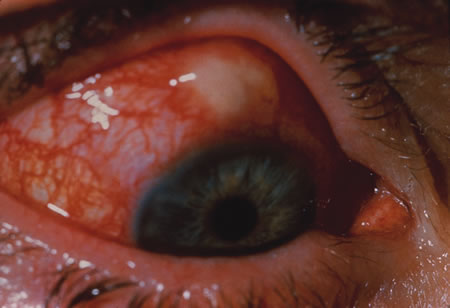
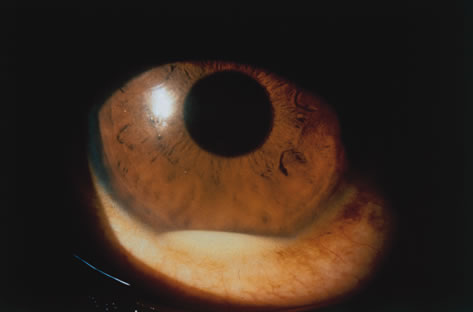
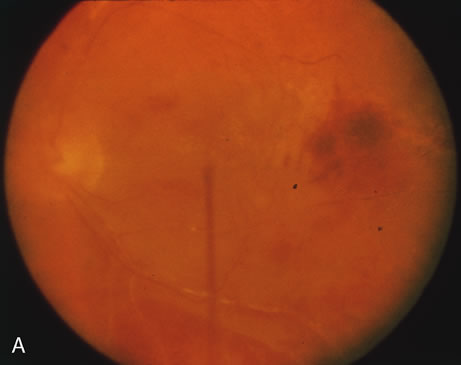
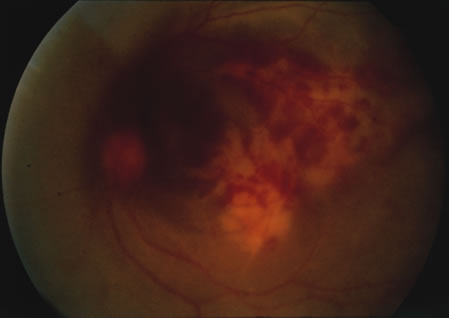
1. Henkind P, Gold DH: Ocular manifestations of rheumatic disorders: Natural and iatrogenic. Rheumatology 4:13, 1973 2. Jabs DA: Rheumatic diseases. In Ryan S (ed): The Retina, 3rd edition, Vol. II, pp. 1410–1433. St. Louis, Mosby, 2001 3. Hochberg MC: Adult and juvenile rheumatoid arthritis: Current epidemiologic concepts. Epidemiol Rev 3:27, 1981 4. Gabriel SE, Crowson CS, O'Fallon W: The epidemiology of rheumatoid arthritis in Rochester, MN, 1955–1985. Arthritis Rheum 42:415, 1999 5. Drosos AA, Alamanos I, Voulgari PV, et al: Epidemiology of adult rheumatoid arthritis in northwest Greece 1987–1995. J Rheumatol 24:2129, 1997 6. Doran MF, Pond GR, Crowson CS, et al: Trends in incidence and mortality in rheumatoid arthritis in Rochester, Minnesota, over
a forty-year period. Arthritis Rheum 46:625, 2002 7. Harris ED: Clinical features of rheumatoid arthritis. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 967–1000. Philadelphia, WB Saunders, 2001 8. Arnett FC, Edworthy SM, Block DA, et al: The American Rheumatism Association 1987 revised criteria for the classification
of rheumatoid arthritis. Arthritis Rheum 31:315, 1988 9. Zvaifler NJ: Immunopathology of joint inflammation in rheumatoid arthritis. Adv Immunol 16:265, 1973 10. Decker JL, Malone DG, Haraqui B, et al: Rheumatoid arthritis: Evolving concepts of pathogenesis and treatment. Ann Intern Med 101:810, 1984 11. Smith JB, Haynes MK: Rheumatoid arthritis: A molecular understanding. Ann Intern Med 136:908, 2002 12. Firestein GS: Etiology and pathogenesis of rheumatoid arthritis. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 921–966. Philadelphia, WB Saunders, 2001 13. Masin AT, Maldonado-Cocco JA, Kaplan SB, et al: Prospective study of the early course of rheumatoid arthritis in young
adults: Comparison of patients with and without rheumatoid factor positivity
at entry and identification of variables correlating with outcome. Semin Arthritis Rheum 4:299, 1976 14. McDermot EM, McDevitt H: The immunogenetics of rheumatic diseases. Bull Rheum Dis 38:1, 1988 15. Stasny P: Association of the B-cell alloantigen Drw4 with rheumatoid arthritis. N Engl J Med 298:869, 1978 16. Hurd ER: Extra-articular manifestations of rheumatoid arthritis. Semin Arthritis Rheum 8:151, 1979 17. Hedfors E, Klareskog L, Lindblad S, et al: Phenotypic characterization of cells within subcutaneous rheumatoid nodules. Arthritis Rheum 26:1333, 1983 18. MacDonald WJ Jr, Crawford MH, Klippel JH, et al: Echocardiographic assessment of cardiac structure and function in patients
with rheumatoid arthritis. Am J Med 63:890, 1977 19. Schneider HA, Yonker RA, Katz P, et al: Rheumatoid vasculitis: Experience with 13 patients and review of literature. Semin Arthritis Rheum 14:280, 1985 20. Scott DGI, Bacon PA, Tribe CR: Systemic rheumatoid vasculitis. A clinical and laboratory study of 50 cases. Medicine 60:288, 1981 21. Goldberg J, Pinals RS: Felty syndrome. Semin Arthritis Rheum 10:52, 1980 22. Harris ED: Treatment of rheumatoid arthritis. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1001–1022. Philadelphia, WB Saunders, 2001 23. American College of Rheumatology Subcommittee on Rheumatoid Arthritis Guidelines: Guidelines
