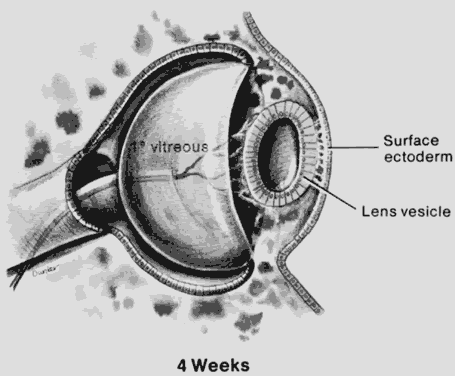
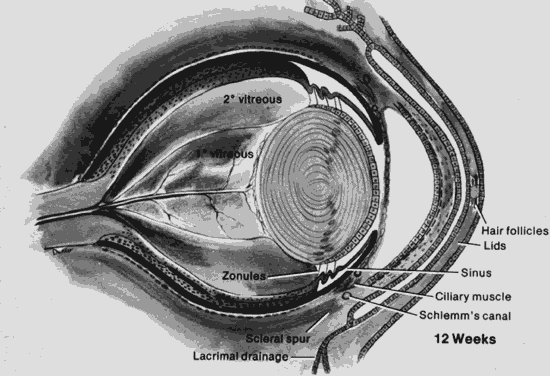
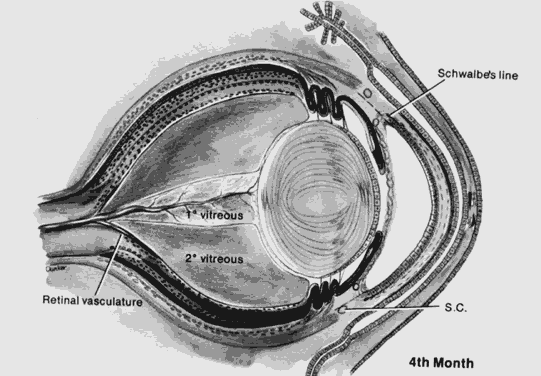
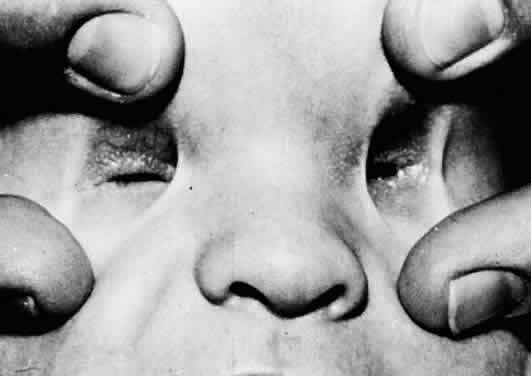
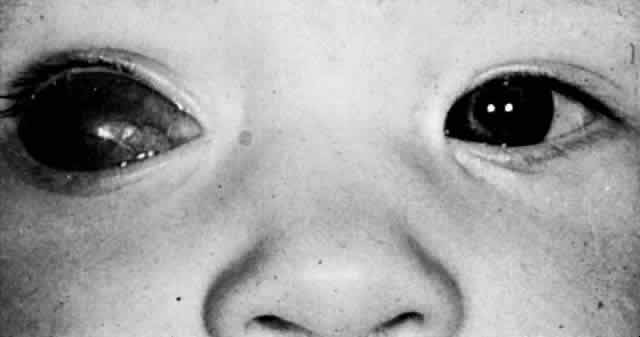
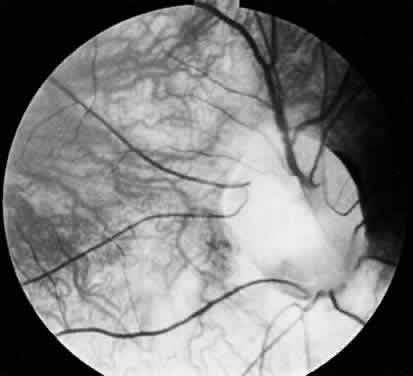
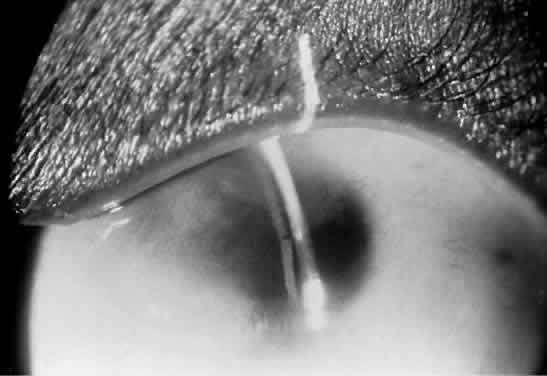
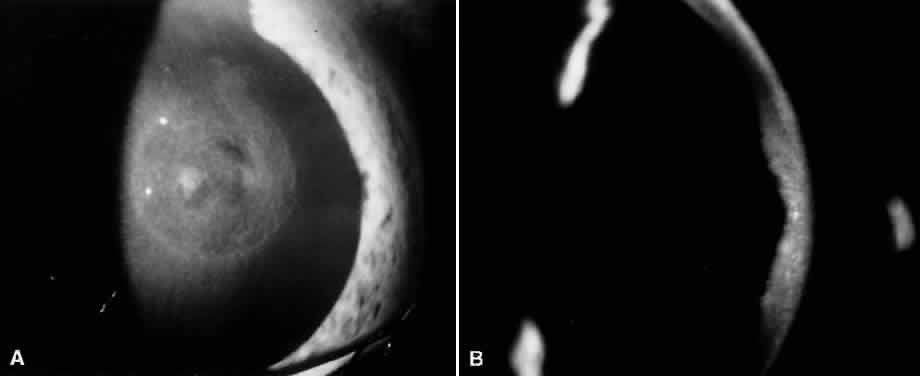
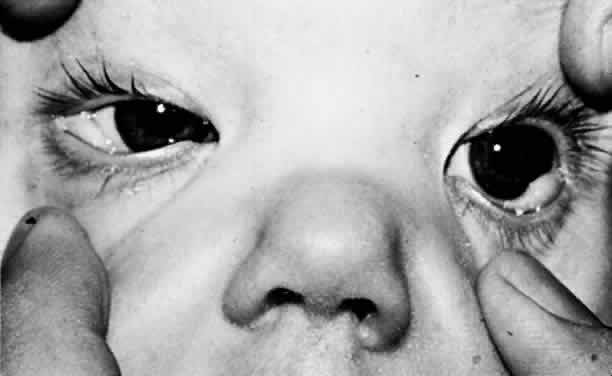
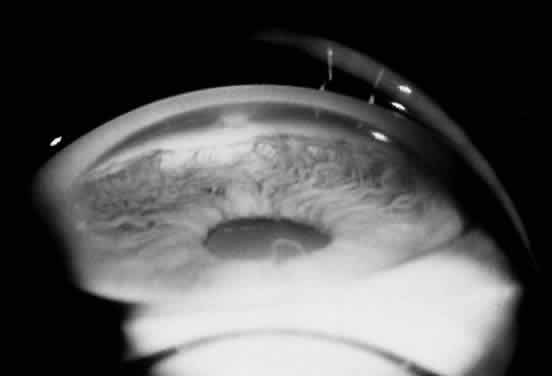
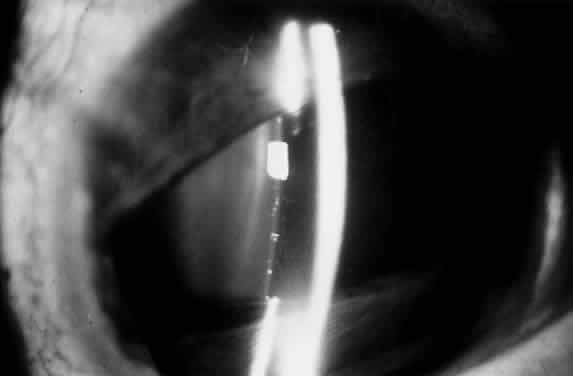
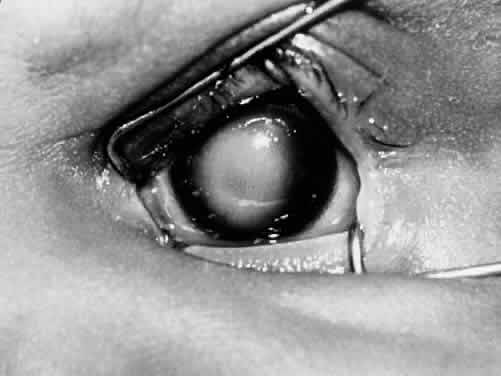
1. Mann I: The Development of the Human Eye. New York, Grune & Stratton, 1969 2. Mann I: Developmental Abnormalities of the Eye, 2nd ed. Philadelphia, JB
Lippincott, 1957 3. Coulombre AJ: Regulation of ocular morphogenesis. Invest Ophthalmol 8:25, 1969 4. Duke-Elder S, Cook C: Normal and abnormal development. In Duke-Elder S (ed): System
in Ophthalmology, Vol 3, Part 2, Congenital Deformities. St
Louis, CV Mosby, 1963 5. Pagon RA: Ocular coloboma. Surv Ophthalmol 25:223, 1981 6. Manschot WA: Primary congenital aphakia. Arch Ophthalmol 69:71, 1963 7. Fulton AB, Craft JL, Howard RO et al: Human retinal dysplasia. Am J Ophthalmol 85:690, 1978 8. Hay ED: Development of the vertebrate cornea. Int Rev Cytol 63:263, 1980 9. Johnson MC, Noden DM, Hazelton RD et al: Origins of avian ocular and periocular tissues. Exp Eye Res 29:27, 1979 10. Zinn KM, Mockel-Pohl S: Fine structure of the developing cornea. Int Ophthalmol Clin 15:19, 1975 11. Peterson RA, Robb RM: The natural course of congenital obstruction of the nasolacrimal duct. J Pediatr Ophthalmol Strabismus 15:246, 1978 12. Clementi M, Turolla L, Mammi I et al: Clinical anophthalmia: An epidemiological study in northeast Italy based
on 368,256 consecutive births. Teratology 46:551, 1992 13. Sassani JW, Yanoff M: Anophthalmos in an infant with multiple congenital anomalies. Am J Ophthalmol 83:43, 1977 14. Daxecker F, Felber S: Magnetic resonance imaging features of congenital anophthalmia. Ophthalmologica 206:139, 1993 15. O'Keefe M, Webb M, Pashby RC et al: Clinical anophthalmos. Br J Ophthalmol 71:635, 1987 16. Zeiter MS: Congenital microphthalmos: A pedigree of four affected siblings and an
additional report of 44 sporadic cases. Am J Ophthalmol 55:910, 1963 17. Bianchine JW: A family with microphthalmia, anophthalmia and concomitant oligophrenia. Birth Defects 7:205, 1971 18. Pearce WG, Nigam W, Rootman J: Primary anophthalmos: Histological and genetic features. Can J Ophthalmol 9:141, 1974 19. Joseph R: A pedigree of anophthalmos. Br J Ophthalmol 41:541, 1957 20. Roberts JAF: Sex-linked microphthalmia sometimes associated with mental deficiency. Br Med J 2:1213, 1937 21. Fryns JP, Legius E, Moerman P et al: Apparently new “anophthalmia-plus” syndrome in sibs. Am J Med Genet 58:113, 1995 22. Graham CA, Redmond RM, Nevin NC: X-linked clinical anophthalmos: Localization of the gene to Xq27–Xq28. Ophthalmic Paediatr Genet 12:43, 1991 23. Kallen B, Castilla EE, Lancaster PA et al: The cyclops and the mermaid: An epidemiological study of two types of rare
malformation. J Med Genet 29:30, 1992 24. Gartner S: Cyclopia. Arch Ophthalmol 37:220, 1947 25. Torczynski E, Jakobiec FA, Johnston MC et al: Synophthalmia and cyclopia: A histopathologic, radiographic and organogenetic
