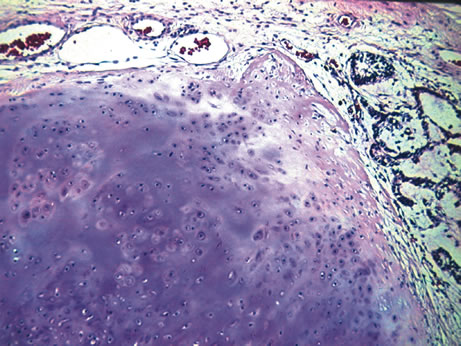
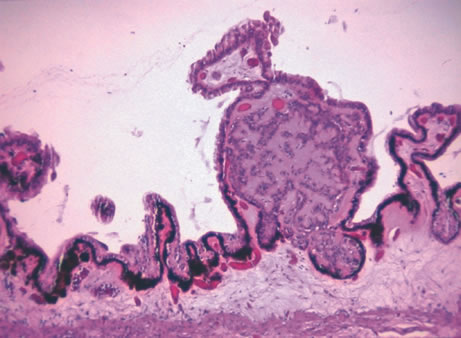
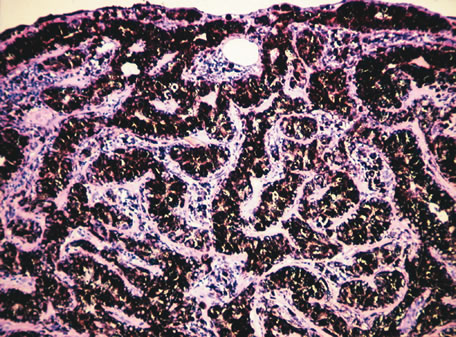
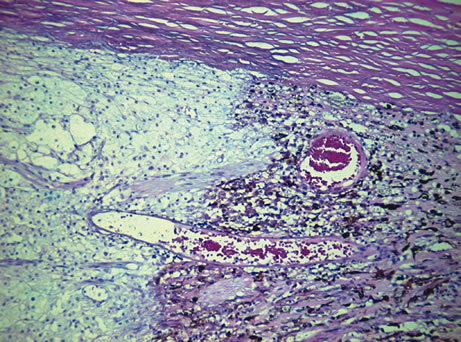
1. Johnston MC, Bhakdinaronk A, Reid YC: An expanded role of the neural crest in oral and pharyngeal development. In Bosma JD (ed): Fourth Symposium on Oral Sensation and Perception: Development in the Fetus
and Infant, pp 37–52. Washington, DC, U.S. Government Printing Office, 1974:37–52. 2. Johnston MC, Noden DM, Hazelton RD et al: Origins of avian ocular and periocular tissues. Exp Eye Res 29:27, 1979. 3. Bahn CF, Falls HF, Varley GA et al: Classification of corneal endothelial disorders based on neural crest origin. Ophthalmology 91:558, 1984. 4. Ozanics V, Jakobiec FA: Prenatal development of the eye and its adnexa. In Duane TD, Jaeger EA (eds): Biomedical Foundations of Ophthalmology, Vol 1, pp 1–23. Philadelphia: Harper & Row, 1987. 5. Tripathi BJ, Tripathi RC: Neural crest origin of human trabecular meshwork and its implications for
the pathogenesis of glaucoma. Am J Ophthalmol 107:671, 1989. 6. Kaiser-Kupfer MI: Neural crest origin of trabecular meshwork cells and other structures of
the anterior chamber. Am J Ophthalmol 107:671, 1989. 7. Beebe DC: Development of the ciliary body: A brief review. Trans Ophthalmol Soc UK 105:123, 1986. 8. Stroeva OG: The role of the lens epithelium in the induction of iris and ciliary body
tissue. Dokl Akad Nauk SSSR 151:464, 1963. 9. Sellheyer K: Spitznas M. Morphology of the developing choroidal vasculature in the human fetus. Graefes Arch Clin Exp Ophthalmol 226:461, 1988. 10. Sellheyer K: Development of the choroid and related structures. Eye 4:255, 1990. 11. Pagon RA: Ocular coloboma. Surv Ophthalmol 25:223, 1981. 12. Mann I: The Development of the Human Eye, pp 1–29. New York, Grune and Stratton, 1928. 13. Pagon RA, Graham JM Jr, Zonana J et al:. Coloboma, congenital heart disease, and choanal atresia with multiple
anomalies: CHARGE association. J Pediatr 99:223, 1981 14. Chestler RJ, France TD: Ocular findings in CHARGE syndrome: Six case reports and a review. Ophthalmology 95:1613, 1988. 15. Font RL, Marines HM, Cartwright J Jr et al: Aicardi syndrome: A clinicopathologic case report including electron microscopic
observations. Ophthalmology 98:1727, 1991. 16. Fine BS, Yanoff M: Ocular Histology: A Text and Atlas, pp 167–212. New York, Harper & Row, 1972. 17. Nelson LB, Spaeth GL, Nowinski TS et al: Aniridia: A review. Surv Ophthalmol 28:621, 1984. 18. Grant WM, Walton DS: Progressive changes in the angle in congenital aniridia with development
of glaucoma. Am J Ophthalmol 78:842, 1974. 19. Miller RW, Fraumeni JF Jr, Manning MD: Association of Wilms' tumor with aniridia, hemihypertrophy and other
congenital malformations. N Engl J Med 270:922, 1964. 20. Shannon RS, Mann JR, Harper E et al: Wilms's tumour and aniridia: Clinical and cytogenetic features. Arch Dis Child 57:685, 1982. 21. Rose EA, Glaser T, Jones C et al: Complete physical map of the WAGR region of 1lp13 localizes a candidate