for the management of rheumatoid arthritis: 2002 update. Arthritis Rheum 46:328, 2002 24. Sharp JT, Wolfe F, Mitchell DM, et al: The progression of erosions and joint space narrowing scores in rheumatoid
arthritis during the first twenty-five years of disease. Arthritis Rheum 34:660, 1991 25. Plant MJ, Jones PW, Saklatvala J: Patterns of radiological profession in early rheumatoid arthritis: Results
of an 8 year prospective study. J Rheumatol 25:417, 1998 26. Clark P, Casas E, Tugwell P, et al: Hydroxychloroquine compared with placebo in rheumatoid arthritis: A randomized
controlled trial. Ann Intern Med 119:1067, 1993 27. Esdaile JM, Suissa S, Shiroky JB, et al: A randomized trial of hydroxychloroquine in early rheumatoid arthritis: the
HERA study. Am J Med 98:156, 1995 28. Tsakonas E, Fitzgerald AA, Fitzcharles MA, et al: Consequences of delayed therapy with second-line agents in rheumatoid
arthritis: A 3-year follow-up on the Hydroxychloroquine
in Early Rheumatoid Arthritis (HERA) study. J Rheumatol 27:623, 2000 29. Williams HJ, Ward JR, Dahl SL, et al: A controlled trial comparing sulfasalazine, gold sodium thiomalate, and
placebo in rheumatoid arthritis. Arthritis Rheum 31:702, 1988 30. Hannonen P, Möttönen T, Hakola M, et al: Sulfasalazine in early rheumatoid arthritis: a 48-week double-blind, prospective, placebo-controlled study. Arthritis Rheum 36:1501, 1993 31. Weinblatt ME, Reda D, Henderson W, et al: Sulfasalazine treatment for rheumatoid arthritis: A metaanalysis of 15 randomized
trials. J Rheumatol 26:2123, 1999 32. Landewe RBM, Goei Thè HS, van Rijthoven AW: A randomized, double-blind, 24-week controlled study of low-dose
cyclosporine versus chloroquine for early rheumatoid arthritis. Arthritis Rheum 37:637, 1994 33. Williams HJ, Wilkens RF, Samuelson CO, et al: Comparison of low-dose oral pulse methotrexate and placebo in the
treatment of rheumatoid arthritis: A controlled clinical trial. Arthritis Rheum 28:721, 1985 34. Weinblatt ME, Coblyn JS, Fox DA, et al: Efficacy of low-dose methotrexate in rheumatoid arthritis. N Engl J Med 312:818, 1985 35. Suarez-Almazor ME, Belseck E, Shea B, et al: Methotrexate for treating rheumatoid arthritis (Cochrane Review). In: The Cochrane Library. Issue 4. Oxford, Update Software, 2001 36. Kremer JM: Safety, efficacy, and mortality in a long-term cohort of patients
with rheumatoid arthritis taking methotrexate: Follow-up after
a mean of 13.3 years. Arthritis Rheum 40:984, 1997 37. Strand V, Cohen S, Schiff M, et al: Treatment of active rheumatoid arthritis with leflunomide compared with
placebo and methotrexate. Arch Intern Med 159:2542, 1999 38. Sharp JT, Strand V, Leung H, et al: Treatment with leflunomide slows radiographic progression of rheumatoid
arthritis: Results from three randomized controlled trials of leflunomide