analysis. Doc Ophthalmol 44:311, 1977 26. O'Rahilly R, Muller F: Interpretation of some median anomalies as illustrated by cyclopia and
symmelia. Teratology 40:409, 1989 27. Lurie IW, Kirillova IA, Nedzved MK et al: Which brain defects accompany cyclopia?. Genet Counsel 3:127, 1992 28. Van Allen MI, Ritchie S, Toi A et al: Trisomy 4 in a fetus with cyclopia and other anomalies. Am J Med Genet 46:193, 1993 29. Dolfus MA, Marx P, Langlois J et al: Congenital cystic eyeball. Am J Ophthalmol 66:504, 1968 30. Bard JBL, Ross ASA: The morphogenesis of the ciliary body of the avian eye. Dev Biol 92:87, 1982 31. Cross HE, Yoder F: Familial nanophthalmos. Am J Ophthalmol 81:300, 1976 32. Kimbrongh RL, Trempe CS, Brockhurst RJ et al: Angle-closure glaucoma in nanophthalmos. Am J Ophthalmol 88:572, 1979 33. Brockhurst RJ: Vortex vein decompression for nanophthalmic uveal effusion. Arch Ophthalmol 98:1987, 1980 34. Trelstad RL, Silbermann NN, Brockhurst RJ: Nanophthalmic sclera ultrastructure of histochemical and biochemical observations. Arch Ophthalmol 100:1935, 1982 35. Yue BY, Duvall J, Goldberg MF et al: Nanophthalmic sclera: Morphologic and tissue culture studies. Ophthalmology 93:534, 1986 36. Kawamura M, Tajima S, Azuma N et al: Biochemical studies of glycosaminoglycans in nanophthalmic sclera. Graefes Arch Clin Exp Ophthalmol 233:58, 1995 37. Shiono T, Shoji A, Mutoh T et al: Abnormal sclerocytes in nanophthalmos. Graefes Arch Clin Exp Ophthalmol 230:348, 1992 38. Syogren T, Carsson T: Microphthalmos and anophthalmos with or without coincident oligophrenia: A
clinical and genetic statistical study. Acta Psychiatr Scand 56(suppl):1, 1949 39. Leydhecker F: Eine Familie mit Mikrophthalmia congenita. Graefes Arch Ophthalmol 139:790, 1938 40. Weiss AH, Kousseff BG, Ross EA: Simple microphthalmos. Arch Ophthalmol 107:1625, 1989 41. Warburg M: Norrie's disease: A congenital progressive oculoacousticocerebral
degeneration. Acta Ophthalmol 89:1, 1966 42. Gorlin RJ, Meskin LM, Geme JW: Oculodentodigital dysplasia. J Pediatr 63:69, 1963 43. Sugar MS, Thompson JP, Davis JD: The oculodentodigital dysplasia syndrome. Am J Ophthalmol 44:1448, 1966 44. Kung HW: Hypoplastic anemia with multiple congenital defects. Pediatrics 10:286, 1952 45. Silver HK, Blair WC, Kempe CH: Fanconi syndrome. Am J Dis Child 83:14, 1952 46. Aristikartis M: A case report of bilateral microphthalmos with cysts. Arch Ophthalmol 82:480, 1969 47. Lieb W, Rochels R, Gronemeyer U: Microphthalmos with colobomatous orbital cyst: Clinical, histological, immunohistological, and
electronmicroscopic findings. Br J Ophthalmol 74:59, 1990 48. Waring GO, Roth AM, Rodrigues MM: Clinicopathologic correlation of microphthalmos with cyst. Am J Ophthalmol 82:714, 1976 49. Lim G, Bejec LC: Congenital cystic microphthalmos. Philippine J Ophthalmol 5:93, 1973 50. Weyman MF: Microphthalmos with cyst formation. Am J Ophthalmol 8:214, 1925 51. Kok-van Alphen CC, Manshot WA, Frederiks E et al: Microphthalmos and orbital cyst. Ophthalmologica 167:389, 1973 52. Makley TA Jr, Battles M: Microphthalmos with cyst: Report on two cases in the same family. Surv Ophthalmol 13:200, 1969 53. Ladenheim J, Metrick S: Congenital microphthalmos with cyst formation. Am J Ophthalmol 41:1059, 1956 54. Yanoff M, Ballian LR, Niederer BS: Ocular and cerebral abnormalities in chromosome 18 deletion defect. Am J Ophthalmol 70:391, 1970 55. von Hippel E: Über die Beyrehunder von Mikrophthalmos mit Unterlidcyste zu allgemeinen
