EMBRYOLOGIC DEVELOPMENT AND GLAUCOMA
Traditionally, it has been thought that much of the anterior segment of the globe is derived from mesoderm, the primary component of the middle embryologic layer. Recent studies suggest that the cells of the neural crest, which arise at the margins of the embryologic neural plate just before the closure of the neural tube, contribute large portions of the eye, orbit, and head, thereby functioning as mesoderm in this region.7–17 These studies demonstrate that specific ocular tissues that may be derived from the neural crest include corneal stroma and endothelium, anterior iris stroma, iris melanocytes, ciliary body, sclera, intraocular vascular pericytes, and portions of the trabecular meshwork, including endothelium. The following description of the embryology of the anterior segment is derived from these studies.
The neural crest cells are believed to move into the developing eye in three waves that pass between the surface ectoderm and the lens. The first wave of cells differentiates into the corneal endothelium by the eighth developmental week and produces the trabecular meshwork and its endothelium. The corneal endothelium is responsible for the production of Descemet's membrane. The second wave infiltrates between the corneal epithelium and endothelium to form keratocytes. The third wave produces the iris stroma. Subsequently, the iris pigment epithelium is derived from neural ectoderm. The endothelium of Schlemm's canal and vascular endothelium are derived from vascular mesoderm and not from neural crest.
Neural crest cells also contribute to important structures of the orbit and head, including the meninges, pituitary gland, bones and cartilage of the upper face, dental papillae, and connective tissue supporting cells of the orbit.
The widespread contributions of neural crest-derived cells in the embryology of the eye have given neural crest a central role in the pathogenesis of several forms of glaucoma. Further, abnormalities in neural crest cell migration and differentiation may explain many of the developmental abnormalities of the face and head associated with various glaucoma syndromes. Table 1 attempts to correlate anterior segment disorders with the presumed abnormality in neural crest cell function. This table will be referenced later in this chapter as several glaucoma syndromes are discussed.
TABLE 19-1. Classification of Anterior Segment Disorders Based on Neural
Crest Origins
| Cell Abnormality | Secondary Disorder |
| Deficient neural crest formation (brain-eye-face malformations) | |
| Abnormal crest cell migration | Congenital glaucoma |
| Posterior embryotoxin | |
| Axenfeld's anomaly | |
| Rieger's syndrome | |
| Peters' syndrome | |
| Sclerocornea | |
| Abnormal crest cell proliferation | Iris nevus syndrome |
| Chandler's syndrome | |
| Essential iris atrophy | |
| Abnormal crest cell terminal induction (final differentiation) | Congenital hereditary endothelial dystrophy |
| Posterior polymorphous dystrophy | |
| Fuchs' combined dystrophy | |
| Acquired abnormalities | Metaplasia |
| Abiotrophy | |
| Proliferation |
CONGENITAL GLAUCOMA
The term congenital glaucoma designates glaucoma that is detected in the neonatal and infantile period. The topic has been reviewed extensively by deLuise and Anderson, and their work has been most helpful in the preparation of this section.18 The incidence of congenital glaucoma is approximately 1 in 10,000 births.19 Usually it is sporadic; however, autosomal inheritance has been reported.18,20 It is bilateral in 64% to 88% of cases,21,22 and most affected persons are boys (60% to 70%).18 It is diagnosed within the first year of life in 86% of cases, with 40% diagnosed at birth and 34% between birth and 6 months of age.18
There have been many theories relating to its pathogenesis. The surface of the trabecular meshwork in congenital glaucoma has been described as having a peculiar sheen on gonioscopic examination. This finding has suggested to some authors the presence of an abnormal mesodermally derived membrane, Barkan's membrane, covering the surface of the trabecular meshwork and preventing the efflux of aqueous. The effectiveness of goniotomy surgery and the ineffectiveness of miotics in the treatment of congenital glaucoma have been cited in support of this theory. There is, however, little histologic proof for this theory.
Except in the anterior chamber cleavage syndromes, absence of Schlemm's canal is probably rare as an etiology of congenital glaucoma. Compression and obliteration of Schlemm's canal is most often seen as a secondary finding in advanced cases in which there is marked distortion of normal anterior segment anatomy.
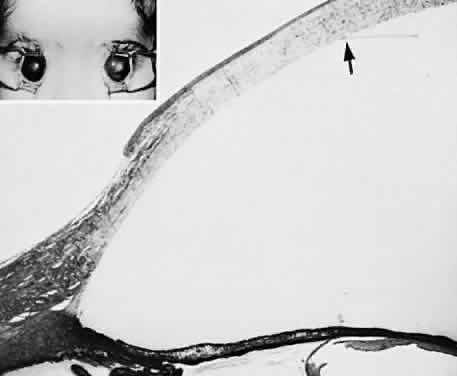
Until recently, the anterior chamber angle was thought to arise through a process of cleavage of mesodermal tissue. Arrest in this process was said to result in an “embryonic” configuration of the anterior chamber angle (Fig. 1). This immature angle is characterized histologically by an anterior insertion of the iris root on the ciliary body, anteriorly displaced ciliary processes, insertion of the ciliary meridional muscles into the trabecular meshwork instead of into (or over) the scleral roll (when more prominent, as in the older adult, the scleral roll is called the scleral spur), and the presence of mesenchymal tissue in the anterior chamber angle.18 More severe abnormalities in this cleavage process were thought to result in the anterior chamber cleavage syndromes: Axenfeld's, Rieger's, and Peters' syndromes.23 Rieger's syndrome may be associated with facial, dental, otologic, dermatologic, and neurologic disorders, including pituitary abnormalities, and also may be associated with trisomy of chromosome 13 or with partial trisomy 16q (see also Chap. 9 for further information on these syndromes). The anterior cleavage syndromes now are recognized as being caused by abnormalities in neural crest development.24
|
Of particular public health importance is the recently recognized association of Peters' syndrome and fetal alcohol syndrome, which is secondary to maternal alcohol ingestion early in the first trimester of pregnancy. It is the most common known cause of mental retardation in the United States.25 Affected infants have a characteristic appearance that includes microcephaly, microphthalmia and other ocular anomalies, a poorly developed philtrum, short palpebral fissures, a thin upper lip, and flattening of the upper maxillary area. Other associated findings are growth retardation and central nervous system and intellectual abnormalities.26,27 Approximately 90% of children with fetal alcohol syndrome have ocular abnormalities,28 which include optic nerve hypoplasia,28–30 tortuosity of retinal blood vessels,28,29 strabismus,28,31 microphthalmos,32 and ptosis.28 Fetal alcohol syndrome also may be associated with DiGeorge syndrome.33
Although an “embryonic” anterior chamber angle is found in congenital glaucoma, it is also seen in age-matched persons who do not have the disorder (see Fig. 1). Further, difficulties in interpreting histologic sections in this age group are compounded by the few truly meridional sections encountered in the usual serial sectioning of the globe. Thus, sectioning artifact enhances the appearance of an “embryonic” configuration in the anterior chamber angle.
The recognition of the contribution of neural crest-derived cells in the embryology of the anterior chamber angle may provide the means for unifying several glaucoma syndromes, particularly those associated with congenital glaucoma.
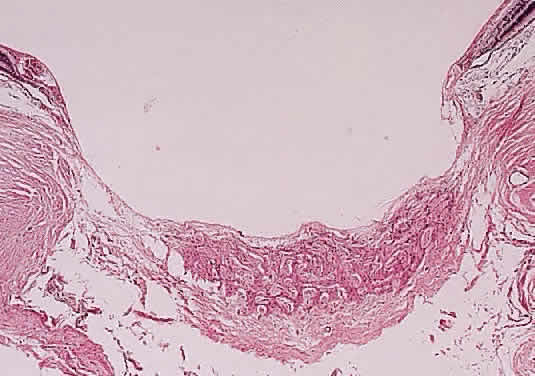
A syndrome of primary glaucoma associated with iridotrabecular dysgenesis and ectropion uveae has been described.34 In these patients, the iris pigment epithelium proliferates onto the anterior iris stroma in utero so that congenital ectropion uveae is present (Fig. 2). Glaucoma may be present in infancy or may develop later in life. Histologically, the central corneal stroma is normal; however, there is posterior embryotoxin (prominence and anterior displacement of Schwalbe's ring) with iris processes. Migration and hyerplasia of the pigment epithelium onto the anterior iris surface is accompanied by endothelialization of the iris surface peripheral to the area where pigment epithelium is present. Angle closure with iris adherent to a normally differentiated trabecular meshwork also is seen. The iris stroma is hypoplastic. The relation of this syndrome to the anterior chamber cleavage syndromes and to the iridocorneal endothelial (ICE) syndrome has not been established. This syndrome may bridge the groups of disorders characterized in Table 1 as secondary to abnormal crest cell migration and proliferation.
|
Congenital ectropion uveae may be found in patients with neurofibromatosis, or it may be associated with other syndromes, including Prader-Willi syndrome.35–39 Table 2 outlines other ocular disorders that may be associated with congenital glaucoma. The possible relation of abnormalities in neural crest cell migration to the developmental disorders in Table 2 remains to be established.
