDREICH-LIKE ATAXIA WITH RETINITIS PIGMENTOSA
Another rare recessively inherited form of ataxia associated with retinitis pigmentosa has recently been described. These patients present in adulthood with Friedreich-like ataxia, dysarthria, hyporeflexia, and decreased proprioceptive and vibratory sensation, as well as markedly decreased serum vitamin E levels. In later stages, patients can develop the fundus changes of retinitis pigmentosa and abnormal ERGs. Molecular genetic analysis has revealed a mutation in the α-tocopherol-transfer protein (α-TTP) gene. Oral administration of vitamin E restored serum vitamin E levels to normal and appeared to halt or slow the progression of the neurologic abnormalities and retinitis pigmentosa in three patients followed for 1, 4, and 10 years, respectively.127,128
USHER SYNDROME
About 15% to 20% of affected individuals with retinitis pigmentosa have associated hearing loss, sometimes referred to as Usher syndrome in recognition of the British ophthalmologist, C.H. Usher, who emphasized that this condition was recessively inherited.129,130 Patients with Usher syndrome type I typically have night blindness in the first or second decade, profound congenital deafness (i.e., greater than 70 dB loss of all frequencies) with unintelligible speech, and vestibular ataxia, whereas those with Usher syndrome type II usually report night blindness in the second to fourth decade, have a partial early onset hearing loss with intelligible speech, and do not show ataxia.131 Patients with Usher syndrome type II have been shown to have abnormalities in the cilia of sperm and abnormalities in the connecting cilia in many remaining photoreceptors in autopsy eyes.132,133 Patients with Usher syndrome type III have onset of hearing loss in midadulthood that can progress to profound deafness; some have vestibular ataxia.84,134
Molecular genetic analyses have revealed gene loci for Usher syndrome type I (further subdivided as types A-F), for Usher syndrome type II (further subdivided as types A-C), and for Usher syndrome type III. Patients with Usher syndrome type IB have mutations in a long-tailed, unconventional myosin designated as myosin VIIA.135 It has been estimated that mutations in the MYO7A gene on chromosome 11q account for approximately 75% of cases of Usher syndrome type I.136 Patients with Usher syndrome type IIA have been found to have mutations in the USH2A gene on chromosome 1q.137 This gene normally encodes a protein called usherin that has several laminin epidermal growth factor-like and fibronectin type III motifs; it is possibly involved in cell adhesion to Bruch's membrane. It has been estimated that mutations in the USH2A gene account for about 85% of cases of Usher syndrome type II.137 Some patients have been identified with autosomal recessive retinitis pigmentosa without hearing loss who have a mutation in the USH2A gene, Cys759Phe.138 Mutations in the USH3 gene on chromosome 3q have been identified in a population from Finland and Italy.84 The frequency of Usher syndrome type III in the United States is unknown.
About 2% to 6% of congenitally deaf children will develop Usher syndrome type I. If a child presents with profound deafness and a balance disorder manifested by late onset of walking usually after 15 months of age, the possibility of Usher syndrome type I should be considered.139 The diagnosis of retinitis pigmentosa as part of Usher syndrome can be made in early life with ERG testing. No tests of retinal function are yet available to identify carriers of Usher syndrome.
Patients with Usher syndrome appear to have a slowly progressive retinal degeneration. Rates of progression on a year-to-year basis remain to be defined. Patients with Usher syndrome type II may reasonably be treated with vitamin A palmitate 15,000 IU per day because these patients were included among those with autosomal recessive retinitis pigmentosa in a clinical trial of vitamin A for retinitis pigmentosa.96
LAURENCE-MOON-BARDET-BIEDL SYNDROME
The Laurence-Moon-Bardet-Biedl syndrome includes retinitis pigmentosa, mental retardation, polydactylism, truncal obesity, and hypogonadism as the most frequent features.140–147 Some authors have subdivided this syndrome into two disorders: polydactylism is found primarily in patients with the Bardet-Biedl form, whereas neurologic findings are found primarily in patients with the Laurence-Moon form.148,149 However, some patients have been described with both polydactylism and neurologic abnormalities and, therefore, would seem to qualify for inclusion in both syndromes.150,151 Variability of clinical expression is well known, and some patients may have this condition without mental retardation or polydactylism.152–156 Genetic heterogeneity exists as the condition has been linked to six distinct loci. Renal disease can be a part of this syndrome,157 and, therefore, patients should have their blood pressure and urine checked periodically, with appropriate treatment instituted for their hypertension and renal disease if detected.
Retinal degeneration is the most common feature of the Laurence-Moon-Bardet-Biedl syndrome, occurring in about 90% of cases.157 The macula is often involved early in this condition. The fundus in early life may be granular without pigment formation, so that the diagnosis is only established after electroretinographic testing.158 A child born with polydactylism to normal parents should be a suspect for this syndrome. Once this syndrome is identified in one individual, parents would know that they have a 25% chance with each succeeding childbirth of having another child with this condition. In some, presumably rare, instances a second abnormal gene locus is required and the chance of having a second affected child may be less than 25%.159 No treatment is known for the retinal degeneration that occurs as part of this syndrome.