Missbildungen besonders zum Lindauschen Symptomenkomplex. Graefes Arch Ophthalmol 132:25, 1934 56. Jesberg DO, Schepans CL: Retinal detachment associated with coloboma of the choroid. Arch Ophthalmol 65:163, 1961 57. Jensen PE, Kalina RE: Congenital anomalies of the optic disk. Am J Ophthalmol 82:27, 1976 58. Francois J: Colobomatous malformations of the ocular globe. Int Ophthalmol Clin 8:797, 1968 59. Pollock JA, Newton TH, Hoyt WF: Transsphenoidal and transethmoidal encephaloceles: A review of clinical
and roentgen features in 8 cases. Radiology 90:442, 1968 60. Corbett JJ, Savino PJ, Schatz NJ et al: Cavitary developmental defects of the optic disc: Visual loss associated
with optic pits and colobomas. Arch Neurol 37:210, 1980 61. Streletz LJ, Schatz NJ: Transsphenoidal encephalocele associated with colobomas
of the optic disc and hypopituitary dwarfism. In Smith JL, Glaser
JS (eds): Neuro-Ophthalmology Symposium of the University of Miami
and the Bascom Palmer Eye Institute, Vol 7, pp 78–86. St. Louis, CV
Mosby, 1973 62. Cross HE: Ocular colobomas. In Bergsma D (ed): Birth Defects Compendium, p 791. New
York, Alan R Liss, 1979 63. Meyer SJ, Holstein T: Spherophakia with glaucoma and brachydactyly. Am J Ophthalmol 24:247, 1941 64. Urbanek J: Glaucoma juvenile inversion. Z Augenheilkd 77:171, 1930 65. Blaxter PL: Spherophakia. Trans Ophthalmol Soc UK 88:621, 1969 66. Willi M, Kut K, Cother E: Pupillary block glaucoma in the Marchesani syndrome. Arch Ophthalmol 90:504, 1973 67. Cross HE, Jensen AD: Ocular manifestations in the Marfan syndrome and homocystinuria. Am J Ophthalmol 75:405, 1973 68. Schar E: Renal disease, inner ear deafness and ocular changes: A new heredofamilial
syndrome. Arch Intern Med 97:627, 1956 69. Bessiere E, Riviera J, Leuret JP: Le rebellar, an association of Klinefelter's disease and congenital
anomalies, comptodactyly, microphakia. Bull Soc Ophthalmol Fr 62:197, 1962 70. Jensen AD, Cross HE, Paton D: Ocular complications in the Weill-Marchesani syndrome. Am J Ophthalmol 77:261, 1974 71. Johnson VP, Grayson M, Christian JC et al: Dominant microspherophakia. Arch Ophthalmol 85:534, 1971 72. Arnott EJ, Crawford MD, Toghill PJ: Anterior lenticonus and Alport's syndrome. Br J Ophthalmol 50:390, 1966 73. Chugh KS, Sakhuja V, Agarwal A et al: Hereditary nephritis (Alport's syndrome): Clinical profile and inheritance
in 28 kindreds. Nephrol Dial Transplant 8:690, 1993 74. Streeten BW, Robinson MR, Wallace R et al: Lens capsule abnormalities in Alport's syndrome. Arch Ophthalmol 105:1693, 1987 75. Cheong HI, Kashtan CE, Kim Y et al: Immunohistologic studies of type IV collagen in anterior lens capsules
of patients with Alport syndrome. Lab Invest 70:553, 1994 76. Francois J: Congenital Cataracts. Assen, Charles C Thomas, 1963 77. Hunter WS, Zimmerman LE: Unilateral retinal dysplasia. Arch Ophthalmol 74:23, 1965 78. Lahav M, Albert DM, Wyand S: Clinical and histopathologic classification of retinal dysplasia. Am J Ophthalmol 75:648, 1973 79. Seefelder R: Zur Frage der Netzhautanomalien in sonst normalen fötalen menschlichen