Wilms' tumor gene. Cell 60:495, 1990. 22. Eagle RC Jr, Shields J: Iridocorneal endothelial syndrome with contralateral guttata endothelial
dystrophy. Ophthalmology 94:862, 1987. 23. Alvarado JA, Murphy CG, Juster RP et al: Pathogenesis of Chandler's syndrome, essential iris atrophy and the
Cogan-Reese syndrome, II: Estimated age at disease onset. Invest Ophthalmol Vis Sci 27:873, 1986. 24. Hirst LW, Quigley HA, Stark WJ et al: Specular microscopy of iridocorneal endothelial syndrome. Am J Ophthalmol 89:11, 1980. 25. Hetherington J Jr: The spectrum of Chandler's syndrome. Trans Am Acad Ophthalmol Otolaryngol 85:240, 1978. 26. Shields JA: Primary cysts of the iris. Trans Am Ophthalmol Soc 79:771, 1981. 27. Spencer WH: Ophthalmic Pathology: An Atlas and Textbook, p 1474. 4th ed. Philadelphia, WB Saunders, 1990. 28. Donaldson DD: The significance of spotting of the iris in Mongoloids. Brushfield spots. Arch Ophthalmol 4:26, 1961. 29. Spencer WH: Ophthalmic Pathology: An Atlas and Textbook, p 1544. 4th ed. Philadelphia, WB Saunders, 1990. 30. Spencer WH: Ophthalmic Pathology: An Atlas and Textbook, p 1612. 4th ed. Philadelphia, WB Saunders, 1990. 31. Duke JR, Dunn SN: Primary tumors of the iris. Arch Ophthalmol 59:204, 1958. 32. Ashton N: Primary tumours of the iris. Br J Ophthalmol 48:650, 1964. 33. Arentsen JJ, Green WR: Melanoma of the iris: Report of 72 cases treated surgically. Ophthalmic Surg 6:23, 1975. 34. Ashton N, Wybar K: Primary tumor of the iris. Ophthalmologica 151:97, 1966. 35. Callender GR, Wilder HC, Ash JE: Five hundred melanomas of the choroid and ciliary body: Followed five years
or longer. Am J Ophthalmol 25:562, 1942. 36. Jakobiec FA, Silbert G: Are most iris “melanomas” really nevi? Arch Ophthalmol 99:2117, 1981. 37. Makley TA Jr: Management of melanomas of the anterior segment. Surv Ophthalmol 19:135, 1974. 38. Rones B, Zimmerman LE: The prognosis of primary tumors of the iris treated by iridectomy. Arch Ophthalmol 60:193, 1958. 39. Stallard HB: Surgery of malignant melanoma of iris. Br J Ophthalmol 35: 774, 1951. 40. Yanoff M, Fine BS: Ocular Pathology. 2nd ed. Philadelphia, Lippincott, 1982. 41. Eoss AJ, Pecorella I, Alexander RA et al: Are most intraocular “leiomyomas” really melanocytic lesions? Ophthalmology 101:919, 1994. 42. Spencer WH: Ophthalmic Pathology: An Atlas and Textbook, p 1672. 4th ed. Philadelphia, WB Saunders, 1990. 43. Spencer WH: Ophthalmic Pathology: An Atlas and Textbook, p 1350. 4th ed. Philadelphia, WB Saunders, 1990. 44. Spencer WH: Ophthalmic Pathology: An Atlas and Textbook, p 32. 4th ed. Philadelphia, WB Saunders, 1990. 45. Shields JA, Kline MW, Augsburger JJ: Primary iris cysts: A review of the literature and report of 62 cases. Br J Ophthalmol 68:152, 1984. 46. Kunimatsu S, Araie M, Ohara K, Hamada C: Ultrasound biomicroscopy of ciliary body cysts. Am J Ophthalmol 127:48, 1999. 47. Bullock JD: Developmental vitreous cysts. Arch Ophthalmol 91:83, 1974. 48. Feman SS, Straatsma BR: Cyst of the posterior vitreous. Arch Ophthalmol 91:328, 1974. 49. Reese AB: Persistence and hyperplasia of primary vitreous: Retrolental fibroplasia—Two