TABLE 19-2. Ocular Disorders Associated With Congenital Glaucoma
Corneal anomalies
Sclerocornea
Microcornea
Megalocornea
Corneal staphyloma
Corneal (ectodermal-mesodermal) dysgenesis
Axenfeld's anomaly
Rieger's syndrome
Peters' syndrome
Iridotrabecular dysgenesis-ectropion uveae syndrome
Microphthalmos
Simple
Associated
Iris anomalies
Aniridia
Colobomata
Polycoria, microcoria
Lens anomalies
Congenital aphakia
Lenticonus
Lentiglobus
Spherophakia
Ectopia lentis
Homocystinuria
Marfan syndrome
Weill-Marchesani syndrome
Ehlers-Danlos syndrome
(Modified from Bardelli AM, Hadjistilianou T: Congenital glaucoma associated with other abnormalities in 150 cases. Glaucoma 9:10, 1987)
Congenital glaucoma is a common feature of several systemic disorders, summarized in Table 3. The multiple ocular anomalies and systemic disorders associated with congenital glaucoma suggest the probability that multiple factors may result in impaired aqueous outflow.
TABLE 19-3. Systemic Disorders Associated With Congenital Glaucoma
Chromosomal defects
10p-
Partial trisomy 16q
Ring chromosome 6
Trisomy 13
Inborn errors of metabolism
Homocystinuria
Hurler's syndrome
Lowe's syndrome
Refsum's syndrome
Infectious
Rubella
Phakomatoses
Neurofibromatosis
Nevus of Ota
Sturge-Weber syndrome
Pluriformative syndromes
Coffin-Siris syndrome
Down's syndrome
Hallermann-Streiff-François syndrome
Marafan syndrome
Meyer-Schwickerath syndrome
Pierre Robin syndrome
Rubenstein-Taybi syndrome
Weill-Marchesani syndrome
(Modified from Bardelli AM, Hadjistilianou T: Congenital glaucoma associated with other abnormalities in 150 cases. Glaucoma 9:10, 1987)
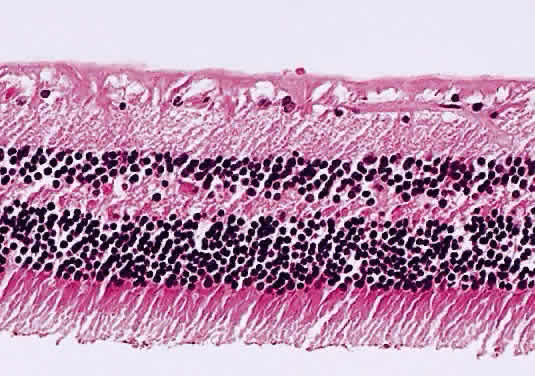
Prolonged elevation of IOP results in characteristic histopathologic changes in the eyes of children 10 years of age or younger. These effects are independent of the specific etiology of the IOP elevation. Ocular stretching, particularly at the corneoscleral limbal region, results in limbal ectasia and the clinical appearance of buphthalmos (Fig. 3). If the thinned and stretched limbus is also lined by uvea (e.g., with peripheral anterior synechiae), a limbal staphyloma is present. When this area extends posteriorly to involve the ciliary body region of sclera, it is called an intercalary staphyloma.
|
The stretching of the entire anterior segment of the globe is also reflected in corneal enlargement. The 95% range of normal corneal diameters is as follows: age 1 month, 9.4 to 11 mm; 6 months, 10.5 to 11.7 mm; 12 months, 10.8 to 12 mm.40 A corneal diameter of 11 mm or less is found in only 24% of patients with primary congenital glaucoma younger than 3 months of age, and in only 9% of those older than 3 months of age so afflicted.41
Progressive corneal enlargement ruptures Descemet's membrane (Haab's striae). These breaks, which are usually horizontal in the central area and concentric toward the limbus, are generally in the lower half of the cornea and are usually associated with corneal edema (see Fig. 3). As in glaucoma in the adult, optic nerve cupping is a characteristic and diagnostic finding in congenital glaucoma. In congenital glaucoma, however, cupping is an early finding and is frequently reversible in its early stages.
Late manifestations of congenital glaucoma include fibrosis of the iris root and trabecular meshwork, disappearance of Schlemm's canal, and generalized atrophy of the ciliary body, choroid, and retina.
PRIMARY GLAUCOMA
Angle-Closure Glaucoma (Narrow Angle; Acute Congestive)
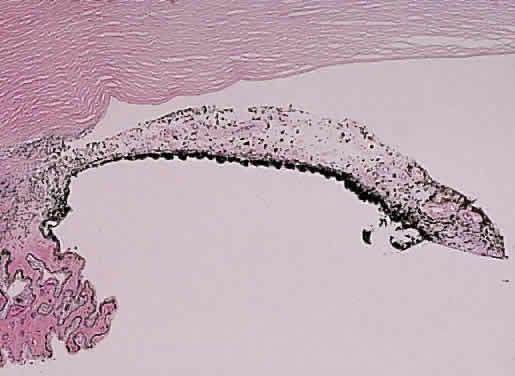
Primary angle-closure glaucoma (PACG) results from an anatomically anomalous anterior segment of the globe (Fig. 4). This configuration predisposes to the apposition of the iris to the surface of the filtering trabecular meshwork. Occlusion of the filtering trabeculum by the iris results in obstruction to aqueous outflow and subsequent IOP elevation, which may be precipitous (Fig. 5). Congdon and associates42 recently extensively reviewed the epidemiology of PACG. The Bedford Glaucoma Survey detected 10 cases of PACG among 5941 persons for an incidence of 0.17%.43 There is, however, considerable racial variation in the incidence of the disorder and its specific manifestations. Acute PACG has been noted to be uncommon among American blacks, in whom subacute or chronic angle-closure glaucoma is more often noted.42 A similar rarity in acute angle closure has been noted in African blacks.44 In contrast, Inuits may have the highest incidence of PACG, with at least 3% of those older than 40 years affected.42,45,46
|
|
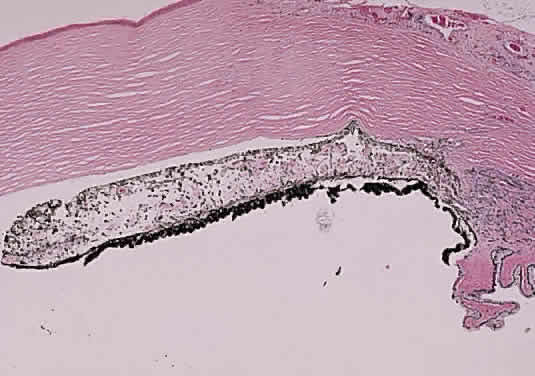
PACG may result from either a classic pupillary block/iris bombé mechanism or from a plateau iris, which is a nonpupillary block mechanism. Eyes that display a pupillary block mechanism have a shallow anterior chamber, increased lens thickness, and decreased axial length.47–52 The resulting congenital anatomic predisposition to angle closure is compounded by further lens thickening resulting from advancing age. In such persons, when the pupil is dilated to the midposition, resistance to the passage of aqueous between the lens and iris results in a ballooning forward of the iris ( bombé), permitting the iris to contact the filtering trabecular meshwork and closing the filtering angle.
In other patients, the anterior chamber is of near-normal depth centrally; however, the peripheral iris has an unusual contour and an extremely anterior insertion on the ciliary body. Dilating the pupil in these patients causes the iris to be drawn up toward the filtering trabeculum, in theory, without pupillary block precipitating the event.53,54 Such patients are said to have a plateau iris configuration to their filtering angle. Recently, the ultrasound biomicroscope has demonstrated the presence of anteriorly placed ciliary processes in patients with plateau iris.55,56
PACG has also been described in association with several syndromes and systemic disorders. Ritch57 has emphasized the significance of angle closure in some patients with pseudoexfoliation (PXE) syndrome, who also may exhibit extremely elevated IOP in the absence of angle closure. An occludable angle has been reported in 9.3% of PXE patients.58 Angle closure has been found in association with childhood cystinosis, in which the typical crystals were noted histologically to thicken the iris and were presumed to precipitate the attack in that manner.59 Iridoschisis60 and Fuchs' dystrophy also are associated with angle closure.61,62
Before an attack of PACG, the trabecular meshwork is normal. After repeated attacks (often at a subclinical level), the anterior chamber angle, although open, may become damaged (sclerosed or fibrotic), simulating chronic simple (open-angle) glaucoma. Similarly, patients with normal-tension (low-tension) glaucoma should be examined carefully to rule out the possibility that subclinical attacks of angle-closure glaucoma have resulted in optic nerve damage, although interval IOPs may return to normal levels.
When IOP rises suddenly, as in acute angle-closure glaucoma, swelling of the iris root and occlusion of the anterior arterial circle of the iris or of one or more of its branches may result in occlusion of the arterial supply to the iris stroma and secondary iris necrosis. Subsequently, segmental iris atrophy, usually in its upper half, may be seen. Extreme atrophy occasionally results in a full-thickness iris hole that may cure the pupillary block. Histologically, there are marked atrophy of the stromal layer of the iris and varying degrees of pigment epithelial atrophy.
Iris sphincter ischemia during the period of acute IOP elevation compounds the attack by fixing the pupil in the mid-dilated position and may render it unresponsive to topical miotics given to break the attack. Later, secondary dilator and sphincter muscle atrophy may result in an irregular pupil. This muscular atrophy or iris necrosis can be demonstrated histologically (Fig. 6). Glaukomflecken are anterior subcapsular, tiny gray-white lens opacities on slit-lamp examination after acute angle-closure glaucoma (see Fig. 6).63 They probably result from interference with lens epithelial cell metabolism, either as a direct pressure effect or through alterations in the normal constituents of the aqueous during the angle-closure attack. Histologically, small areas of epithelial cell necrosis are seen accompanied by tiny areas of adjacent subcapsular cortical degeneration.
|

Visualization of the optic nerve during an attack of angle-closure glaucoma frequently is limited because of corneal edema. If the cornea is cleared with topical glycerol, however, optic disc edema often is present. In experimental acute elevation of IOP, blockage of axonal transport within the optic nerve can be demonstrated, even in the presence of hyperbaric oxygenation. Mechanical compression of axonal fibers with blockade of axoplasmic flow at the level of the lamina cribrosa has been postulated as the cause for this finding, rather than optic nerve head ischemia.