Augen. Graefes Arch Ophthalmol 73:216, 1909 80. Silverstein AM, Osburn BI, Prendergast RA: The pathogenesis of retinal dysplasia. Am J Ophthalmol 72:13, 1971 81. Reese AB, Blodi FC: Retinal dysplasia. Am J Ophthalmol 33:23, 1950 82. Reese AB, Straatsma BR: Retinal dysplasia. Am J Ophthalmol 45:199, 1958 83. Miller M, Robbins J, Fishman R et al: A chromosomal anomaly with multiple ocular defects including retinal dysplasia. Am J Ophthalmol 55:901, 1963 84. Cogan DG, Kuwabara T: Ocular pathology of the 13–15 trisomy syndrome. Arch Ophthalmol 72:246, 1964 85. Zimmerman LE, Font RL: Congenital malformations of the eye. JAMA 196:684, 1966 86. Fradkin AH: Norrie's disease: Congenital progressive oculoacousticocerebral degeneration. Am J Ophthalmol 72:947, 1971 87. Howard RO, Bey WR, Albert DM et al: Retinoblastoma and chromosome abnormality. Arch Ophthalmol 92:490, 1974 88. Haddad R, Font RL, Reeser F: Persistent hyperplastic primary vitreous: A clinicopathologic study of 62 cases
and review of the literature. Surv Ophthalmol 23:123, 1976 89. van Sien S, Sullivan WB: Congenital retinal fold: A case report. Am J Ophthalmol 39:643, 1955 90. Guerry D: Congenital retinal folds: Report of two cases. Am J Ophthalmol 27:1132, 1944 91. Anderson SR: A typical falciform retinal fold. Acta Ophthalmol 30:325, 1952 92. Dessoff J: Blue sclerotics, fragile bones and deafness. Arch Ophthalmol 12:60, 1934 93. Ruedman AD: Osteogenesis imperfecta congenita and blue sclera. Arch Ophthalmol 49:6, 1953 94. Francois J: Affections of the cornea. In: Heredity in Ophthalmology. St. Louis, CV
Mosby, 1961 95. Howard RO, Abrahams IW: Sclerocornea. Am J Ophthalmol 71:1254, 1971 96. Goldstein JE, Cogan DG: Sclerocornea and associated congenital anomalies. Arch Ophthalmol 67:761, 1962 97. Kanai A, Wood TC, Polack FM et al: The fine structure of sclerocornea. Invest Ophthalmol Vis Sci 10:687, 1971 98. Petroutsos G, Patey A, Savoldelli M et al: Sclerocornee: Etude ultrastruccturale et morphologique. J Fr Ophthalmol 6:769, 1983 99. Kenyon KR, Fogle JA, Grayson M: Dysgeneses, dystrophies, and degenerations
of the cornea. In Duane TD (ed): Clinical Ophthalmology. Philadelphia, JB
Lippincott, 1982 100. Kokott W: Cornea plana und mikrocornea periplana. Klin Monatsbl Augenheilkd 98:372, 1937 101. Tahvanainen E, Villanueva AS, Forsius H et al: Dominantly and recessively inherited cornea plana congenita map to the
same small region of chromosome 12. Genome Res 6:249, 1996 102. Tahvanainen E, Forsius H, Karila E et al: Cornea plana congenita gene assigned to the long arm of chromosome 12 by
linkage analysis. Genomics 26:290, 1995 103. Lowe RF: Aetiology of anatomical basis of primary angle-closure glaucoma: Biometrical
comparisons between normal eyes and eyes with primary angle-closure
glaucoma. Br J Ophthalmol 54:161, 1970 104. Grayson M: Diseases of the Cornea. St. Louis, CV Mosby, 1979 105. Vail DT: Adult hereditary anterior megalophthalmus sine glaucoma: A definite disease