entities. Arch Ophthalmol 41:527, 1949. 50. Reese AB: Persistent hyperplastic primary vitreous. Am J Ophthalmol 40:317, 1955. 51. Manschot WA: Persistent hyperplastic primary vitreous. Arch Ophthalmol 59:188, 1958. 52. Haddad R, Font RL, Reeser F: Persistent hyperplastic primary vitreous. A clinicopathologic study of 62 cases
and review of the literature. Surv Ophthalmol 23:123, 1978. 53. Reese AB, Payne F: Persistence and hyperplasia of the primary vitreous. Am J Ophthalmol 29:1, 1964. 54. Font RL, Yanoff M, Zimmerman LE: Intraocular adipose tissue and persistent hyperplastic primary vitreous. Arch Ophthalmol 82:43, 1969. 55. Cogan DG, Kuwabara T: Ocular pathology of the 13-15 trisomy syndrome. Arch Ophthalmol 72:246, 1964. 56. Miller M, Robbins J, Fishman R et al: A chromosomal anomaly with multiple ocular defects, including retinal dysplasia. Am J Ophthalmol 55:901, 1963. 57. Sergovich F, Madronich JS, Barr ML et al: The D trisomy syndrome: A case report with a description of ocular pathology. Can Med Assoc J 89:151, 1963. 58. Alien RA, Miller DH, Straatsma BR: Cysts of posterior ciliary body (pars plana). Arch Ophthalmol 66:302, 1961. 59. Zimmerman LE, Fine BS: Production of hyaluronic acid by cysts and tumors of the ciliary body. Arch Ophthalmol 72:365, 1964. 60. Gartner J: Fine structure of pars plana cysts. Am J Ophthalmol 73:971, 1972. 61. Baker TR, Spencer WH: Ocular findings in multiple myeloma: A report of two cases. Arch Ophthalmol 91:110, 1974. 62. Johnson BL, Storey JD: Proteinaceous cyst of the ciliary epithelium: I. Their clear nature and
immunoelectrophoretic analysis in case of multiple myeloma. Arch Ophthalmol 84:166, 1970. 63. Johnson BL: Proteinaceous cysts of the ciliary epithelium: II. Their occurrence in
nonmyelomatous hypergammaglobulinemic conditions. Arch Ophthalmol 84:171, 1970. 64. Spencer WH: Ophthalmic Pathology: An Atlas and Textbook, pp 21–24. 4th ed. Philadelphia: WB Saunders, 1990. 65. McDonnell PJ, Green WR, Maumenee AE et al: Pathology of intraocular lenses in 33 eyes examined postmortem. Ophthalmology 90:386, 1983. 66. Crawford JB: A histopathologic study of the position of the Shearing intraocular lens
in the posterior chamber. Am J Ophthalmol 91:458, 1981. 67. Kenyon KR, Pederson JE, Green WR et al: Fibroglial proliferation in pars planitis. Trans Ophthalmol Soc UK 1975;95:391–396 68. Pederson JE, Kenyon KR, Green WR et al: Pathology of pars planitis. Am J Ophthalmol 86:762, 1978. 69. Green WR, Kincaid MC, Michels RG et al: Pars planitis. Trans Ophthalmol Soc UK 101:361, 1981. 70. Prieto JF, Dios C, Gutierrez JM et al: Pars planitis: Epidemiology, treatment, and association with multiple sclerosis. J Ocul Immunol Inflamm 9.2:93, 2001. 71. Zierhut M, Foster CS: Multiple sclerosis, sarcoidosis and other diseases in patients with pars