Untreated angle-closure glaucoma results in the formation of peripheral anterior synechiae in which the peripheral iris is permanently adherent to the trabecular meshwork, resulting in chronic angle-closure glaucoma (see Fig. 5) (more information on this topic is given later in this chapter).
The treatment of PACG is to make an opening in the iris, either through a cutting procedure (iridectomy) or with the aid of a laser (iridotomy).64 The goal of these procedures is to create a pressure-relief communication between the anterior and posterior chambers of the eye so as to prevent the buildup of pressure behind the iris, thereby eliminating iris bombé. If peripheral anterior synechiae are forming, or if iridotomy fails to correct the tendency to angle closure, and plateau iris is suspected, laser gonioplasty may be required.65–67
Open-Angle Glaucoma (Chronic Simple Glaucoma; Idiopathic)
Primary open-angle glaucoma (POAG) is a bilateral condition that may be asymmetric both in time of onset and in severity when each eye is compared. By definition, the filtration angle is open to gonioscopic examination. The diagnosis is to some extent one of exclusion because secondary causes must be ruled out before the diagnosis is made. The incidence of the disorder shows wide racial and ethnic variation. Its overall incidence is probably 1% to 3% and increases with increasing patient age: by age 75 years, 5% of the population is affected.68 It is documented to be a more severe problem among American blacks, of whom more than 11% are affected by age 80 years.5 Conversely, it is found less frequently than PACG in Inuits and Asians.
POAG is considered a genetically determined disorder. A positive family history for POAG is found in 50% of patients with POAG.69 The exact mode of inheritance is not definitely established for most glaucomas; however, some juvenile and adult open-angle glaucomas have been mapped to the long arm of chromosome 1.70–72
The nature of the obstruction to aqueous outflow in POAG is not known, although it is commonly believed to be located in the area of the juxtacanalicular connective tissue adjacent to Schlemm's canal.73–78 Unfortunately, few early cases of POAG have been examined histologically. One must be cautious in interpreting specimens from more advanced cases because changes may reflect secondary effects resulting from elevated IOP or from the medication used to treat it. Even greater caution must be used in interpreting specimens removed at surgery for glaucoma: the possibility of surgically induced artifact must be added to “end stage” and previous medical therapy as obfuscating factors. Thus, reports of “sclerosis” of the trabecular meshwork, compression of Schlemm's canal, or changes in the number of macrovacuoles in the endothelial lining of Schlemm's canal may reflect “end stage” changes or artifacts related to fixation.
The relation of changes in the trabecular extracellular matrix (including acid mucopolysaccharides, glycoproteins, glycosaminoglycans, and collagen fibrils) to the pathophysiology of POAG remains to be determined.79–81 A significant decrease in the number of trabecular endothelial cells, when compared to age-matched controls, has been reported in patients with POAG.82,83 This finding can be viewed as an acceleration of the usual age-related decline in the number of trabecular endothelial cells.83,84 Similarly, in POAG, some authors have reported histologic evidence for premature accentuation of the usual age-related development of the scleral spur, characterized by compaction of the uveal meshwork against the scleral roll, “hyalinization” of the adjacent ciliary muscle, and atrophy of the iris root.84 The latter changes are on occasion accompanied by proliferation of the endothelial cells lining Schlemm's canal into its lumen.
In the normal person, an age-related decrease in aqueous facility of outflow is accompanied by a parallel decrease in aqueous production. The net result is a quantitative balance between aqueous production and drainage. POAG can be viewed as an imbalance in the usual parallel relation of these aging changes. From this perspective, it is easy to speculate that POAG itself may really represent a multifactoral problem with a final common pathway of elevated IOP in most patients.
SECONDARY ANGLE-CLOSURE GLAUCOMA
Secondary angle-closure glaucoma (SACG) can be subdivided in a manner similar to PACG as follows:
- SACG with pupillary block
- Untreated PACG
- Phacogenic
- Phacomorphic
- Secondary to lens subluxation or dislocation
- Phacomorphic
- Posterior synechiae induced
- Inflammatory
- Aphakic or pseudophakic pupillary block
- Ciliary block (malignant glaucoma)
- Inflammatory
- Untreated PACG
- SACG without pupillary block
- Secondary to sheetlike cellular or vascular proliferations in the anterior
segment
- Neovascular glaucoma (rubeosis iridis)
- ICE syndrome
- Epithelial downgrowth
- Stromal ingrowth
- Endothelialization of the anterior chamber angle
- Neovascular glaucoma (rubeosis iridis)
- Secondary to anterior displacement of anterior segment structures
- Postoperative failure of formation of the anterior chamber
- Tumor or cyst related
- Retinopathy of prematurity
- Persistent hyperplastic primary vitreous
- Postoperative failure of formation of the anterior chamber
- Miscellaneous
- Iridoschisis
- Iridoschisis
- Secondary to sheetlike cellular or vascular proliferations in the anterior
segment
Untreated PACG
Repeated attacks of PACG may produce peripheral anterior synechiae and SACG. Even without the formation of synechiae, repeated subclinical attacks of PACG may damage the trabecular meshwork, giving rise to a secondary open-angle glaucoma (SOAG). Histologically, peripheral anterior synechiae, which may be broad-based, may be seen (see Fig. 5).
Phacomorphic Phacogenic Glaucoma
Phacomorphic angle-closure glaucoma usually is seen in the final stages of lens cortex liquefaction. Sudden osmotically induced lens swelling displaces the iris anteriorly, inducing pupillary block. To a more modest degree, the lens probably plays a contributory role in inducing pupillary block in most cases of PACG. Occasionally, a more gradual chronic angle closure with peripheral anterior synechiae formation can result from lens-induced anterior displacement of the iris. Depending on the asymmetry of cataract formation in a given patient, there may be a significant difference in the anterior chamber depth when each eye is compared in phacomorphic glaucoma.
Phacomorphic Glaucoma Secondary to Lens Subluxation or Dislocation
Lens subluxation or dislocation may result from several causes. Blunt trauma may rupture zonules. Several syndromes and systemic disorders are associated with abnormal lens mobility. These disorders include Marfan syndrome, homocystinuria, and Weill-Marchesani (microspherophakia) syndrome.85–97 Ectopia lentis is present in up to 56% of patients with aniridia.98 Of particular importance to the clinician is the significant incidence of lens subluxation in PXE syndrome, which was discussed in the section on PACG.
In all of these disorders, an abnormally mobile lens may shift position, block the pupil, and precipitate angle closure. The small lens in the Weill-Marchesani syndrome may dislocate into the anterior chamber, causing an unusual inverse pupillary block configuration.
The clinician may be consulted for fluctuating vision by patients with unusual lens motility. It is particularly important, therefore, to be sensitive to the possibility of homocystinuria, because such patients may die from surgical and anesthetic complications.99–103 The early diagnosis of homocystinuria also is important because treatment with vitamins and dietary measures can be helpful in reducing complications if treatment is instituted early in infancy.104
Recently, genetically determined abnormalities in fibrillin expression and in microfibrillar structure have been found in Marfan syndrome.105–107
Posterior Synechiae-Induced Angle-Closure Glaucoma
Posterior synechiae usually are inflammatory adhesions between the iris and the lens. Less commonly, such adhesions may form between the iris and vitreous face (aphakic pupillary block) or between the iris and an intraocular lens (pseudophakic pupillary block). All such glaucomas result from iris bombé secondary to adhesions preventing the egress of aqueous from the posterior to the anterior chamber. All are treated initially by pupillary dilatation to try to break adhesions, or by laser iridotomy or laser pupillary mydriasis if pharmacologic pupillary dilatation fails. Obviously, pupillary dilatation might be contraindicated in the presence of an iris-plane intraocular lens.