entity, with special reference to extraction of the cataract. Arch Ophthalmol 6:39, 1931 106. Meire FM, Bleeker WE, Oehler M et al: X-linked megalocornea: Ocular findings and linkage analysis. Opthalmic Paediatr Genet 12:153, 1991 107. Francois J: La gonioscopie, III, Coloboma de l'iris. Adv Ophthalmol 4:44, 1955 108. Malbran E, Dodds R: Megalocornea and its relation to congenital glaucoma. Am J Ophthalmol 49:908, 1960 109. Stephenson WV: Anterior megalophthalmos and arachnodactyly. Am J Ophthalmol 8:315, 1954 110. Mackey DA, Buttery RG, Wise GM et al: Description of X-linked megalocornea with identification of the gene locus. Arch Ophthalmol 109:829, 1991 111. Krachmer JH, Rodrigues MM: Posterior keratoconus. Arch Ophthalmol 96:1867, 1978 112. Mannis MJ, Lightman J, Plotnik RD: Corneal topography of posterior keratoconus. Cornea 11:351, 1992 113. Jacobs HB: Posterior conical cornea. Br J Ophthalmol 41:31, 1957 114. Haney W, Falls HF: The occurrence of congenital keratoconus posticus circumscriptus. Am J Ophthalmol 52:53, 1961 115. Wolter JR, Haney W: Histopathology of keratoconus posticus circumscriptus. Arch Ophthalmol 69:357, 1963 116. Butler TH: Keratoconus posticus. Trans Ophthalmol Soc UK 50:551, 1930 117. Greene PB: Keratoconus posticus circumscriptus: Report of a case. Arch Ophthalmol 34:432, 1945 118. Benjamin SN, Allen HF: Classification for limbal dermoid choristomas and branchial arch anomalies. Arch Ophthalmol 87:305, 1972 119. Baum JL, Feingold M: Ocular aspects of Goldenhar's syndrome. Am J Ophthalmol 75:250, 1973 120. Hogan MJ, Zimmerman LE: Ophthalmic Pathology. Philadelphia, WB Saunders, 1962 121. Maumenee AE: Congenital hereditary corneal dystrophy. Am J Ophthalmol 50:1114, 1960 122. Kenyon KR, Maumenee AE: Further studies of congenital hereditary endothelial dystrophy of the cornea. Am J Ophthalmol 76:419, 1973 123. Judisch GF, Maumenee IH: Clinical differentiation of recessive congenital hereditary endothelial
dystrophy and dominant hereditary endothelial dystrophy. Am J Ophthalmol 85:606, 1978 124. Arffa RC: Disorders of the endothelium. In Grayson's Diseases of the
Cornea, 2nd ed. St. Louis, Mosby-Year Book, 1991 125. Ostler HB: Diseases of the cornea. In: Diseases of the External Eye and
Adnexa: A Text and Atlas. Baltimore, Williams & Wilkins, 1993 126. Bahn CF, Falls HF, Varley GA et al: Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology 91:558, 1984 127. Sekundo W, Marshall GE, Lee WR et al: Immunoelectron labelling of matrix components in congenital hereditary
endothelial dystrophy. Graefes Arch Clin Exp Ophthalmol 232:337, 1994 128. Alkemade PPH: Dysgenesis Mesodermalis of the Iris and the Cornea. Assen, Charles
C Thomas, 1969 129. Wilson ME: Congenital iris ectropion and a new classification for anterior segment
dysgenesis. J Pediatr Ophthalmol Strabismus 27:48, 1990 130. Townsend WM: Congenital anomalies of the cornea. In Kaufman HE, Barron
BA, McDonald MB, Waltman SR (eds): The Cornea. New York, Churchill Livingstone, 1988 131. Waring GO, Rodrigues MM, Laibson PR: Anterior-chamber cleavage syndrome: A stepladder classification. Surv Ophthalmol 20:3, 1975 132. Yue BY, Baum JL, Silbert JE: Synthesis of glycosaminoglycans by cultures of normal human corneal endothelial
and stromal cells. Invest Ophthalmol Vis Sci 17:523, 1978 133. Townsend WM, Font RL, Zimmerman LE: Congenital corneal leukomas: II. Histopathologic findings in 19 eyes with