planitis. J Dev Ophthalmol 23:41, 1992. 72. Takahashi T, Takase H, Urano T et al: Clinical features of human T-lymphocytic virus type I uveitis: a
long term follow-up. J Ocul Immunol Inflamm 8:234, 2000. 73. Breeveld J, Rothova A, Kupier H: Intermediate uveitis and Lyme borreliosis. Br J Ophthalmol 76.3:181, 1992. 74. Mizuno K, Watanabe T: Sarcoid granulomatous cyclitis. Am J Ophthalmol 81:82, 1976. 75. Mizuno K, Takahashi J: Sarcoid cyclitis. Ophthalmology 93:511, 1986. 76. Schachat AP (ed): Retina pp 1758–1759. 3rd ed. St. Louis, Mosby, 2001. 77. Albert DM, Diaz-Rohena R: A historical review of sympathetic ophthalmia and its epidemiology. Surv Ophthalmol 34:1, 1989. 78. Croxatto JO, Rao NA, McLean IW et al:. Atypical histopathologic features in sympathetic ophthalmia: A study of
a hundred cases. Int Ophthalmol 4:129, 1982. 79. Hedges TR III, Albert DM: The progression of the ocular abnormalities of herpes zoster: Histopathologic
observations of nine cases. Ophthalmology 89:165, 1982. 80. Seward DN: Tuberculoma of the ciliary body. Med J Aust 1:297, 1973. 81. Sabates R, Smith T, Apple D: Ocular histopathology in juvenile rheumatoid arthritis. Ann Ophthalmol 11:733, 1979. 82. Merriam JC, Chylack LT Jr, Albert DM: Early-onset pauciarticular juvenile rheumatoid arthritis. A histopathologic
study. Arch Ophthalmol 101:1085, 1983. 83. Zimmerman LE: Verhoeff's “terato-neuroma”: A critical reappraisal
in light of new observations and current concepts in embryonic tumors. Am J Ophthalmol 72:1039, 1971. 84. Broughton WL, Zimmerman LE: A Clinicopathologic study of 56 cases of intraocular medulloepitheliomas. Am J Ophthalmol 85:407, 1978. 85. Bateman JB, Foos RY: Coronal adenomas. Arch Ophthalmol 97:2379, 1979. 86. Brown HH, Glasgow BJ, Foos RY: Ultrastructural and immunohistochemical features of coronal adenomas. Am J Ophthalmol 112:34, 1991. 87. Dryja TP, Zakov ZN, Albert DM: Adenocarcinoma arising from the epithelium of this iris and ciliary body. Int Ophthalmol Clin 20:177, 1980. 88. Bowers JF: Melanocytoma of the ciliary body. Arch Ophthalmol 71:649, 1964. 89. Blodi FC: Leiomyoma of the ciliary body. Am J Ophthalmol 33:939, 1950. 90. Meyer SL, Fine BS, Font RL et al: Leiomyoma of the ciliary body. Electron microscopic verification. Am J Ophthalmol 66:1061, 1968. 91. Jakobiec FA, Font RL, Tso MO et al: Mesectodermal leiomyoma of the ciliary body. A tumor of presumed neural
crest origin. Cancer 39:2102, 1977. 92. Shields JA, Shields CL, Eagle RC Jr et al: Observations on seven cases of intraocular leiomyoma. Arch Ophthalmol 112:521, 1994. 93. Weiss SW, Langloss JM, Enzinger FM: Value of S-100 protein in the diagnosis of soft tissue tumors with