in patients with active rheumatoid arthritis. Arthritis Rheum 43:495, 2000 39. Kremer JM, Genovese MC, Cannon GW, et al: Concomitant leflunomide therapy in patients with active rheumatoid arthritis
despite stable doses of methotrexate. Ann Intern Med 137:726, 2002 40. O'Dell JR, Haire CE, Erikson N, et al: Treatment of rheumatoid arthritis with methotrexate alone, sulfasalazine
and hydroxychloroquine, or a combination of all three medications. N Engl J Med 334:1287, 1996 41. O'Dell JR, Leff R, Paulsen G, et al: Treatment of rheumatoid arthritis with methotrexate and hydroxychloroquine, methotrexate
and sulfasalazine, or a combination of the three medications: Results
of a two year, randomized, double-blind, placebo-controlled
trial. Arthritis Rheum 46:1164, 2002 42. Moreland LW, Schiff MH, Baumgartner SW, et al: Etanercept therapy in rheumatoid arthritis: A randomized controlled trial. Ann Intern Med 130:478, 1999 43. Bathon JM, Martin RW, Fleischmann RM, et al: A comparison of etanercept and methotrexate in patients with early rheumatoid
arthritis. N Engl J Med 343:1586, 2000 44. Lipsky PE, van der Heijde DM, St. Clair EW, et al: Infliximab and methotrexate in the treatment of rheumatoid arthritis. N Engl J Med 343:1594, 2000 45. Maini RN, Breedveld FC, Kalden JR, et al: Therapeutic efficacy of multiple intravenous infusions of anti-tumor
necrosis factor α monoclonal antibody combined with low-dose weekly methotrexate
in rheumatoid arthritis. Arthritis Rheum 41:1552, 1998 46. Cohen S, Hurd E, Cush J, et al: Treatment of rheumatoid arthritis with anakinra, a recombinant human interleukin-1 receptor antagonist, in combination with methotrexate. Results
of a twenty-four-week, multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum 46:614, 2002 47. Woodland J, Chaput de Saintongue DM, Evans SJ, et al: Azathioprine in rheumatoid arthritis: Double-blind study of full
versus half doses versus placebo. Ann Rheum Dis 40:355, 1981 48. Csuka ME, Carrera FG, McCarty DJ: Treatment of intractable rheumatoid arthritis with combined cyclophosphamide, azathioprine, and
hydroxychloroquine: A follow-up study. JAMA 255:2315, 1986 49. Van Rijthoven AW, Dijkmans BA, Goei Thè HS, et al: Cyclosporin treatment for rheumatoid arthritis: A placebo-controlled, double
blind, multicentre study. Ann Rheum 45:726, 198650. 50. Townes AS, Sowa JM, Shulman LE: Controlled trial of cyclophosphamide in rheumatoid arthritis. Arthritis Rheum 19:563, 1976 51. O'Dell JR, Paulsen G, Haire CE, et al: Treatment of early seropositive rheumatoid arthritis with minocycline: Four-year
follow up of a double-blind, placebo-controlled
trial. Arthritis Rheum 42:1691, 1999 52. Kloppenburg M, Breedveld FC, Terwiel JP, et al: Minocycline in active rheumatoid arthritis: A double-blind, placebo-controlled
trial. Arthritis Rheum 37:629, 1994 53. Watson PG, Hazelman BL: The Sclera and Systemic Disorders. Philadelphia, WB Saunders, 1976 54. Foster CS, Forstot SL, Wilson LA: Mortality rate in rheumatoid arthritis patients developing necrotizing