central defect in Descemet's membrane. Am J Ophthalmol 77:192, 1974 134. Traboulsi EI, Maumenee IH: Peters' anomaly and associated congenital malformations. Arch Ophthalmol 110: 1739, 1992 135. Yang LLH, Lambert SR, Fernhoff PM, Stulting RD: Peters' anomaly: Associated congenital malformations and etiology. Invest Ophthalmol Vis Sci 36(suppl):S41, 1995 136. Wilson FM: Congenital anomalies. In Smolin G, Thoft RA (eds): The Cornea: Scientific
Foundations and Clinical Practice, 2nd ed, p 457. Boston, Little, Brown & Co, 1987 137. Pearce WG, Tripathi RC, Morgan G: Congenital endothelial corneal dystrophy: clinical, pathological and genetic
study. Br J Ophthalmol 53:577, 1969 138. Bourne WM: Partial corneal involvement in the iridocorneal endothelial syndrome. Am J Ophthalmol 94:774, 1982 139. Kolker AE, Hetherington J Jr: Becker-Shaffer's Diagnosis and Therapy
of the Glaucomas. St. Louis, CV Mosby, 1970 140. Hetherington J: Congenital glaucoma. In Duane TD (ed): Clinical Ophthalmology, Vol 3, Chap 51. Philadelphia, JB Lippincott, 1982 141. Barkan O: Pathogenesis of congenital glaucoma. Am J Ophthalmol 40:1, 1955 142. Worst J: The cause and treatment of congenital glaucoma. Trans Am Acad Ophthalmol Otolaryngol 68:766, 1964 143. Kupfer C, Kaiser-Kupfer MI: Observations on the development of the anterior chamber angle with reference
to the pathogenesis of congenital glaucoma. Am J Ophthalmol 88:424, 1979 144. Tripathi BJ, Tripathi RC: Neural crest origin of human trabecular meshwork and its implications for
the pathogenesis of glaucoma. Am J Ophthalmol 107:583, 1989 145. Cross HE: Ectopia lentis et pupillae. Am J Ophthalmol 88:381, 1979 146. Nelson LB, Spaeth GL, Nowinski TS et al: Aniridia: A review. Surv Ophthalmol 28:621, 1984 147. Dowling JL, Albert DM, Nelson LB et al: Primary glaucoma associated with iridotrabecular dysfunction and ectropion
uvae. Ophthalmology 92:912, 1985 148. Quigley HA, Stanish FS: Unilateral congenital iris pigment epithelial hyperplasia associated with
late onset glaucoma. Am J Ophthalmol 86:182, 1978 149. Ritch R, Forbes M, Hetherington J et al: Congenital ectropion uvae with glaucoma. Ophthalmology 91:326, 1984 150. Shields MB: Axenfeld-Rieger syndrome: A theory of mechanism and distinction from the
iridocorneal endothelial syndrome. Trans Am Ophthalmol Soc 81:736, 1983 151. Shaw MW, Falls HF, Neel JV: Congenital aniridia. Am J Hum Genet 12:389, 1960 152. Layman PR, Anderson RA, Flynn JT: Frequent occurrence of hypoplastic optic discs in patients with aniridia. Am J Ophthalmol 77:513, 1974 153. Elsas FJ, Maumenee IH, Kenyon KR et al: Familial aniridia with preserved ocular function. Am J Ophthalmol 83: 718, 1977 154. Miller RW, Fraameni JF, Manning MD: Association of Wilms' tumor with aniridia, hemihypertrophy and other congenital
malformations. N Engl J Med 170:922, 1964 155. Fraumeni JF: The aniridia-Wilms' tumor syndrome. Birth Defects 5:198, 1969 156. Riccardi VM, Sujansky E, Smith AC et al: Chromosomal imbalance in the aniridia-Wilms' tumor association: 11p interstitial
deletion. Pediatrics 61:604, 1978 157. DiGeorge AM, Harley RD: Association of aniridia, Wilms' tumor and genital abnormalities. Arch Ophthalmol 75: 796, 1966 158. Flanagan JC, DiGeorge AM: Sporadic aniridia and Wilms' tumor. Am J Ophthalmol 67:558, 1969 159. Fraumeni JF, Glass AG: Wilms' tumor and aniridia. JAMA 206:825, 1968 160. Francke U, Holmes LB, Atkins L et al: Aniridia-Wilms' tumor association: Evidence for specific deletion of 11P13. Cytogenet Cell Genet 24:185, 1979 161. Francke U, Riccardi VM, Hittner HM et al: Interstitial deletion (11P) as a cause of the aniridia-Wilms' tumor association: Band