CILIARY BLOCK (MALIGNANT) GLAUCOMA. Ciliary block (malignant) glaucoma is a dreaded complication of cataract extraction, iridectomy, and filtration surgery, particularly that after PACG, although “spontaneous” attacks have been reported.108–110 Its onset may follow shortly after surgery or may lag for many years.111 Malignant glaucoma was extensively reviewed by Luntz and Rosenblatt in 1987,112 by Levene in 1986,113 and by Shaffer and Hoskins in 1978.114 Initially, it was hoped that the avoidance of traditional surgical iridectomy through the use of laser iridotomy would prevent malignant glaucoma associated with PACG. Nevertheless, unilateral and even bilateral attacks have been reported after laser iridotomy.115–117 Malignant glaucoma has resulted from laser suture lysis after filtration surgery118,119 and YAG laser posterior capsulotomy after cataract surgery.120 It is ironic that the latter procedure often is used to treat pseudophakic malignant glaucoma.121–124 Melamed and colleagues125 noted malignant glaucoma after glaucoma seton surgery. An usual form of malignant glaucoma has accompanied keratomycotic infections.126–128
In ciliary block glaucoma, the lens or vitreous face becomes incarcerated in the ciliary ring, forcing misdirection of aqueous into the vitreous compartment. A cycle of increasing compression of the lens or vitreous face into the ciliary ring and anterior iris displacement, followed by increasing misdirection of aqueous into the vitreous compartment, is established.129,130 Ultrasound biomicroscopy has confirmed the pathophysiology of this entity.131–133 Changes or abnormalities in vitreous permeability also may play some role in its pathogenesis.134
Treatment of malignant glaucoma is directed at providing access to the anterior chamber for aqueous trapped behind the lens within the vitreous. Such therapy includes cycloplegia, hyperosmolar agents, and vitreous face disruption. Some laser techniques already have been discussed. Pars plana vitrectomy may be helpful if less invasive measures are unsuccessful.135–137
Neovascular Glaucoma (Rubeosis Iridis)
Neovascular glaucoma is characterized by the proliferation of a fibrovascular membrane on the anterior iris and onto the trabecular meshwork (Fig. 7). In the initial stages, as the sheet of tissue proliferates over the trabecular meshwork surface, neovascular glaucoma is a SOAG. Later, myofibroblasts within this fibrovascular membrane contract, drawing the iris into contact with the trabeculum and closing the filtration angle through the creation of peripheral anterior synechiae.138
|
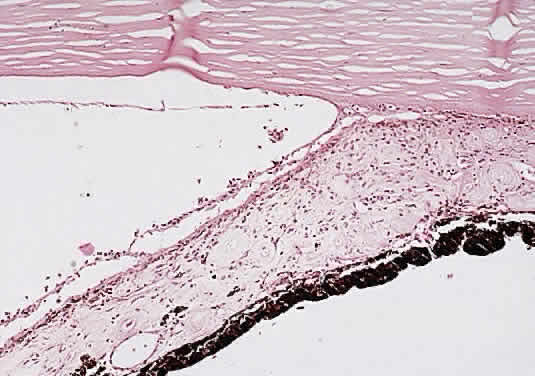
In a review of 208 patients with neovascular glaucoma, Brown and associates139 found retinal venous obstructive disease in 36.1%, diabetic retinopathy in 32.2%, and carotid artery obstructive disease in 12.9%. They concluded that 97% of involved eyes they studied demonstrated extensive retinal ischemia. In such cases, the ischemic retina is believed to produce a vasoproliferative substance that stimulates and supports vascular proliferation. Other causes of neovascular glaucoma include chronic retinal disease, uveal melanoma, longstanding retinal detachment, previous ocular injury, or Fuchs' heterochromic iridocyclitis (FHI).
The fragile vessels produced by neovascularization are prone to bleed, creating a source of spontaneous hyphema and compounding the secondary glaucoma. Because of its association with iris neovascularization and secondary hyphema, retinoblastoma must be excluded in any child with “spontaneous” hyphema.140,141 Other diagnoses to be considered in a child with spontaneous hyphema include child abuse, medulloepithelioma, iris juvenile xanthogranuloma, pseudogliomas, and blood dyscrasia.142–148
Iridocorneal Endothelial Syndrome
ICE syndrome encompasses and unifies the disorders previously categorized as Chandler's syndrome, iris nevus (Cogan-Reese) syndrome, and essential iris atrophy.149–153 Although certain patients may exhibit one or more features of a specific entity, including iris holes, endothelial dystrophy, iris surface nodules, and corectopia,154–156 there is enough overlap between them that they seem to represent different manifestations of the same or very similar developmental abnormalities.149–153 For clarification, each entity will be described in its classic form; however, a given patient may display elements of more than one of these disorders in the same eye.
Table 4 compares Axenfeld-Rieger and ICE syndromes and posterior polymorphous dystrophy. Posterior polymorphous dystrophy may show characteristics similar to the ICE syndrome such as endothelial abnormalities on specular microscopy, corneal edema, iridocorneal adhesions, endothelialization of the anterior chamber angle, and glaucoma. The corneal endothelium in posterior polymorphous dystrophy, however, displays epithelial-like characteristics, in contrast to the ICE syndrome.157 Nevertheless, some authors also have suggested epithelial characteristics for the cells in Chandler's syndrome.158,159 Posterior polymorphous dystrophy also differs from the ICE syndrome in that the former is usually familial, is bilateral, and tends not to be progressive.157
TABLE 19-4. Distinctions Among Axenfeld-Rieger (A-R) and Iridocorneal Endothelial (ICE) Syndromes
and Posterior Polymorphous Dystrophy (PPMD)
| Characteristics | A-R Syndrome | ICE Syndrome | PPMD |
| Laterality | Bilateral | Unilateral | Bilateral |
| Age of presentation | Birth | Young adult | Birth |
| Sex predilection | None | Women | None |
| Familial | Frequently | Rarely | Typically |
| Associated nonocular disorders | Frequent | No | No |
| Corneal edema | No | Frequent | Occasional |
| Corneal endothelium | Normal | Abnormal | Abnormal |
| Origin of membrane | Retention of primordial tissue | Proliferation from abnormal corneal endothelium | Proliferation from abnormal corneal endothelium |
| Mechanism of secondary glaucoma | Maldevelopment of aqueous outflow system | Outflow obstruction by membrane or peripheral synechiae | Either maldevelopment (as in A-R) or membrane induced (as in ICE) |
Table 1 provides a possible differentiation of Axenfeld-Rieger and ICE syndromes and posterior polymorphous dystrophy on the basis of the hypothetical underlying abnormality in neural crest cell embryology or activity. Viewed from this perspective, Axenfeld-Rieger syndrome would be related to abnormal neural crest cell migration, the ICE syndrome to abnormal crest cell proliferation, and posterior polymorphous dystrophy to abnormal crest cell terminal induction or differentiation. Nevertheless, the specific causes for the abnormal behavior of corneal endothelium present in these syndromes have not been established with certainty. Alvarado and associates160–163 have suggested that herpes simplex virus infection plays a causative role in the pathogenesis of the ICE syndrome.
Essential iris atrophy is usually unilateral and most often affects women in the third decade of life. The initial clinical finding is the formation of a peripheral anterior synechia that distorts the pupil in the direction of the synechia. There is progressive pupillary distortion, and ectropion uveae develops. (Ectropion uveae refers to the migration of the posterior pigment epithelium of the iris onto the anterior iris surface, usually as the result of traction from contracting iris surface membranes.) The iris findings may lead to a misdiagnosis of iris melanoma.164
Characteristically, full-thickness iris holes develop in the quadrant opposite the distorted pupil. The etiology of the iris holes is not known. Because they develop in the iris least involved with endothelial proliferation, it is tempting to postulate a “splinting effect” by endothelial proliferation protecting the involved iris from the traction-induced holes seen in the segment of iris not covered by endothelium. A vascular etiology for the iris holes has been suggested and disputed.165–167
Peripheral anterior synechia formation progresses circumferentially, closing the filtration angle and resulting in intractable glaucoma. Iris nodules may appear late in the disorder and are a clinical marker for iris endothelialization. They display a progressive change in color from yellow initially to dark brown when larger.
Histologically, the iris stroma is atrophic. Eventually, full-thickness iris holes involve the pigment epithelium. As in all three of the components of the ICE syndrome, endothelial proliferation is present. In essential iris atrophy, endothelialization of the iris surface and filtration angle is a characteristic finding. The endothelium then deposits a new basement membrane beneath the original endothelium and Descemet's membrane.168
Chandler's syndrome is almost always unilateral. The glaucoma is usually mild; however, poor endothelial function results in corneal edema even with modest pressure elevation. Iris surface abnormalities and peripheral anterior synechiae are least prominent in this syndrome. Stromal thinning is limited and holes are rare. Histologically, endothelialization of the anterior chamber angle and anterior iris may be present.
The iris nevus syndrome, like the other components of the ICE syndrome, most often is unilateral and is most often seen in women. Commonly described features of the iris nevus syndrome include peripheral anterior synechiae and associated iris stromal atrophy, a matted appearance to the iris stroma accompanied by a whorl-like iris surface displaying loss of crypts, and the presence of fine iris nodules.156–170 Iris nodules are not covered by the proliferating endothelium. Other associated findings include ectropion uveae, heterochromia, and secondary glaucoma. Histologically, the iris surface contains a diffuse nevus and is covered by an endothelial proliferation and ectopic Descemet's membrane that also covers the anterior chamber angle.
Epithelial Downgrowth
Epithelial downgrowth is an uncommon complication of anterior segment surgery. It was noted in 0.06% of 45,500 anterior segment surgical procedures reported by Terry and colleagues from the Massachusetts Eye and Ear Infirmary.171 Over the past decade, the incidence of the disorder at that institution was 0.08%, with decreased vision, red eye, and pain being the most common presenting complaints.172 Epithelial downgrowth is probably more common after keratoplasty (0.27%) than after cataract surgery (0.091% to 0.11%).173 It is, however, a dreaded complication with a poor visual prognosis.
Typically, epithelial downgrowth presents as a horizontally oriented area of endothelial haze that progressively migrates inferiorly on the posterior cornea.174 This finding may be accompanied by chronic inflammation and ocular hypotony progressing to intractable glaucoma. Corneal striae and stromal vascularization may be seen overlying the area of corneal involvement. Epithelium may proliferate on the anterior iris before it is visible elsewhere;175 however, eventually iris surface architecture effacement is seen. Histologically, a multilayered epithelial sheet, which often has characteristics suggesting a conjunctival origin, is seen to proliferate on the posterior cornea, angle structures, and iris surface, and it may even involve the vitreous face and ciliary body.176–178
Poor wound apposition, fistulas, postoperative hypotony, and nonspecific postoperative complications such as inflammation and hemorrhage are all thought to contribute to the establishment of an epithelial beach-head within the anterior chamber.171–181 Sassani and associates confirmed earlier work by Cameron and colleagues and others that emphasized the importance of healthy endothelium in inhibiting epithelial migration, thereby acting as a defense against epithelial downgrowth.182–185
There can be several mechanisms for the glaucoma in epithelial downgrowth. Desquamating epithelial cells may plug the trabecular meshwork, resulting in SOAG.171 If the epithelial sheet causes posterior synechiae, pupillary block angle-closure glaucoma can be produced. Ultimately, secondary angle closure without pupillary block results from progressive synechial closure of the filtering angle by the proliferating epithelial sheet.172
Specular microscopy can be of assistance in the diagnosis of epithelial downgrowth,186,187 and laser photocoagulation of the iris surface can be helpful in delineating the extent of epithelial membrane proliferation. Cytologic examination of aqueous or vitreous fluids from aspirates or of scrapings from the corneal endothelial surface or from the iris surface is confirmatory.