particular reference to benign and malignant Schwann cell tumors. Lab Invest 49:299, 1983. 94. Croxatto JO, Malbran ES: Unusual ciliary body tumor. Mesectodermal leiomyoma. Ophthalmology 89:1208, 1982. 95. Takagi T, Ueno Y, Matsuya N: Mesectodermal leiomyoma of the ciliary body. An ultrastructural study. Arch Ophthalmol 103:1711, 1985. 96. Barr CC, Green WR, Payne JW et al: Intraocular reticulum-cell sarcoma: Clinicopathologic study of four
cases and review of the literature. Surv Ophthalmol 19:224, 1975. 97. Grossniklaus HE, Gass JD: Clinicopathologic correlations of surgically excised Type 1 and Type 2 submacular
choroidal neovascular membranes. Am J Ophthalmol 126:59, 1998. 98. Aicardi J, LeFebvre J, Lerique-Koechlin A: A new syndrome: Spasm in flexion, callosal agenesis, ocular abnormalities. Electroencephalogr Clin Neurophysiol 19:609, 1965. 99. Hughes AE, Luterg AJ, Silvestri G: Five localizations of the gene for central areolar choroidal dystrophy
on chromosome 17p. J Med Genet 135:770, 1998. 100. Hoyng CB, Heutin KP, Testers L et al: Autosomal dominant central areolar choroidal dystrophy caused by a mutation
coder 142 in the periphery/RDS gene. Am J Ophthalmol 124:623, 1996. 101. Ferry AP, Llovera I, Shafer DM: Central areolar choroidal dystrophy. Arch Ophthalmol 88:39, 1972. 102. Krill AE, Archer D: Classification of the choroidal atrophies. Am J Ophthalmol 72:562, 1971. 103. Takki K: Gyrate atrophy of the choroid and retina associated with hyperornithinaemia. Br J Ophthalmol 58:3, 1974. 104. Kaiser-Kupfer MI, Valle D, Del Valle LA: A specific enzyme defect in gyrate atrophy. Am J Ophthalmol 85:200, 1978. 105. Kaiser-Kupfer MI, Caruso RC, Valle D: Gyrate atrophy of the choroid and retina: Long term reduction of orinthine
slows retinal degeneration. Arch Ophthalmol 109:1539, 1991. 106. Wilson DJ, Weleber RG, Green WR: Ocular clinicopathologic studies of gyrate atrophy. Am J Ophthalmol 111:24, 1991. 107. Cameron JD, Fine BS, Shapiro I: Histopathologic observations in choroideremia with emphasis on vascular
changes of the uveal tract. Ophthalmology 94:197, 1987. 108. Hamilton WK, Ewing CC, Ives EJ et al: Sorsby's fundus dystrophy. Ophthalmology 96:1755, 1989. 109. Capon MRC, Marshall J, Kraft JI et al: Sorsby's fundus dystrophy: A light and electrom microscopic study. Ophthalmology 96:1769, 1989. 110. Klein BA: Ischemic infarcts of the choroid (Elschnig spots). A cause of
retinal separation in hypertensive disease with renal insufficiency. A
clinical and histopathologic study. Am J Ophthalmol 66:1069, 1968. 111. Morse PH: Elschnig's spots and hypertensive choroidopathy. Am J Ophthalmol 66:844, 1968. 112. Amalric P: Acute choroidal ischemia. Trans Ophthalmol Soc UK 91:305, 1971. 113. Foulds WS, Lee WR, Taylor WO: Clinical and pathologic aspects of choroidal ischemia. Trans Ophthalmol Soc UK 91:323, 1971. 114. Condon PI, Serjeant GR, Ikeda M: Unusual chorioretinal degeneration in sickle cell disease. Possible sequelae