localization and a hereditable basis (abstr). Am J Hum Genet 30:81A, 1978 162. Hittner HM, Riccardi VM, Francke U: Aniridia caused by hereditable chromosome 11 deletion. Ophthalmology 86:1173, 1979 163. Ton CCT, Hirvonen H, Miwa H et al: Positional cloning and characterization of a paired box—and homeobox—containing
gene from the aniridia region. Cell 67:1059, 1991 164. Cresswell TM: A case of congenital miosis. Br J Ophthalmol 8:278, 1924 165. Polomeno RC, Milot J: Congenital miosis. Can J Ophthalmol 14:43, 1979 166. Veirs ER, Brown W: Congenital miosis. Arch Ophthalmol 65:59, 1961 167. Lambert SR, Amaya L, Taylor D: Congenital idiopathic microcoria. Am J Ophthalmol 106:590, 1988 168. Holth S, Berner O: Congenital miosis or pinhole pupils owing to developmental faults of the
dilator muscle. Br J Ophthalmol 7:401, 1923 169. Jensen OA: Persistent hyperplastic primary vitreous. Acta Ophthalmol 46:418, 1968 170. Pruett RC, Schepens CL: Posterior hyperplastic primary vitreous. Am J Ophthalmol 69:535, 1970 171. Wolter JR, Flaherty NW: Persistent hyperplastic vitreous. Am J Ophthalmol 47:491, 1959 172. Manschot WA: Persistent hyperplastic primary vitreous. Arch Ophthalmol 59:188, 1958 173. Font RL, Yanoff M, Zimmerman LE: Intraocular adipose tissue and persistent hyperplastic primary vitreous. Arch Ophthalmol 82:43, 1969 174. Bullock JO: Developmental vitreous cysts. Arch Ophthalmol 91:83, 1974 175. Francois J: Prepapillary cyst developed from remnants of the hyaloid artery. Br J Ophthalmol 34:365, 1950 176. Apple DJ, Rabb MF, Walsh PM: Congenital anomalies of the optic disc. Surv Ophthalmol 27:3, 1982 177. Nelson LB, Maumenee IH: Ectopia lentis. Surv Ophthalmol 27:143, 1982 178. Frisen L, Holmegaard L: Spectrum of optic nerve hypoplasia. Br J Ophthalmol 62:7, 1978 179. Hoyt CS, Billson FA: Optic nerve hypoplasia: Changing perspectives. Aust NZ J Ophthalmol 14:325, 1986 180. Manshot WA: Eye findings in hydroencephaly. Ophthalmologica 162:151, 1971 181. Ellenberger C Jr, Runyon TE: Holoprosencephaly with hypoplasia of the optic nerves, dwarfism and agenesis
of the septum pellucidum. Am J Ophthalmol 70:960, 1970 182. Hoyt WF, Kaplan SL, Grumbach MM et al: Septo-optic dysplasia and pituitary dwarfism. Lancet 1:893, 1970 183. Harris RJ, Haas L: Septo-optic dysplasia with growth hormone deficiency (de Morsier syndrome). Arch Dis Child 47:973, 1972 184. Edwards WC, Layden WE: Optic nerve hypoplasia. Am J Ophthalmol 70:950, 1970 185. Scheie HG, Adler FH: Aplasia of the optic nerve. Arch Ophthalmol 26:61, 1941 186. Barkovich AJ, Lyon G, Evrard PL: Formation, maturation, and disorders of white matter. AJNR 13:447, 1992 187. Brodsky MC: Septo-optic dysplasia: A reappraisal. Semin Ophthalmol 6:227, 1991 188. Hatten ME, Mason CA: Mechanisms of glial guided neuronal migration in vitro and in vivo. Experientia 46:907, 1990 189. Hoyt CS, Good WV: Do we really understand the difference between optic nerve hypoplasia and