The clinical progression of epithelial downgrowth to intractable glaucoma may be gradual although inexorable. The clinical course of the patient must be weighed against the radical therapy usually required to treat the disorder.
Stromal Ingrowth
Stromal ingrowth most frequently is seen after failed corneal transplants;188,189 however, it may follow other surgical procedures, such as cataract surgery, especially if they have been complicated by vitreous loss and incarceration.190,191 Although many of these membranes represent direct proliferation of keratocytes through a dehiscence in Descemet's membrane, some retrocorneal fibrous membranes may represent fibrous metaplasia of corneal endothelium.192,193 Histologically, a fibrous membrane is seen to extend from a corneal wound to proliferate over the posterior cornea. Peripheral anterior synechiae result in SACG.
Endothelialization of the Anterior Chamber Angle
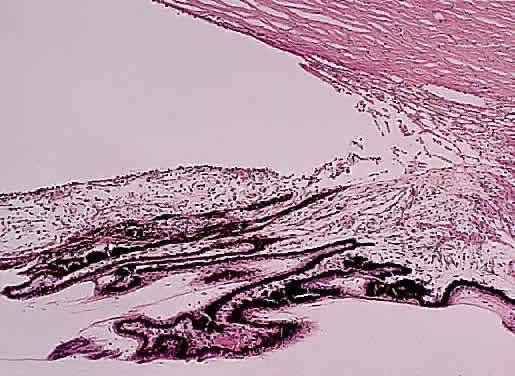
Endothelialization of the anterior chamber angle is seen on histologic examination in 20% of enucleated eyes.194 Endothelial proliferation extends over anterior segment structures, including the iris and the anterior chamber angle (or pseudoangle in the presence of peripheral anterior synechiae, which are noted frequently) (Fig. 8). Endothelialization may be found accompanying rubeosis iridis.195 In three such cases, the endothelial cells exhibited myoblastic features.195
|
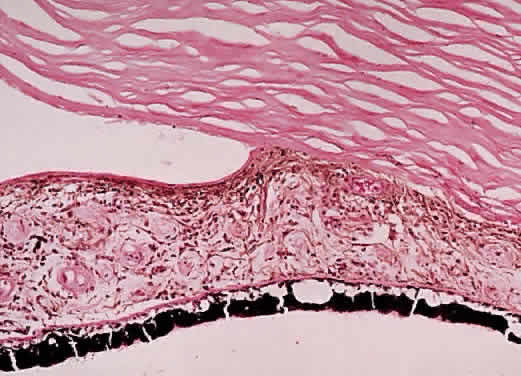
Postoperative Failure of Anterior Chamber Reformation
Postoperative “flat” anterior chamber is seen in various surgical procedures, particularly those performed for glaucoma, in which overfiltration and wound leaks are common causes.196–199 Such overfiltration or wound leaks are particularly likely to occur if adjunctive antimetabolites are used.200–202 The advent of viscoelastic agents and releasable sutures has decreased the frequency of these complications.203–208 Less commonly, malignant glaucoma may simulate simple delayed chamber formation.209–212
Persistent flat anterior chamber may result in peripheral anterior synechiae that may be very broad. Corneal-lens contact usually requires immediate surgical reformation of the anterior chamber to prevent the rapid formation of cataract.213,214 Persistent hypotony secondary to aqueous loss also may lead to choroidal “detachment,” which can further contribute to anterior chamber shallowing.215
Iris and Ciliary Body Cysts
Cysts of the iris and anterior ciliary body, particularly if multiple, may displace the iris lens diaphragm anteriorly, resulting in angle closure.216 Angle closure in the case of multiple cysts may be acute. The cysts may be idiopathic or acquired, such as those associated with trauma or late syphilitic interstitial keratitis.217–219 Unlike acquired cysts, primary cysts tend to be stationary, although important exceptions occur.219,220 The latter lesions also accounted for 38% of lesions misdiagnosed as iris melanoma.164 Late syphilitic interstitial keratitis also may be associated with angle closure secondary to peripheral anterior synechia formation.217,221,222 Recently, the ultrasound biomicroscope has been demonstrated to be useful in the evaluation of cystic lesions of the iris.223
Intraocular Tumors
Intraocular tumors, if large, may displace the iris-lens diaphragm anteriorly in a manner similar to that of iris and ciliary body cysts.224 This displacement is compounded by retinal detachment, if present. Posterior synechiae also may result in pupillary block angle closure. Eventually, peripheral anterior synechiae may form. Large tumors also may be associated with angle closure secondary to rubeosis iridis, or on a phacomorphic basis secondary to cataract formation. Tumors that infiltrate the iris (e.g., diffuse iris melanoma) may close the angle secondary to iris thickening.
Retinopathy of Prematurity and Persistent Hyperplastic Primary Vitreous
Hartnett and associates225 have stated that unsuspected glaucoma may be a cause of decreased vision in severe retinopathy of prematurity. Progressive anterior segment crowding may lead to angle closure in these patients.226 They found peripheral anterior synechiae of at least 180° in 12% of 27 eyes in 17 infants with stage IV or V retinopathy of prematurity.227 Numerous other angle abnormalities also were noted on gonioscopic examination of these infants. Even adults with a history of retinopathy of prematurity should have periodic examinations to exclude the development of additional ocular abnormalities, particularly glaucoma.226
Persistent hyperplastic primary vitreous is a congenital ocular disorder characterized by microphthalmos, cataract, and leukocoria.228 Progressive cataract formation also may contribute to angle closure, and repeated hemorrhages may result in organization and iridocorneal adhesions.
Iridoschisis
Iridoschisis affects persons in the seventh decade. It is bilateral and without sex predilection. Unlike the ICE syndrome, the pupil is not displaced (Fig. 9); rather, a separation develops within the iris stroma that elevates it, particularly in its inferior half. Initially, the stromal fibers maintain their pupillary and peripheral attachments. Later, they become detached at one end, which floats free in the anterior chamber. Eventually, glaucoma develops in 50% of affected patients secondary to peripheral anterior synechia formation. Histologic examination of the involved iris has demonstrated stromal atrophy; however, no vascular or neural abnormalities have been documented.229 Thus, the specific etiology of this disorder is not known. Its relation to PACG was discussed earlier in this chapter.60 It also may be familial, or it may follow trauma, keratoconus, syphilitic interstitial keratitis, or microphthalmos.230–235
|
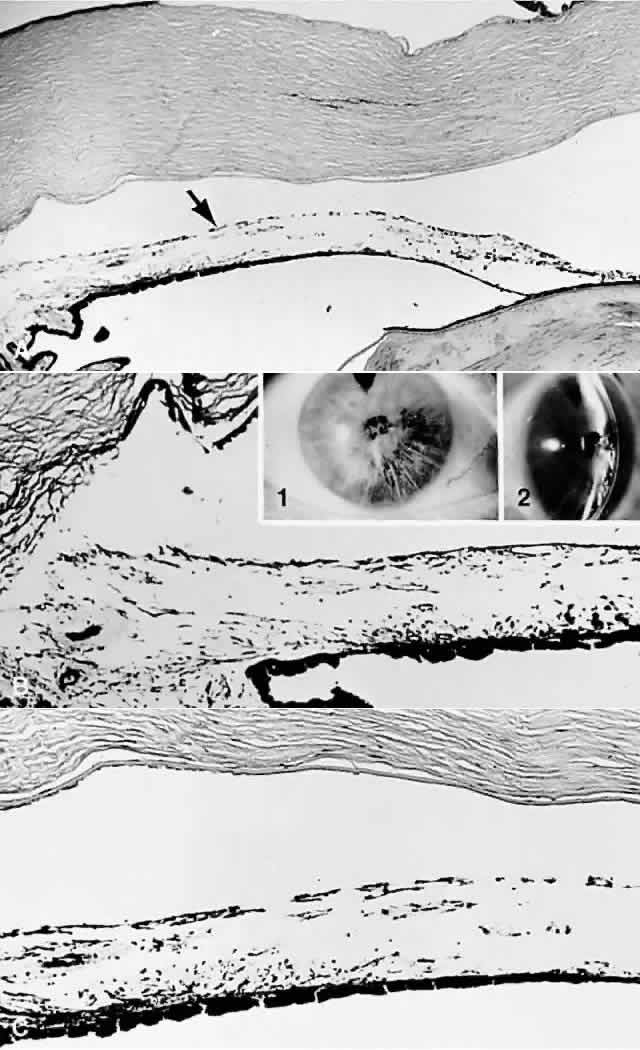
SECONDARY OPEN-ANGLE GLAUCOMA
In SOAG, the angle is open anatomically but is functioning poorly for reasons other than POAG. The classification scheme for SOAG that follows is somewhat arbitrary; nevertheless, it may be helpful to conceptualize these entities, at least in their pure form. It is beyond the scope of this chapter to discuss all the causes of SOAG cited, so only selected topics will be described.