scleritis or peripheral ulcerative keratitis. Ophthalmology 91:1253, 1984 55. Thorne JE, Jabs DA: Ocular manifestations of vasculitis. Rheum Dis Clin North Am 27:761, 2001 56. Jabs DA, Mudun A, Dunn JP, et al: Episcleritis and scleritis: Clinical features and treatment results. Am J Ophthalmol 130:469, 2000 57. Sainz de la Maza M, Jabbur NS, Foster CS: Severity of scleritis and episcleritis. Ophthalmology 101:389, 1994 58. Matsuo T, Kono R, Matsuo N, et al: Incidence of ocular complications in rheumatoid arthritis and the relation
of keratoconjunctivitis sicca with its systemic activity. Scand J Rheumatol 26:113, 1997 59. Sainz de la Maza M, Foster CS, Jabbur NS: Scleritis associated with rheumatoid arthritis and with other systemic
immune-mediated diseases. Ophthalmology 101:1281, 1994 60. Sanford-Smith JH: Intermittent superior oblique tendon sheath syndrome. Br J Ophthalmol 53:412, 1969 61. Killian PJ, McClain B, Lawless OJ: Brown's syndrome: An unusual manifestation of rheumatoid arthritis. Arthritis Rheum 20:1080, 1977 62. Wright KW, Silverstein D, Marrone AC, et al: Acquired inflammatory superior oblique tendon sheath syndrome. Arch Ophthalmol 100:1752, 1982 63. Nabili S, McCarey DW, Browne B, et al: A case of orbital myositis associated with rheumatoid arthritis. Ann Rheum Dis 61:938, 2002 64. Scherbel Al, MacKenzie AH, Nousek JE, et al: Ocular lesions rheumatoid arthritis and related disorders with particular
reference to retinopathy: A study of 741 patients treated with and
without chloroquine drugs. N Engl J Med 273:360, 1965 65. Shearer RB, Dubois EL: Ocular changes induced by long term hydroxychloroquine (Plaquenil) therapy. Am J Ophthalmol 64:245, 1967 66. Finbloom DS, Silver K, Newsome DA, et al: Comparison of hydroxychloroquine and chloroquine use and the development
of retinal toxicity. J Rheumatol 12:692, 1985 67. Tobin DR, Krohel GB, Rynes RI: Hydroxychloroquine: Seven year experience. Arch Ophthalmol 100:81, 1982 68. Johnson MW, Vine AK: Hydroxychloroquine therapy in massive doses without retinal toxicity. Am J Ophthalmol 104:139, 1987 69. Thorne JE, Maguire AM: Resolution of hydroxychloroquine maculopathy. Br J Ophthalmol 83:1201, 1999 70. Prashker MJ, Meenan RF: The total costs of drug therapy for rheumatoid arthritis. A model based
on costs of drug, monitoring, and toxicity. Arthritis Rheum 38:318, 1995 71. Mazzuca SA, Yung R, Brandt KD, et al: Current practices for monitoring ocular toxicity related to hydroxychloroquine (Plaquenil) therapy. J Rheumatol 21:59, 1994 72. Arnett FC: Seronegative spondyloarthropathies. Bull Rheum Dis 37:1, 1988 73. Brewerton DA, Caffrey M, Nicholls , et al: Acute anterior uveitis and HLA-27, Lancet 2:994, 1973 74. Zervas J, Tsokos G, Papadakis G, et al: HLA-B27 frequency in Greek patients with acute anterior uveitis. Br J Ophthalmol 61:699, 1977 75. Rosenbaum JT: Characterization of uveitis associated with spondyloarthritis. J Rheumatol 16:792, 1989 76. Rosenbaum JT: Acute anterior uveitis and spondyloarthropathies. Rhem Dis Clin North Am 18:143, 1992 77. Brewerton DA, Hart FD, Nicholls A, et al: Ankylosing spondylitis and HLA-27. Lancet 1:904, 1973 78. Schlosstein L, Terasaki PI, Bluestone R, et al: High association of an HL-A antigen W27 with ankylosing spondylitis. N Engl J Med 288:704, 1973 79. Woodrow JC: HL-A 27 and Reiter's syndrome. Lancet 2:671, 1973 80. Bluestone R, Morris RI, Metzger AL, et al: HLA-W27 and the spondylitis of chronic inflammatory bowel disease
