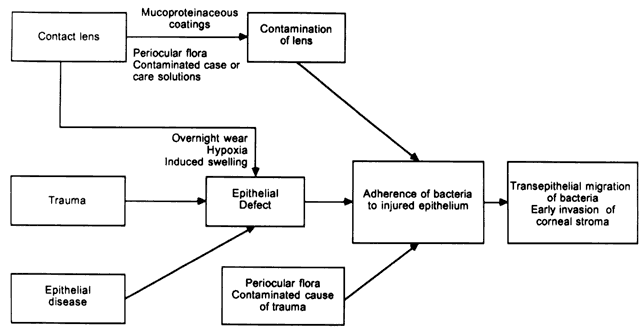
The mechanisms behind the development of epithelial defects in the first two groups is self-evident. In the third group, contact lenses may lead to epithelial injury in a number of ways.10 The cornea can be injured during the process of insertion or removal of the lenses, by trauma from defects in the lenses or deposits on the lenses, by lens-induced hypoxia, or by chemical toxicity from contact lens disinfectants. It is not necessary for defects in the epithelium to be full thickness. Corneas with partial-thickness epithelial defects are more susceptible to adherence by Pseudomonas than are corneas with a fully intact epithelium,11 and extended wear of contact lenses modifies superficial epithelial cells so that these cells are more susceptible to attachment by Pseudomonas.12 Overnight wear of currently available soft lenses, which do not have adequate oxygen transmissibility to prevent hypoxia, causes superficial desquamation of epithelium and a greater propensity for adherence by Pseudomonas.13 Most important, however, is corneal swelling induced by overnight wear of contact lenses. The cornea normally swells 2% to 4% during sleep.14 In the presence of a contact lens, however, overnight swelling is increased to an average of 15%, and gross stromal edema can be present on awakening.14–16 In some patients, induced corneal swelling can be sufficient to cause the development of bullae; these can rupture, leading to the development of epithelial defects. In experimental models of contact lens-associated Pseudomonas keratitis in which there was no corneal trauma, the degree of induced corneal swelling under closed-eye conditions was the most important variable associated with the development of corneal infection.17,18 In fact, in the absence of corneal trauma, prolonged lid closure is required for infection to develop.18 This is the most likely mechanism responsible for the proven association between the overnight wear of contact lenses and the increased risk of bacterial keratitis.19,20
For bacteria to infect a mucosal surface, they must adhere to that surface to resist the natural mechanisms that inhibit bacterial colonization. In the eye, these include the protective effects of the cilia, the mechanical washing effect of the tear film and blinking, the coating of the cornea with ocular mucous,21,22 and the presence of antibacterial substances in the tears, including lysozyme, lactoferrin, betalysin, and IgA antibodies. Bacteria do not adhere well to intact corneal epithelium or to stroma completely denuded of epithelium; they readily adhere to injured or diseased epithelium at the edge of an epithelial defect.23 For this reason, a defect in the epithelium that exposes epithelial receptors is a prerequisite for the development of a bacterial corneal infection.
It is logical, therefore, to assume that the pathogenicity of a bacterial species for the cornea should be related to its ability to adhere to injured corneal epithelium. The most common causes of bacterial corneal ulceration are P. aeruginosa, S. aureus, and S. pneumoniae.1 Reichert and Stern24 demonstrated that these three organisms exhibit a greater degree of in vitro adherence to human corneal epithelium than do other bacteria that are not common corneal pathogens. Panjwani and colleagues25 reached similar conclusions regarding the adherence of S. aureus and P. aeruginosa to rabbit corneal epithelium.
The process of bacterial adherence generally involves a chemical or structural interaction between a component or appendage of the bacterial cell wall, called an adhesin, and a receptor on the epithelial cell or other structure to which the bacteria adhere. For example, teichoic acids, which are essential components of the cell wall of gram-positive cocci, are responsible for adherence of staphylococci and streptococci to epithelial cells.26,27 In the case of gram-negative organisms, filamentous cell wall appendages called fimbriae (commonly called pili, although this term is best reserved for structures involved in the transfer of genetic material) are often the adhesins.