SOAG can be subclassified as follows:
- SOAG caused by cells or debris in the angle
- Hyphema
- Uveitis
- Pigmentary glaucoma
- Pseudoexfoliation
- Hemolytic and “ghost cell” glaucoma
- Phacolytic glaucoma
- Nondenatured lens material-induced glaucoma
- Melanomalytic or melanocytomalytic glaucoma
- Tumor seeding of the trabecular meshwork
- Schwartz-Matsuo syndrome
- Hyphema
- SOAG caused by damaged outflow channels
- Previous uveitis
- Blunt trauma
- Repeated hyphema
- Siderosis and hemosiderosis oculi
- Repeated attacks of acute angle-closure glaucoma
- Early rubeosis or other anterior segment cellular proliferative disorder
- Previous uveitis
- SOAG caused by corneoscleral and extraocular disease
- Interstitial keratitis
- Orbital venous thrombosis
- Encircling element after retinal disease
- Retrobulbar mass
- Leukemia
- Mediastinal mass
- Interstitial keratitis
- SOAG secondary to miscellaneous causes
- Steroid-induced glaucoma
- Alpha-chymotrypsin glaucoma
- Glaucomatocyclitic crisis (Posner-Schlossman syndrome)
- Fuchs' heterochromic iridocyclitis
- Steroid-induced glaucoma
SOAG Caused by Cells or Debris in the Angle
HYPHEMA.
Hyphema refers to blood in the anterior chamber. The most common causes of this condition are anterior segment neovascularization (rubeosis iridis) and ocular trauma. Rubeosis iridis was discussed as a cause of SACG. As in all the syndromes involving cellular proliferations in the anterior segment that may result in SACG, rubeosis iridis, in its early stages, may go through a period of SOAG in which the trabeculum may be coated by the cellular proliferating membrane, which has not yet resulted in synechial angle closure.
One must be extremely cautious when presented with a case of apparently spontaneous hyphema in a child. Most frequently, such cases represent occult trauma, particularly child abuse.148,236 Other important causes of spontaneous hyphema in children include juvenile xanthogranuloma,237 retinoblastoma,140,238,239 and medulloepithelioma.146,240
When the globe is struck forcefully, the cornea is compressed, transmitting the concussive force to the aqueous humor. The shock wave thus generated is propagated through the eye and may give rise to open-angle glaucoma through several mechanisms. The force may cause scarring within the trabecular meshwork, resulting in SOAG. The ciliary body face may be lacerated, tearing the anterior arterial circle and resulting in hyphema. Recession of the anterior chamber angle is a marker for such severe trauma. Initially, the sheer mass of blood may block aqueous outflow through the trabecular meshwork. Later, as the clot undergoes dissolution, red blood cell debris and the macrophages that engulf it also may plug the trabeculum, resulting in hemolytic glaucoma (Fig. 10). In a similar fashion, degenerating “ghost” red blood cells from a vitreous hemorrhage may block the trabeculum after they wash into the anterior chamber through a disrupted vitreous face.241,242 Degenerating hemoglobin releases iron, which acts as an enzymatic poison to the trabecular meshwork, further contributing to the possibility of SOAG. Finally, organization of the hyphema may result in SACG.
|
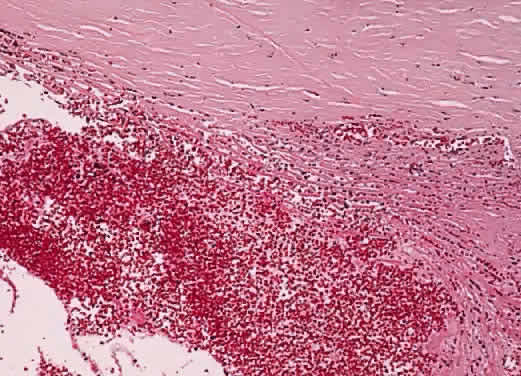
Several days after the hyphema from the initial injury, spontaneous rebleeding may cause the entire cycle to be repeated in the presence of an already compromised aqueous outflow mechanism.243–246 Spontaneous rebleeding may be more common in blacks.247–252 Such rebleeding is associated with a poorer visual prognosis, particularly in children.253 Scarring of the trabeculum may follow recurrent hyphema. Patients who have sickle cell disease may be at particular risk for increased IOP from hyphema because sickled red blood cells pass poorly through the trabecular meshwork.254–256 The damage caused by the increased IOP may be compounded by the tendency for these patients to develop visual loss secondary to ischemic optic neuropathy during such pressure rises.257
Histologically, free blood and clot are seen initially. Later, debris, disrupted red blood cells, and macrophages that may contain hemosiderin and other blood breakdown products are present. In “ghost cell” glaucoma, these more rigid cell remnants appear to obstruct the trabecular meshwork mechanically.241,242
The most severe form of hyphema is the “black ball,” characterized by its darkened color. Histologic examination of the clot from such patients reveals the clots to be composed of concentric layers of fibrin lacking fibroblastic or neovascular proliferations.258
Elevated IOP at the time of hyphema contributes to corneal blood staining; however, this entity has been reported in the presence of relatively low IOP.259 Corneal blood staining represents hemoglobin debris that has traversed the corneal endothelium to reside within the stroma. Histologic examination of involved corneas also has demonstrated intracellular and extracellular hemoglobin particles, keratocyte degeneration, and endothelial cell loss.260,261 Light toxicity exacerbated by the presence of porphyrins has been postulated as contributing to keratocyte and endothelial damage.262,263
UVEITIS. Aqueous production is usually decreased in the presence of iritis, iridocyclitis, or pars planitis. As a result, IOP may be normal or even decreased, even if aqueous outflow is decreased. Later, as aqueous production resumes with appropriate anti-inflammatory therapy, glaucoma may ensue, reflecting damage to the aqueous drainage mechanism.264
There are several mechanisms through which uveitis may lead to glaucoma. SACG resulting from posterior synechia-caused iris bombé was discussed under SACG. Inflammatory cells and debris may plug the trabecular meshwork and trabeculitis may decrease its function, resulting in SOAG. Topical or systemic steroids used in the treatment of uveitis also may cause SOAG in “steroid responders.” Finally, recurrent or chronic inflammation may cause trabecular scarring, thereby decreasing aqueous outflow.264
Two specific “inflammatory” syndromes that result in SOAG will be discussed—glaucomatocyclitic crisis and Fuchs' heterochromic iridocyclitis. SOAG also may result from low-grade chronic anterior uveitis in oculodermal melanocytosis. The melanocytosis itself also may contribute to aqueous outflow obstruction.
Glaucomatocyclitic Crisis (Posner-Schlossman Syndrome). The original designation of this syndrome as the “syndrome of glaucomatocyclitic crises” by Adolph Posner and Abraham Schlossman highlights its characteristic recurrent nature.265,266 It is usually found as a unilateral condition in adults in the third through fifth decades. During acute episodes, patients may experience a rise in IOP to 40 to 60 mmHg. If pressure is sufficiently elevated, halos around light and decreased vision may result from corneal edema. Usually, discomfort is minimal and the involved eye does not appear inflamed on external examination. The pupil often is mildly dilated during the attack. The duration of the episode may be hours to weeks.
A few fine keratic precipitates may be seen initially on slit-lamp examination. Later, the precipitates can resemble mutton fat. Little or no anterior chamber reaction is usually noted, and the angle appears normal on gonioscopic examination. During the acute episode, aqueous outflow facility is decreased and aqueous production has been reported to be elevated. After the acute episode, the IOP may decrease to below normal levels. Some patients have a persistent elevation of IOP and decreased outflow facility. Although optic nerve damage is not characteristic, cupping may occur and filtration surgery is occasionally required. An association between this syndrome and POAG is probable.267,268 A developmental anomaly of the anterior chamber angle and abnormal prostaglandin production have been postulated to be causative for Posner-Schlossman syndrome.269–272 Herpes simplex virus also has been suggested as a factor in the development of the syndrome.273
Light and electron microscopic examination of a trabeculectomy specimen from a patient with recurrent episodes of this disorder revealed numerous mononuclear cells within the trabecular meshwork, as well as erythrocytes.274 The latter were thought to be secondary to the surgery. Giant vacuoles were not seen in the inner wall of Schlemm's canal, even though the preoperative IOP was 60 mmHg. The authors postulated that the macrophages might obstruct aqueous outflow; however, they might also represent a secondary response to the longstanding episode that preceded surgery.
Decreased corneal endothelial cell count has been demonstrated in patients with recurrent attacks of Posner-Schlossman syndrome.275
Fuchs' Heterochromic Iridocyclitis. In 1906, Fuchs276 summarized previously reported cases of heterochromia (difference in iris color between eyes) associated with cataract and reported 38 additional cases. Fuchs' heterochromic iridocyclitis (FHI), the syndrome he defined,277–279 is characterized by iris stromal atrophy, which usually causes the involved iris to be hypochromic relative to the fellow eye. As the condition worsens, increased visibility of the iris pigment epithelium may make the involved iris appear darker. Mild iritis with fine keratic precipitates, iris muscle atrophy, and mydriasis also are seen.
A recent review of 89 eyes of 77 patients noted that the most frequent presenting complaint was decreased vision (96.1%); only 18.2% complained of pain.280 FHI was diagnosed in 50.6% at presentation. Clinical heterochromia was found in 70.1% and was bilateral in 15.6% of patients. Cataract was present in 73%, with 40.4% having already had cataract surgery. Glaucoma was diagnosed in 15.7% at presentation and in 21.3% at review, by which time 9% of the total had required filtration surgery.280
The glaucoma in FHI can be very difficult to control. La Hey and associates281 reported 30 patients with glaucoma (27%) among 111 patients with FHI. Filtration surgery was required in 22 of these 30 patients (73%).