of posterior ciliary occlusion. Br J Ophthalmol 57:81, 1973. 115. Dizon RV, Jampol LM, Goldberg MF et al: Choroidal occlusive disease in sickle cell hemoglobinopathies. Surv Ophthalmol 23:297, 1979. 116. Fasternberg DM, Fetkenhour CL, Choromokos E et al: Choroidal vascular changes in toxemia of pregnancy. Am J Ophthalmol 89:362, 1980. 117. Folk VC, Weingeist TA: Fundus changes in toxemia. Ophthalmology 88:1173, 1981. 118. Cogan DG: Ocular involvement in disseminated intravascular coagulopathy. Arch Ophthalmol 93:1, 1975. 119. Samples JR, Buettner H: Ocular involvement in disseminated intravascular coagulopathy. Ophthalmology 90:914, 1983. 120. Jakobiec FA, Marboe CC, Knowles DM et al: Human sympathetic ophthalmia: An analysis of the inflammatory infiltrate
by hybridoma-monoclonal antibodies, immunochemistry, and correlative
electron microscopy. Ophthalmology 90:76, 1983. 121. Marak GE Jr: Recent advances in sympathetic ophthalmia. Surv Ophthalmol 24:141, 1979. 122. Rao NA, Robin J, Hartmann D, Sweeney JA et al: The role of the penetrating wound in the development of sympathetic ophthalmia: experimental
observations. Arch Ophthalmol 101:102, 1983. 123. Rao NA, Wacker WB, Marak GE Jr: Experimental allergic uveitis: Clinicopathologic features associated with
varying doses of S antigen. Arch Ophthalmol 97:1954, 1979. 124. Sugita S, Sagawa K, Mochizuki M et al: Melanocyte lysis by cytotoxic T lymphocytes recognizing the MART-1 melanoma
antigen in HLA-A2 patients with Vogt-Koyanagi-Harada
disease. Int Immunol 8:799, 1996. 125. Read RW, Holland GN, Rao NA et al: Revised diagnostic criteria for Vogt-Koyangi-Harada disease: Report
of an international committee on nomenclature. Am J Ophthalmol 131:647, 2001. 126. Rao NA, Hidayat AA: Endogenous mycotic endophthalmitis: Variations in clinical and histopathologic
changes in candidiasis compared with aspergillosis. Am J Ophthalmol 132:244, 2001. 127. Wilder H: Toxoplasma chorioretinitis in adults. Arch Ophthalmol 48:127, 1952. 128. Desmontis G, Remington JS, Couvreur J: Congenital toxoplasmosis. In Stern L, Vert P (eds): Neonatal Medicine, pp 992–678. New York, Masson, 1987. 129. Stagno S, Dykes AC, Amos CS et al: An outbreak of toxoplasmosis linked to cats. Pediatrics 65:706, 1980. 130. Akstein RB, Wilson LA, Teutsch SM: Acquired toxoplasmosis. Ophthalmology 89:1299, 1982. 131. Michelson JB, Shields JA, McDonald R et al: Retinitis secondary to acquired systemic toxoplasmosis with isolation of
parasite. Am J Ophthalmol 86:548, 1978. 132. Hausmann N, Richard G: Acquired ocular toxoplasmosis. Ophthalmology 98:1647, 1991. 133. Rothova A: Ocular involvement in toxoplasmosis. Br J Ophthalmol 77:371–377, 1993. 134. Holland GN, Engstrom RE Jr, Glasgow BJ et al: Ocular toxoplasmosis in patients with acquired immunodeficiency syndrome. Am J Ophthalmol 106:653, 1988. 135. Sternberg P Jr, Knox DL, Finkelstein D et al: Acute retinal necrosis syndrome. Retina 2:145, 1982. 136. Greven CM, Ford J, Stanton C et al: Progressive outer retinal necrosis secondary to varicella zoster virus