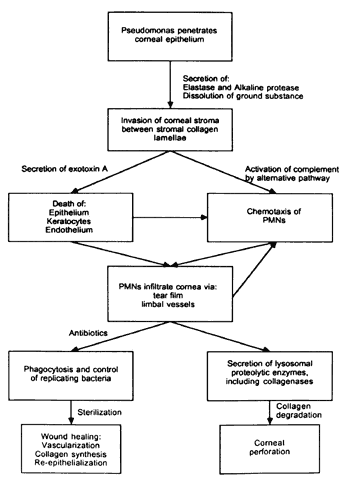
Although adherence of Pseudomonas to injured epithelium is known to be the initial step in the pathogenesis of corneal ulceration, the exact nature of the interaction between bacterial and epithelial cells is complex and incompletely understood. It is likely that fimbriae are important Pseudomonas adhesins, especially when adhering to traumatized corneas.28 Cell surface glycoproteins appear to be the major receptors for Pseudomonas fimbriae, especially those containing moieties of mannose,29–33 N-acetylglucosamine,29 galactose β(1,3) N-acetylgalactosamine,29 and most importantly, sialic (N-acetylneuraminic) acid.29,33–35 Adherence is not completely correlated, however, with the presence of fimbriae: Mutant hyperfimbriated strains have diminished adherence and virulence in comparison to normal strains of Pseudomonas.36 It is possible that adherence is also related to other structures expressed by the rpoN gene, which regulates transcription of mRNA responsible for the synthesis of fimbrial proteins.37 Cell membrane lipopolysaccharides are also Pseudomonas adhesins,28,38 and it is likely that their epithelial receptors are neutral glycosphingolipids, especially asialo-GM1.38,39
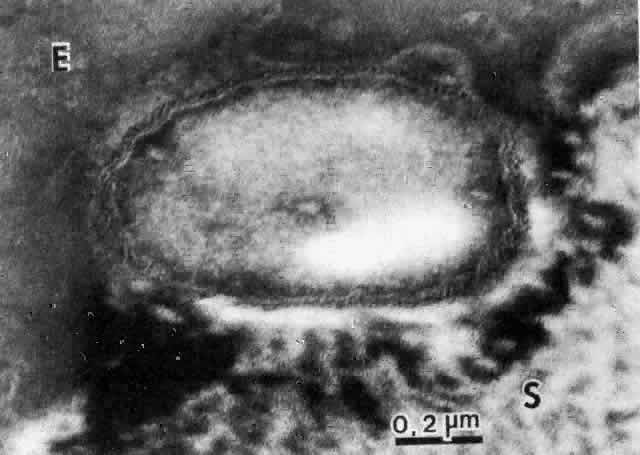
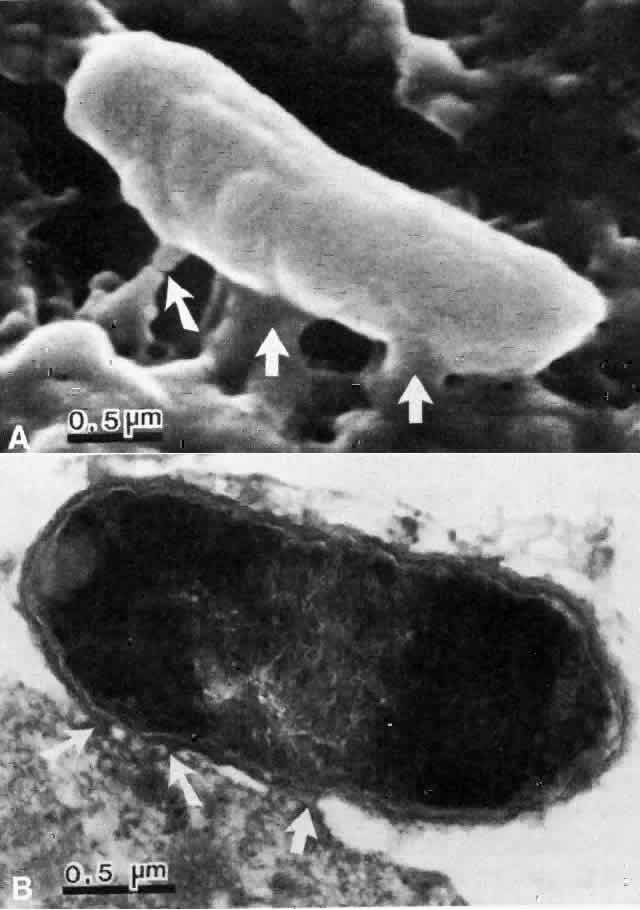
The physical events involved in the adhesion and early penetration of P. aeruginosa to the rabbit cornea have been studied by Stern and co-workers40 using scanning and transmission electron microscopy. Bacteria inoculated onto the surface of a cornea with an epithelial defect adhere to the injured epithelium within 15 minutes. Adherence is the result of an interaction between the bacterial and epithelial cell walls, with fusion of the two structures (Fig. 1). In some cases, fimbriae or other filamentous bacterial appendages are the adhesive structures. Within the subsequent 15 minutes, excavations in the epithelial cells appear to form around the bacteria. It is unknown whether this is the result of bacterial enzymes or an active process in which bacteria are engulfed into the epithelial cell. Forty-five minutes after inoculation, these excavations begin to fill in with the bacteria disappearing into the substance of the epithelial cell. One hour after inoculation, bacteria are no longer found on the epithelial surface. Instead, they are seen within epithelial cells or migrating through epithelial cells into the anterior stroma, and they are replicating at this stage (Fig. 2). Pseudomonas, therefore, appears to reach the anterior stroma by a process of adhesion and transepithelial migration. The invasion of epithelial cells by Pseudomonas renders the bacteria transiently resistant to host defenses and the effects of topical antibiotics.41 Entry of Pseudomonas into the epithelial cell involves active metabolic processes (e.g., tyrosine kinase and cellular actin microfilaments) and may be mediated by binding to integrins.42 The entire process of adherence and early stromal penetration in the experimental rabbit eye occurs in only 1 hour.
|
|