atrophy? Eye 6:210, 1992 190. Skarf B, Hoyt CS: Optic nerve hypoplasia in children. Arch Ophthalmol 102:255, 1984 191. Kindler P: Morning glory syndrome: Unusual congenital optic disk anomaly. Am J Ophthalmol 69:376, 1970 192. Pollock S: The morning glory disc anomaly: Contractile movement, classification, and
embryogenesis. Doc Ophthalmol 65:439, 1987 193. Beyer WB, Quencer RM, Osher RH: Morning glory syndrome: A functional analysis including fluorescein angiography, ultrasonography, and computerized tomography. Ophthalmology 89:1362, 1982 194. Caprioli J, Lesser RL: Basal encephalocele and morning glory syndrome. Br J Ophthalmol 67:349, 1983 195. Goldhammer Y, Smith JL: Optic nerve anomalies in basal encephaloceles. Arch Ophthalmol 93:115, 1975 196. Hope-Ross M, Hohnston SS: The morning glory syndrome associated with sphenethmoidal encephalocele. Ophthalmic Paediatr Genet 2:147, 1990 197. Koenig SP, Naidich TP, Lissner G: The morning glory syndrome associated with sphenoidal encephalocele. Ophthalmology 89:1368, 1982 198. Traboulsi EU, O'Neill JF: The spectrum in the morphology of the so called “morning glory disc
anomaly.” J Pediatr Ophthalmol Strabismus 25:93, 1988 199. Gardner TW, Zaparackas ZG, Naidach TP: Congenital optic nerve colobomas: CT demonstration. J Comput Assist Tomogr 8:95, 1984 200. Mafee MF, Jampol LM, Langer BG, Tso MOM: Computed tomography of optic nerve colobomas, morning glory anomaly, and
colobomatous cyst. Radiol Clin North Am 25:693, 1987 201. Greear JN Jr: Pits or crater like holes in the optic disk. Arch Ophthalmol 28:467, 1942 202. Ferry AP: Macular detachment associated with congenital pit of the optic nerve head. Arch Ophthalmol 70:346, 1963 203. Sugar HA: An explanation for the acquired macular pathology associated with congenital
pits of the optic disc. Am J Ophthalmol 57:833, 1964 204. Gass JD: Serous detachment of the macula secondary to optic pit of the nerve head. Am J Ophthalmol 67:821, 1969 205. Brockhurst R: Optic pits and posterior retinal detachment. Trans Am Ophthalmol Soc 73:264, 1975 206. Kranenburg EW: Craterlike holes in the optic disc and central serous retinopathy. Arch Ophthalmol 64:912, 1960 207. Francois J: Syndrome malformatif avec cryptophthalmic. Acta Genet Med Gemellol 18:18, 1969 208. Sugar HS: The cryptophthalmos-syndactyly syndrome. Am J Ophthalmol 66:897, 1968 209. Waring GO, Shields JA: Partial unilateral cryptophthalmos with syndactyly, brachycephaly and renal
anomalies. Am J Ophthalmol 79:437, 1975 210. Goldberger E: Epicanthus and its variants among Caucasians. Arch Ophthalmol 16:506, 1936 211. Usher CH: The Bowman lecture: On a few hereditary eye affections. Trans Ophthalmol Soc UK 55:164, 1935 212. Berke RN: Congenital ptosis: A classification of two hundred cases. Arch Ophthalmol 41:188, 1949 213. Briggs H: Hereditary congenital ptosis with report of 64 cases conforming to the
mendelian rule of dominance. Am J Ophthalmol 2:408, 1919 214. Roden FH, Barkan H: Hereditary congenital ptosis: Report of a pedigree and review of literature. Am J Ophthalmol 18:213, 1935 215. Rank BK, Thomson JA: The genetic approach to hereditary congenital ptosis. Aust NZ J Surg 28:274, 1959 216. Berke RN, Wadsworth JAC: Histology of levator muscle in congenital and acquired ptosis. Arch Ophthalmol 53:413, 1955 217. Cassidy JV: Developmental anatomy of the nasolacrimal duct. Arch Ophthalmol 47:141, 1952 |