and psoriasis. Ann Rheum Dis 34:31, 1975 81. Morris RI, Metzger Al, Bluestone R, et al: HLA-W27: A useful discriminator in the arthropathies of inflammatory
bowel disease. N Engl J Med 290:1117, 1974 82. Reveille JD, Ball EJ, Khan MA: HLA-B27 and genetic predisposing factors in spondyloarthropathies. Curr Opin Rheumatol 13:265, 2001 83. Khan MA: Update on spondyloarthropathies. Ann Intern Med 136:896, 2002 84. Keat A: Reiter's syndrome and reactive arthritis in perspective. N Engl J Med 309:1606, 1983 85. Yu DT, Choo SY, Schaack T: Molecular mimicry in HLA-B27 related arthritis. Ann Intern Med 111:581, 1989 86. Scofield RH, Warren WL, Koelsch G, et al: A hypothesis for the HLA-B27 immune dysregulation in spondyloarthropathy: contributions
from enteric organisms, B27 structure, peptides
bound by B27, and convergent evolution. Proc Natl Acad Sci USA 90:9330, 1993 87. van der Linden S, van der Heijde D: Ankylosing spondylitis. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1039–1053. Philadelphia, WB Saunders, 2001 88. Russell ML: Ankylosing spondylitis: The case for the underestimated female. J Rheumatol 12:4, 1985 89. Jimenez-Balderas FJ, Mintz G: Ankylosing spondylitis: Clinical course in women and men. J Rheumatol 20:2069, 1993 90. Tay-Kearney ML, Schwam BL, Lowder C, et al: Clinical features and associated systemic diseases of HLA-B27 uveitis. Am J Ophthalmol 121:47, 1996 91. Kidd BL, Cawley MI: Delay in diagnosis of spondyloarthritis. Br J Rheumatol 27:230, 1988 92. Carette S, Graham D, Little H, et al: The natural disease course of ankylosing spondylitis. Arthritis Rheum 26:186, 1983 93. Creemers MCW, Franssen MJAM, van de Putte LBA, et al: Methotrexate in severe ankylosing spondylitis: An open study. J Rheumatol 22:1104, 1995 94. Braun J, Brandt J, Listing J, et al: Treatment of active ankylosing spondylitis with infliximab: A randomized
controlled multicenter trial. Lancet 359:1187, 2002 95. Van Den Bosch F, Kruithof E, Baeten D, et al: Randomized double-blind comparison of chimeric monoclonal antibody
to tumor necrosis factor alpha (infliximab) versus placebo
in active spondyloarthropathy. Arthritis Rheum 46:755, 2002 96. Wilkinson M, Bywater EGL: Clinical features and course of ankylosing spondylitis. Ann Rheum Dis 17:209, 1958 97. Kimura SJ, Hogan MJ, O'Connor GR, et al: Uveitis and joint diseases: A review of 191 cases. Trans Am Ophthalmol Soc 64:291, 1966 98. Rosenbaum JT: Uveitis: An internist's view. Arch Intern Med 149:1173, 1989 99. Belmont JB, Michelson JB: Vitrectomy in uveitis associated with ankylosing spondylitis. Am J Ophthalmol 94:300, 1982 100. Yu DTY, Fan PT: Reiter's syndrome and undifferentiated spondyloarthropathy. In Ruddy S, Harris ED, Sledge CB, et al: (eds) Kelly's Textbook of Rheumatology, 6th ed, pp. 1055–1069. Philadelphia, WB Saunders, 2001 101. Leirisalo-Repo M: Prognosis, course of disease, and treatment of the spondyloarthropathies. Rheum Dis Clin North Am 24:737, 1998 102. Butler MJ, Russell AS, Percy JS, et al: A follow-up study of 48 patients with Reiter's syndrome. Am J Med 67:808, 1979 103. Lee DA, Barker SM, Su WPD, et al: The clinical diagnosis of Reiter's syndrome: Ophthalmic and non-ophthalmic