Vascular anomalies of the iris have been noted, including iris perfusion defects and leakage of iris vessels, on fluorescein angiography in FHI.282 Such findings have suggested anterior segment ischemia as one causative factor in FHI.282 Indolent iris neovascularization may be present and may give rise to the filiform hemorrhage seen during cataract surgery in these patients. Such hemorrhage usually is self-limited and of no clinical significance. Iris and filtering angle neovascularization, combined with trabeculitis, is thought to result in the associated glaucoma.283 Histologically, hyalinized blood vessels have been observed in iris specimens from affected patients.284 This finding supports the vascular theory for the origin of this condition. Various inflammatory and immunologic abnormalities also have been detected in iris specimens from patients with FHI.285,286 Abnormal melanocytes with few, small, and immature melanin granules accompanying iris plasma cell infiltration, and abnormal adrenergic innervation were noted on one electron microscopic study of two iris specimens.287
Intractable glaucoma has been reported as a consequence of secondary posterior capsulotomy in one patient with this disorder.288 The eye had been free of inflammation and glaucoma for 27 years before the procedure.
Pigment Dispersion Syndrome (Pigmentary Glaucoma)
The pigment dispersion syndrome (PDS) is found most commonly in young adult male myopes.289,290 It was found in 10% of white and black subjects with and without glaucoma.291 In some black patients, the syndrome may involve older, hyperopic women who may not demonstrate iris transillumination and who tend to have a flatter iris insertion into the ciliary body.292 The syndrome is called pigmentary glaucoma when it is accompanied by glaucoma. Migliazzo and colleagues293 noted progression to glaucoma in 35% of their series of PDS patients with ocular hypertension over a mean follow-up period of 17 years.
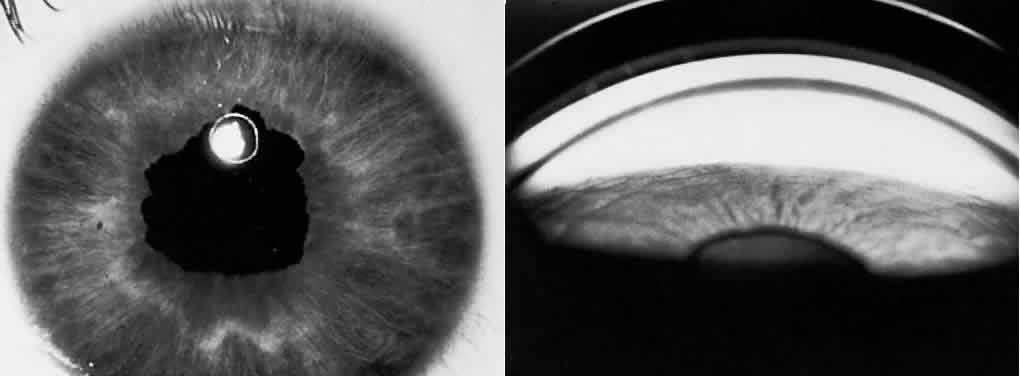
The syndrome is characterized by depigmentation of iris pigment epithelium, producing characteristic iris transillumination, which is demonstrable on slit-lamp examination at the junction of the inner two thirds with the outer one third of the iris (Fig. 11). These areas of depigmentation are accompanied by a band of increased granular iris pigmentation overlying the ring of increased iris transillumination. The finding is most easily seen in blue irises but may be seen fairly easily in brown. This band is presumed to be caused by many pigment-laden macrophages within this area of iris stroma. It has been postulated that the distribution of iris depigmentation corresponds to areas where bundles of zonules chafe the iris pigment epithelium. Iridodonesis may be present. Gonioscopic examination reveals that the trabecular meshwork is heavily pigmented. An increased incidence of retinal detachment is also found in PDS, suggesting the possibility of an underlying pigment epithelial defect.
|
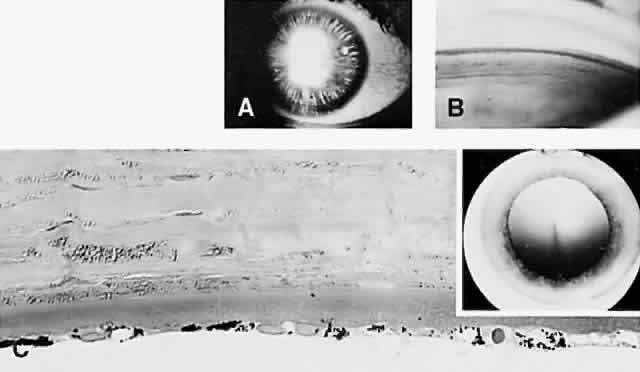
Another typical slit-lamp finding in PDS is the presence of a Krukenberg's spindle, a vertically oriented column of pigment phagocytosed by central and inferior corneal endothelium (see Fig. 11). Pigment may be found within the aqueous after pupillary dilatation or after exercise. Such pigment release may be accompanied by an acute rise in IOP. Significant deposits of pigment also are found on the iris surface, lens, and zonules and within the trabecular meshwork.
The pathophysiology of glaucoma associated with some PDS seems to be related to an “inverse pupillary block” mechanism in which posterior bowing of the iris results in chafing of its posterior surface on bundles of zonules or on ciliary processes, thereby releasing pigment.294–299 However, some have questioned this mechanism.300 The ultrasound biomicroscope has provided strong evidence for this mechanism in a subset of patients who exhibit the characteristic concave iris contour.299–304
Increased IOP in some PDS patients caused by pigment released during exercise is well recognized but not universally accepted.305,306 Posterior iris concavity also tends to increase with exercise in some patients.307 It also may be exacerbated by cataract formation.308 Prophylactic or therapeutic iridotomy has been suggested for patients with posterior iris bowing.297–303 Iris chafing as a cause of pigment dispersion also has been reported in pseudophakia.309–311
Some authors have cited the frequent occurrence of anomalous iris processes in pigmentary glaucoma as evidence that a congenitally anomalous anterior chamber angle is the cause for the glaucoma.299 Ultrasound biomicroscopic examination demonstrated such processes in 75% of untreated PDS patients.299 Other authors believe that the PDS gene may merely be located close to a gene for POAG so that they tend to be inherited together.
Argon laser trabeculoplasty appears to be successful in PDS, with a life-table analysis indicating a success rate of 80% at 1 year, 62% at 2 years, and 45% at 6 years after treatment in one study.312
The incidence of retinal detachment is increased in PDS.313 In one study of 60 PDS patients, lattice retinal degeneration was found in 20%, full-thickness retinal breaks were present in 11.7%, and asymptomatic retinal detachments were found in 3.3% (two patients).314
Histologically, the posterior layer of iris pigment epithelium atrophies in areas that correspond to the foci of iris transillumination.315 The dilator muscle may be atrophic, hypertrophic, or absent. The adjacent iris stroma contains large numbers of pigment-filled macrophages. Neuroepithelial melanin granules are widely distributed within the endothelium of the cornea (corresponding to the Krukenberg's spindle) (see Fig. 11) and within the trabecular meshwork; however, endothelial cell counts have been reported to be similar to those in control patients.316,317
Johnson318 was unable to find differences in trabecular cellularity or morphology in nine normal human eye anterior segments examined for pigment-associated differences in trabecular cellularity or morphology. Epstein and colleagues319 were unable to produce long-term IOP elevation by infusing melanin granules into monkey eyes, and they concluded that factors other than, or in addition to, pigment particle accumulation in the trabecular meshwork must be involved in the mechanism of human pigmentary glaucoma. Similarly, Murphy and associates320 reported that 3.5% of the pigment in PDS was in the juxtacanalicular tissue and 96.5% in the corneoscleral and uveoscleral tissue in the specimens they examined. They concluded that the development of glaucoma in PDS cannot be directly attributed to pigment accumulation in the juxtacanalicular tissue. Clinically, however, increased pigment dispersion, as evidenced by an increase in iris transillumination, an increase in corneal pigmentation, or the appearance of pigment granules on the surface of the lens in the pupil, has been associated with worsening of glaucoma in PDS.321
Alvarado and Murphy322 concluded that trabecular cul-de-sacs provide a major portion of aqueous outflow resistance, and that reduction in the trabecular cul-de-sac area accounts for a major portion of the increase in aqueous outflow resistance in PDS. They also stated that macrophages were largely responsible for clearing the trabecular meshwork of pigment and debris.