in acquired immune deficiency syndrome. Retina 15:14, 1995. 137. Naumann G, Yanoff M, Zimmerman LE: Histogenesis of malignant melanomas of the uvea. Arch Ophthalmol 76:784, 1966. 138. Timmerman LE: Melanocystes, melanocytic nevi and melanocytomas. Invest Ophthalmol 4:11, 1965. 139. Witschel H, Font RL: Hemangioma of the choroid. A clinicopathologic study of 71 cases and a
review of the literature. Surv Ophthalmol 20:415, 1976. 140. Anand R, Augsburger JJ, Shields JA: Circumscribed choroidal hemangiomas. Arch Ophthalmol 107:1338, 1989. 141. Aylword GW, Chang TS, Pautler SE et al: A long-term follow-up of choroidal osteoma. Arch Ophthalmol 116:1337, 1998. 142. Williams AT, Font RL, Van Dyk HJL et al: Osseous choristoma of the choroid simulating a choroidal melanoma. Arch Ophthalmol 96:1875, 1978. 143. Perri P, Pauduano B, Incorvaia C et al: Mesectodermal leiomyoma exclusively involving the posterior choroid. Am J Ophthalmol 134:451, 2002. 144. Harkin JC, Reed RJ: Tumors of the peripheral nervous system. In Armed Forces Institute of Pathology: Atlas of Tumor Pathology. 2nd Series, Fascicle 3. Washington, DC, 1969. 145. Freedman SF, Elner VM, Donev R et al: Intraocular neurilemmoma arising from the posterior ciliary nerve in neurofibromatosis. Ophthalmology 195:1559, 1988. 146. Zimmerman LE: Malignant melanoma. In Spencer WH (ed): Ophthalmic Pathology: An Atlas and Textbook, pp 2072–2141. 3rd ed. Philadelphia, WB Saunders, 1985. 147. Gallagher RP, Elwood JM, Rootman J: Epidemiologic aspects of intraocular malignant melanoma. Cancer Treat Res 43:73, 1988. 148. Cutler SJ, Young JL: Third National Cancer Survey. Incidence Data, pp 1–9. NCI Monograph. Vol 41. Bethesda, National Institutes of Health, 1975. 149. Margo CE, McLean IW: Malignant melanoma of the choroid and ciliary body in black patients. Arch Ophthalmol 102:77, 1984. 150. Scotto J, Fraumenti JF Jr, Lee JA: Melanomas of the eye and other noncutaneous sites: Epidemiologic aspects. J Natl Cancer Inst 56:489, 1976. 151. Gonder JR, Ezell PC, Sheilds, JA et al: Ocular melanocytosis. A study to determine the prevalence rate of ocular
melanocytosis. Ophthalmology 89:950, 1982. 152. Shields JA, Albert DM: Malignant melanoma of the choroid associated with oculodermal melanocytosis. Ophthalmology 88:372, 1981. 153. Gass JD: Observation of suspected choroidal and ciliary body melanomas for evidence
of growth prior to enucleation. Ophthalmology 187:523, 1980. 154. Naumann G, Yanoff M, Zimmerman LE: Histiogenesis of malignant melanomas of the uvea. I. Histopathologic characteristics
of nevi of the choroid and ciliary body. Arch Ophthalmol 1966;76:784–96 155. Yanoff M, Zimmerman LE: Histogenesis of malignant melanomas of the uvea. II. Relationship of uveal