CONGENITAL AMAUROSIS OF LEBER
Congenital amaurosis of Leber is usually an autosomal recessive disorder associated with severe reduction in vision near birth and very reduced ERGs.160–165 Patients characteristically have hyperopia and nystagmus, and fundus examination reveals granularity, white flecks, or intraretinal bone spicule pigment or some combination. When recessively inherited, parents would know that they have a 25% chance with each succeeding childbirth of having a child with this condition. Retardation of mental development has been reported in some patients, possibly secondary to visual impairment.164 Leber congenital amaurosis can occur in patients with a de novo mutation not present in the affected patient's parents; in this dominant form, the chance of having an affected child is 50%, possibly lower if the affected patient is a mosaic.166 Leber congenital amaurosis involving the retina and pigment epithelium should not be confused with Leber optic atrophy, a maternally inherited condition involving the optic nerve associated in some families with an abnormality in mitochondrial deoxyribonucleic acid (DNA).167,168
In 8 of 15 families with one form of this condition studied, mutations in the guanylate cyclase gene on chromosome 17p have been identified, suggesting that cGMP production in photoreceptors is abolished in this form. Consequently, photoreceptor excitation would be expected to be impaired due to closure of cGMP-gated cation channels with hyperpolarization of the plasma membrane. It has been proposed that the cGMP concentration in photoreceptor cells cannot be restored to the dark level, leading to a situation equivalent to constant light exposure during photoreceptor development.169 A model for this disease exists in the rd/rd chicken, which is functionally blind at hatching with an extinguished ERG, absent guanylate cyclase activity, and a mutation in the guanylate cyclase gene.170,171 Mutations in another gene, designated as RPE65, which is involved in the vitamin A cycle via the isomerase in the RPE, account for about 16% of cases of Leber congenital amaurosis in the United States;172,173 mutations in this gene are also a cause of about 2% of cases of autosomal recessive retinitis pigmentosa.173 Other genes found to be abnormal in Leber congenital amaurosis are cone-rod homeobox-containing gene (CRX), aryl hydrocarbon receptor-interacting protein-like 1 (AIPL1), tubby-like protein 1 (TULP1), homologous to crumbs protein in Drosophila (CRB1), and retinitis pigmentosa GTPase regulator interacting protein 1 (RPGRIP1). TULP1 encodes a protein thought to play a role in the escort of proteins in the inner segment. RPGRIP1 encodes a protein normally found in the photoreceptor connecting cilium; defects in this protein may interfere with transport of proteins from the inner to the outer segment. The functions of the proteins encoded by the other abnormal genes are not as well understood.
OTHER FORMS OF RETINITIS PIGMENTOSA
Mutations in the CRB1 gene77 have been observed in an unusual recessively inherited form of retinitis pigmentosa called preserved para-arteriolar retinal pigment epithelium (PPRPE). Like patients with typical Leber congenital amaurosis, these patients are highly hypermetropic. The condition is named for the relative preservation of the RPE adjacent to and underlying retinal arterioles in earlier stages; in later stages the RPE previously spared is lost. The degeneration associated with this condition appears to be rapidly progressive.174
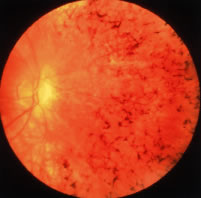
Retinitis punctata albescens can be associated with some signs and symptoms characteristic of retinitis pigmentosa.175 Patients usually present with profound adaptational problems and gradual loss of peripheral vision. Examination with a direct ophthalmoscope reveals multiple punctate white deposits in the macula and around the midperiphery at the level of the pigment epithelium for which this condition was named. Patients have retinal arteriolar attenuation and some develop round areas of atrophy of the RPE in the midperiphery, as well as intraretinal bone spicule pigment in the midperiphery. This raises the possibility that this condition is a variant of retinitis pigmentosa.176,177 This form of retinal degeneration may affect the eyes of a patient asymmetrically; often these patients receive a neurologic evaluation in search of an intracranial abnormality before it is realized that the visual loss is due to a widespread retinal degeneration. Full-field ERGs of such patients are invariably abnormal with reductions in amplitude and delays in implicit times. The condition is usually slowly progressive, although the course appears to vary from one individual to another. This condition has been thought to be inherited by an autosomal recessive mode, but a dominant mode of transmission can occur; therefore, ERG testing of relatives of affected patients is recommended to help establish the mode of transmission. A null mutation in the peripherin/RDS gene has been reported in one family with this condition with a dominant mode of transmission;178 other families with an autosomal recessive mode of transmission have shown mutations in the cellular retinaldehyde binding protein (CRALBP) gene.179
Another atypical form of retinitis pigmentosa is designated as progressive cone-rod degeneration.156,180 Patients with this form typically present with reduced visual acuity, photophobia, color deficiency, and night deficiency and show signs of macular degeneration, retinal arteriolar attenuation, and, in some cases, intraretinal bone spicule pigment in the peripheral fundus. Dark-adaptation testing shows elevated final dark-adaptation thresholds across the fundus, whereas full-field ERGs show a profound loss of cone function and some reduction in rod function across the retina. Patients with the earlier stages of cone-rod degeneration typically have rod ERGs that are more than 30 times larger than cone ERGs. The condition is usually inherited by an autosomal recessive mode but can also be inherited by an autosomal dominant mode. Mutations have been found in the retina-specific ATP-binding cassette transporter (ABCA4) gene,181 the same gene that has been found to be abnormal in patients with juvenile macular degeneration (Stargardt) disease. No treatment is know for patients with cone-rod degeneration.
Other unusual forms of retinitis pigmentosa include those designated as paravenous or unilateral. The paravenous form is usually characterized by slightly reduced full-field ERGs with normal implicit times and intraretinal pigment and atrophy of the pigment epithelium confined to the distribution of the retinal veins in each eye. Unilateral retinitis pigmentosa is characterized by fundus changes of retinitis pigmentosa in one eye with no evidence of retinal degeneration in the other eye. Full-field ERGs are substantially reduced in the affected eye and normal in the fellow eye. Patients with unilateral retinitis pigmentosa do not develop retinitis pigmentosa in the fellow eye at a later time. In my experience, patients with either paravenous or unilateral disease have presented with a negative family history for retinal degeneration, suggesting that these forms of retinitis pigmentosa are not inherited.