Pseudoexfoliation Glaucoma
PXE refers to granular or frostlike material deposited on the lens surface in a characteristic pattern (Fig. 12). The term was first used by Georgiana Dvorak-Theobald323 to distinguish the condition from “true exfoliation,” which is a splitting off of layers of the lens capsule that results most commonly from occupational exposure to infrared irradiation. Typical PXE material has been found in the conjunctiva of patients with “preclinical” PXE324 and in PXE patients.325,326 Similar deposits have been noted in the wall of a short posterior ciliary artery in the orbit,327 in eyelid skin,328 and in other extraocular structures, such as extraocular rectus and oblique muscles, vortex veins, and the optic nerve sheaths, in PXE patients.329,330 Abnormalities of the cornea in PXE have been reported.331 An abnormal blood-ocular barrier in PXE is supported by the findings of increased laser flare-cell measurements in these patients,332 and by the fibrinoid reaction sometimes found in PXE patients after cataract surgery.333,334 The systemic nature of the disorder has been confirmed by the finding of typical material in skin biopsy specimens335 and in the skin, heart, lung, liver, kidney, and meninges at autopsy.336,337
|
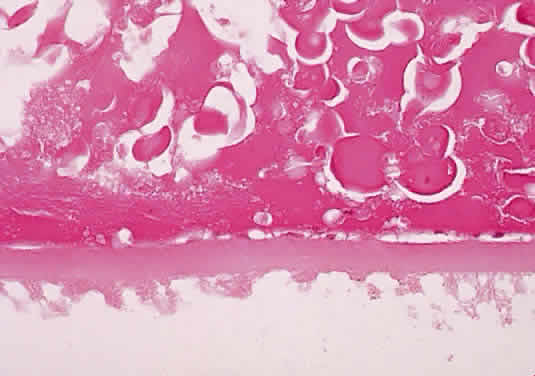
Increased anterior chamber angle pigmentation has been reported in PXE.338 A pigmented line, Sampaolesi's line, is found anterior to Schwalbe's line and may cause errors in estimation of the width of the anterior chamber angle on gonioscopic examination.339 As was stated earlier in this chapter, Ritch and others57,334,340,341 have emphasized the significance of angle closure in some patients with PXE, which also may exhibit extremely elevated IOP in the absence of angle closure. An occludable angle was reported in 9.3% of PXE patients in one study.58
Peripheral and generalized iris transillumination also is typical of PXE.342,343 Generalized iris transillumination has been presented as evidence of vascular insufficiency in these patients.343 Other characteristic clinical findings in PXE are pupillary ruff defects, iris sphincter transillumination, a characteristic whorl-like pattern of particulate pigment deposition on the iris sphincter, particulate pigment deposition on the peripheral iris and trabecular meshwork, and exfoliation material accumulation on the zonules and ciliary body.344,345
There appears to be a considerable racial variation in the incidence of PXE.346–351 Forsius351 reported a variation in prevalence of PXE worldwide of 0% in Eskimos to 21% in Finns over 60 years of age. Aasved352 noted a prevalence of PXE of 4%, 4.7%, and 6.3%, respectively, in Birmingham, Bonn, and Bergen. These differences were not statistically significant. There is an increased incidence of cataract and glaucoma among PXE patients in the Eastern Mediterranean area of Turkey.353 The percentage of PXE patients with glaucoma was 34.3%, and the prevalence of PXE in glaucoma patients was 46.9%. There is a particularly high incidence of PXE in African Bantus, in whom there is a 6.4% prevalence in the age group 30 to 39.354 In contrast, the Framingham Eye Study noted a prevalence of only 0.6% for persons ages 52 to 64.355 The prevalence increased to only 5% in those 75 to 85 years. There appears to be a higher incidence in women. There also is an increased prevalence among Navajo Indians356 and among the male Spanish American population of New Mexico.357 The prevalence of PXE syndrome has been reported to be decreased in some areas of the southeastern United States.358,359 PXE has been reported in association with PDS.360 The prevalence of PXE is decreased in persons with background diabetic retinopathy and is further decreased in those with proliferative diabetic retinopathy.361
Considerable variation in the prevalence of glaucoma in the PXE syndrome also has been reported. Ringvold and associates362 found a 30% prevalence of glaucoma and a 4.2% prevalence of ocular hypertension in those with PXE among 1941 persons older than 64 years. PXE increased in prevalence up to age 75 to 79 years and declined thereafter. Jones and colleagues357 noted that the incidence of glaucoma associated with PXE in New Mexico was 22 times higher. Wollensak and associates363 even questioned whether there is an increased incidence of glaucoma in PXE.
Herry and colleagues364 studied 680 patients with PXE followed from 1940 to 1980. Of these patients, 347 initially had normal IOP. The cumulative probability of an eye with PXE developing elevated IOP was 5.3% in 5 years and increased to 15.4% in 10 years. In a study of 100 consecutive patients in whom PXE was an incidental finding, 78% had normal IOP, 15% were ocular hypertensives, and 7% had glaucoma.365,366 A retrospective study of 206 eyes in 164 patients with PXE found that 65.5% of the eyes that initially were normotensive remained normotensive; however, 34.5% developed ocular hypertension or glaucoma.367
Glaucoma associated with PXE may be more difficult to control and may be associated with more severe optic nerve damage than POAG.352,357,368,369 Henke and Naumann370 also noted an increased prevalence of PXE among 455 enucleated eyes (3.4%), and postulated that PXE may cause increased secondary changes leading to the necessity for enucleation. PXE was found in 87.8% of those undergoing trabeculectomy in a Greek population.371 The severity of glaucoma in PXE may be associated with the patient's blood group.372 Elastosis of the optic nerve head has been reported in PXE compared to POAG.373 One might postulate that such changes could contribute to increased IOP sensitivity in these patients.
The cause of the glaucoma in PXE is not certain. Some authors postulate a plugging of the trabecular meshwork by the PXE material or by pigment released from the iris pigment epithelium.339,374–377 The trabecular meshwork and its endothelium also may undergo secondary and degenerative changes that may compound the simple mechanical effects of the material deposited within it.378,379 It has been suggested that PXE is closely associated with a gene for POAG, and that the PXE is not directly responsible for the glaucoma associated with the disorder.380 Congenital anterior chamber angle anomalies (goniodysgenesis) have been postulated to contribute to the glaucoma in PXE.381
Fragmented zonular fibers have been noted in an electron microscopic study of anterior segments in PXE,382 and increased zonular friability has been reported.383 It is not surprising, therefore, that an increased incidence of cataract surgery complications has been found in PXE.384–387 Naumann385 noted an incidence of vitreous loss of 1.8% in those without PXE and 9% in those with PXE among 1205 cataract extractions. Poor pupillary dilatation also increases the chances of complications during cataract surgery in patients with PXE.385
Morrison and Green378 reviewed the light microscopic findings in PXE and noted that the material is only loosely adherent to most anterior segment structures. It is firmly adherent, however, to the equatorial lens capsule, the posterior epithelium of the iris, and the nonpigmented ciliary epithelium, where the characteristic material (fibrils embedded in an amorphous matrix) is present within epithelial cells and associated with a disorganized, reduplicated basement membrane.378
Streeten and others388–396 have contributed greatly to our knowledge of the composition of the material deposited in PXE and have suggested a similarity to elastic-like material. The fibrils are 20 to 30 nm thick with 10-nm subunits. They may be 800 to 900 nm long and have a characteristic 50-nm periodicity. The material has been produced in cultured tissues from PXE patients.397 The deposition of a precapsular layer composed of microfibrils, amorphous material, and granular inclusions has been cited as a possible precursor to PXE.398
In their review of the light microscopic findings in PXE, Morrison and Green378 noted that glycosaminoglycans, which may comprise the interfibrillar portion of the material, have been found in PXE. They also noted that other studies have demonstrated the similarities between PXE material and zonules, and that the fibrils are related to the microfibrillar portion of elastin. This latter material has the staining properties of oxytalan, the microfibrillar component of elastic tissue.399,400 A similarity to amyloid material also has been postulated by some;401 however, this concept is not widely supported.378 Takei and Mizuno382 proposed that PXE material may represent degenerated zonular fibers. Glycoconjugates of exfoliative material have been characterized.402 Actually, various abnormal materials probably are produced as part of PXE material. Based on immunohistochemical studies, Schlotzer-Schrehardt and colleagues403 have postulated that PXE material is a multicomponent expression of a disordered extracellular matrix synthesis, including the incorporation of the major noncollagenous basement membrane components. Additional histologic findings in this syndrome are discussed in the chapter on the lens.
Phacogenic SOAG
The lens may give rise to SOAG in several ways. Leaking lens protein from a mature lens may incite a marked histiocyte response. This finding was confirmed by Flocks and associates,404 who examined eyes enucleated because of glaucoma associated with hypermature cataracts. They noted that macrophages, swollen with phagocytosed lens material, can plug the trabecular meshwork, resulting in aqueous outflow obstruction. They coined the term “phacolytic glaucoma” to designate the severe rise in IOP that is characteristic of the entity. Similar findings were reported by Yanoff and Scheie405 based on anterior chamber aspirates after needling procedures for soft cataracts. Alternatively, heavy-molecular-weight proteins from a leaking mature lens or fragments of cortical material may themselves block outflow through the trabecular meshwork.406–408 Epstein408 noted that macrophages are not invariably found in phacolytic glaucoma. Both cholesterol crystals and calcium oxalate crystals have been found in phacolytic glaucoma.409,410
SOAG Caused by Uveal Malignant Melanoma
Yanoff224 noted that 20% of 96 eyes containing uveal malignant melanomas presented clinically with glaucoma. Approximately 7% to 10% of enucleated eyes have been reported to contain melanomas that were unsuspected clinically.411–415 Unsuspected melanomas were 10 times more prevalent in persons presenting with glaucoma than in control patients.224 The pathophysiology of the glaucoma included peripheral anterior synechia formation with secondary angle closure, and cellular obstruction of an open anterior chamber angle (Fig. 13). Melanoma should be ruled out in any adult with a blind, painful eye having opaque media.
|
Melanomalytic glaucoma represents mechanical obstruction of the trabecular meshwork by macrophages and debris from a necrotic uveal melanoma.416–418 A similar process has been reported in association with necrotic iris melanocytomas.419 Ring melanoma may result in glaucoma secondary to tumor infiltration of the anterior chamber angle structures (see Fig. 13).420
The mechanisms for angle-closure glaucoma secondary to uveal malignant melanomas were discussed earlier in this chapter. There are several mechanisms for SOAG in these lesions.
Schwartz-Matsuo Syndrome
In general, IOP is lowered in eyes with retinal detachment. Occasionally, however, such eyes present with an acute elevation in IOP, the characteristic finding in Schwartz-Matsuo syndrome.421,422 These cases also typically are accompanied by the presence of cells in the anterior chamber. The cells have been characterized as photoreceptor outer segments.