nevi to malignant melanomas. Cancer 20:493, 1967. 156. Nik ND, Glew WB, Zimmerman LE: Malignant melanoma of the choroid in the nevus of Ota of a black patient. Arch Ophthalmol 76:784, 1966. 157. Lynch HT, Anderson DE, Krush AJ: Heredity and intraocular malignant melanoma. Cancer 21:119, 1968. 158. Walker JP, Weiter JJ, Albert DM et al: Uveal malignant melanoma in three generations of the same family. Am J Ophthalmol 88:723, 1979. 159. Prescher G, Bornfeld N, Becher R: Nonrandom chromosomal abnormalities in primary uveal melanoma. J Natl Cancer Inst 82:1765, 1990. 160. Sisley K, Cottam DW, Rennie IG et al: Non-random abnormalities of chromosomes 3, 6 and 8 associated with
posterior uveal melanoma. Genes Chromosomes Cancer 5:197, 1992. 161. Wiltshire RN, Elner VM, Dennis T et al: Cytogenic analysis of posterior uveal melanoma. Cancer Genet Cytogenet 66:47, 1993. 162. Tobal K, Warren W, Cooper CS et al.: Increased expression and mutation of p53 in choroidal melanoma. Br J Cancer 66:900, 1992. 163. Jay M, McCartney AC: Familial malignant melanoma of the uvea and p53: A Victorian detective
story. Surv Ophthalmol 37:457, 1993. 164. Zimmerman LE, McLean IW: Do growth and onset of symptoms of uveal melanomas indicate subclinical
metastasis? Ophthalmology 91:685, 1984. 165. Font RL, Spaulding AG, Zimmerman LE: Diffuse malignant melanoma of the uveal tract. A clinicopathologic report
of 54 cases. Trans Am Acad Ophthalmol Otolaryngol 72:877, 1968. 166. Callender GR: Malignant melanotic tumors of the eye: a study of histologic types in 111 cases. Trans Am Acad Ophthalmol Otolaryngol 36:131, 1931. 167. Burnier MN Jr, McLean IW, Carmel JW: Immunohistochemical evaluation of uveal melanocytic tumors. Expression
of HMB-45, S-100 protein, and neuron-specific enolase. Cancer 68:809, 1991. 168. Iwamoto S, Burrows RC, Kalina RE et al: Immunophenotypic differences between uveal and cutaneous melanomas. Arch Ophthalmol 130:466, 2002. 169. Wilder HC, Paul EV: Malignant melanoma of the choroid and ciliary body: a study of 2,535 cases. Mil Surg 109:370, 1951. 170. McLean IW, Foster WD, Zimmerman, LE: Prognostic factors in small malignant melanomas of choroid and ciliary
body. Arch Ophthalmol 95:48, 1977. 171. Gamel JW, McCurdy JB, McLean IW: A comparison of prognostic covariates for uveal melanoma. Invest Ophthalmol Vis Sci 33:1919, 1992. 172. Hayton S, Lafreniere R, Jerry LM et al: Ocular melanoma in Alberta: A 38 year review pointing to the importance
of tumor size and tumor histology as predictors of survival. J Surg Oncol 42:215, 1989. 173. Shammas HF, Blodi FC: Prognostic factors in choroidal and ciliary body melanomas. Arch Ophthalmol 95:53, 1977. 174. Affeldt JC, Minckler DS, Azen SP et al.: Prognosis in uveal melanoma with extrascleral extension. Arch Ophthalmol 98:1975, 1980. 175. Folberg R, Pe'er J, Gruman LM et al: The morphologic characteristics of tumor blood vessels as a marker of tumor
progression in primary human uveal melanoma: A matched case-control
study. Hum Pathol 23:1298, 1992. 176. Ferry AP, Font RL: Carcinoma metastatic to the eye and orbit. I. A clinicopathologic study
of 227 cases. Arch Ophthalmol 92:276, 1974. 177. Stephens RF, Shields JA: Diagnosis and management of cancer metastatic to the uvea: A study of 70 cases. Ophthalmology 86:1336, 1979. |