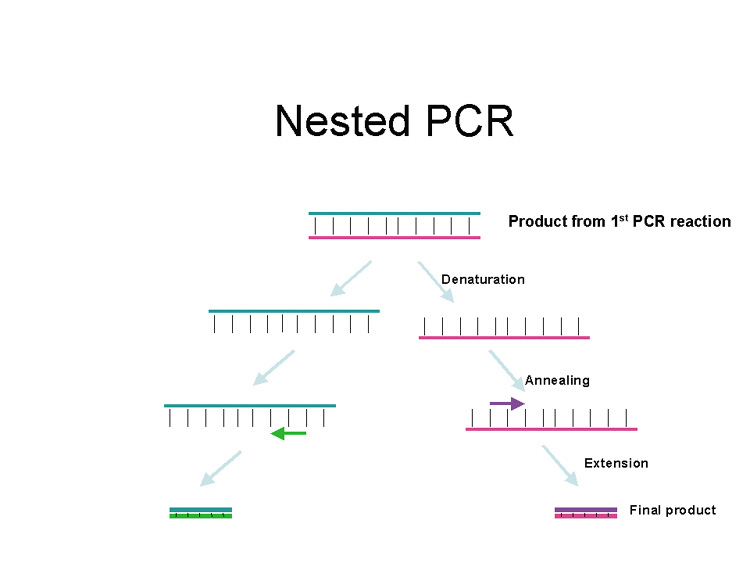
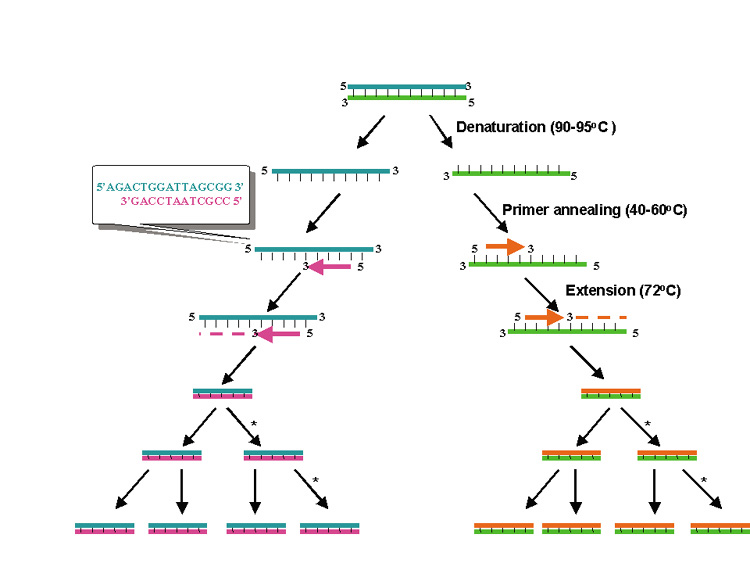
PCR is essentially an in vitro replication of bacterial DNA replication, stripped of all inessential components (Fig. 1). The necessary ingredients are the DNA template (i.e., from a pathogen residing in a biopsy specimen), a pair of 20 to 30 nucleotide oligonucleotide primers specific for the targeted DNA sequence (i.e., a sequence from a viral genome), nucleotide triphosphates (the building blocks for DNA), a thermostable DNA polymerase to catalyze replication, appropriate salts and buffers, and a thermal cycling machine. Once combined, the thermal cycler creates conditions for the three steps of PCR. Each step usually takes between 30 seconds and 2 minutes. The process begins with the denaturation step (typically at approximately 94°C), which separates the double-stranded template DNA into single strands. The temperature is then lowered to the annealing temperature (specific to each pair of primers, typically 45°C to 65°C), which allows the primers to optimally bind to their target sequences. After annealing, the temperature is elevated to an optimal temperature for thermostable DNA polymerase activity (typically between 72°C and 74°C). DNA polymerases capable of functioning at such high temperatures are isolated from thermophilic bacteria (such as those living in geothermal pools). At this temperature, the polymerase catalyzes synthesis of DNA complementary to the template at a rate of approximately 1000 bases per minute; thus a 500 base pair (bp) target may require 30 seconds of extension time. After extension, the temperature is again raised to denature the template DNA from the newly formed complementary strand of DNA. Thus, at the end of the first cycle, the number of target DNA strands has doubled. Both the original template and the new complement strand can then become templates for another round of amplification. PCR is an exponential process; theoretically, after 35 cycles (standard for many protocols), more than 35 billion copies of the original template DNA will have been synthesized. Starting with a single virion (whose DNA weighs 1.6 × 10−16 g), after 35 cycles nearly 20 ng (2.0 × 10−8 g) of 500 bp product will be produced. This quantity of DNA is adequate for further analysis.
|
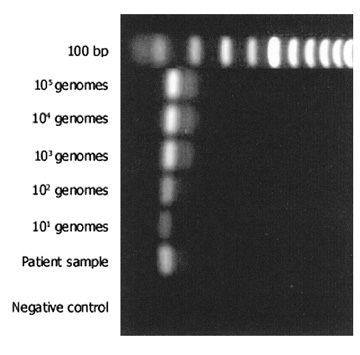
The most commonly used method for detection of the PCR product is electrophoresis in an ethidium bromide-stained agarose gel (Fig. 2). The limit of detection in such a gel is approximately 10 ng of double-stranded DNA. Given the calculations above, PCR should theoretically be able to detect a single genome in a given sample. However, because of imperfect amplification efficiency, maximal sensitivity is generally on the order of 10 genomes. PCR products may also be detected in other ways, using more sensitive dyes such as fluorescent SYBR Green®. This dye may be used in a special variant of PCR (real-time PCR), which allows quantification of product (see later). Sensitivity for detection of PCR products can be improved by use of radioactivity and autoradiography, but these techniques are seldom applied in clinical practice.
|
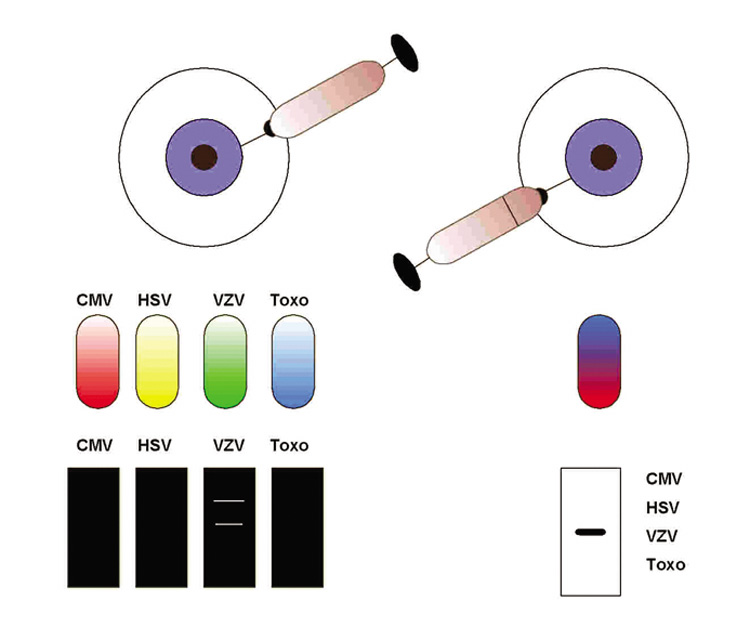
Although detection of a band of the correct size on electrophoresis is generally considered indicative of a positive result, PCR products may be more specifically identified in a number of ways. Restriction endonucleases can digest the PCR product into sequence-specific fragments, the pattern of which, on electrophoresis, forms a distinct fingerprint. Alternatively, hybridization methods allow identification by binding the PCR product to a nylon or nitrocellulose filter paper and then incubating the nylon with a labeled DNA or RNA probe that binds to a specific DNA sequence. The gold standard for demonstration of a specific identity for a PCR product is DNA sequencing, which can be performed either directly from the PCR product or after cloning of that fragment into a plasmid vector.