aspects. Ophthalmology 93:350, 1986 104. Needham AD, Harding SP, Carey P: Bilateral multifocal choroiditis in Reiter syndrome. Arch Ophthalmol 115:684, 1997 105. Conway RM, Graham SL, Lassere M: Incomplete Reiter's syndrome with focal involvement of the posterior
segment. Aust NZ J Ophthalmol 23:63, 1995 106. Mills, RP, Kalina RE: Reiter's keratitis, Arch Ophthalmol 87:447, 1972 107. Mark DB, McCulley JB: Reiter's keratitis. Arch Ophthalmol 100:781, 1982 108. Rowson NJ, Dart JK: Keratitis in Reiter's syndrome. Br J Ophthalmol 76:126, 1992 109. Greenstein J, Janowitz HD, Sachar DB: The extra intestinal complications of Crohn's disease and ulcerative
colitis: A study of 700 patients. Medicine (Baltimore) 55:401, 1976 110. Bernstein CN, Blanchard JF, Rawsthorne P, et al: The prevalence of extaintestinal diseases in inflammatory bowel disease: A
population-based study. Am J Gastroenterol 96:1116, 2001 111. Wollheim FA: Enteropathic arthritis. In Ruddy S, Harris ED, Sledge CB, et al: (eds) Kelly's Textbook of Rheumatology, 6th ed, pp. 1081–1085. Philadelphia, WB Saunders, 2001 112. de Vlam K, Mielants H, Cuvelier C, et al: Spondyloarthropathy is underestimated in inflammatory bowel disease: Prevalence
and HLA association. J Rheumatol 27:2860, 2000 113. Orchard TR: Arthritis associated with inflammatory bowel disease. In Bayless TM, Hanauer SB (eds): Advanced Therapy of Inflammatory Bowel Disease, pp 279–282. Ontario: BC Decker Inc., 2001 114. Baert FJ, D'Haens GR, Peters M, et al: Tumor necrosis factor-α antibody (infliximab) therapy profoundly down-regulates
the inflammation in Crohn's ileocolitis. Gastroenterology 116:22, 1999 115. Crohn BB: Ocular lesions complicating ulcerative colitis. Am J Med Soc 1699:260, 1925 116. Ellis PP, Gentry JH: Ocular complications of ulcerative colitis. Am J Ophthalmol 58:779, 1964 117. Hopkins DJ, Horan E, Burton IL, et al: Ocular disorders in a series of 332 patients with Crohn's disease. Br J Ophthalmol 58:732, 1974 118. Knox DL, Schachat AP, Mustoner E: Primary, secondary and coincidental ocular complications of Crohn's
disease. Ophthalmology 91:163, 1984 119. Lyons JL, Rosenbaum JT: Uveitis associated with inflammatory bowel disease compared with uveitis
associated with spondyloarthropathy. Arch Ophthalmol 115:61, 1997 120. Banares A, Hernandez-Garcia C, Fernandez-Gutierrez B, et al: Eye involvement in the spondyloarthropathies. Rheum Dis Clin North Am 24:771, 1998 121. Schneiderman JH, Sharpe JA, Sutton DMC: Cerebral and retinal vascular complications of inflammatory bowel disease. Ann Neurol 5:331, 1979 122. Duker JS, Brown GC, Brooks L: Retinal vasculitis in Crohn's disease. Am J. Ophthalmol 103:664, 1987 123. Heuer DK, Gager WE, Reeser FH: Ischemic optic neuropathy associated with Crohn's disease. J Clin Neuroophthalmol 2:175, 1982 124. Knox DL, Snip RC, Stark WJ: The keratopathy of Crohn's disease. Am J Ophthalmol 90:862, 1980 125. Bradshaw DJ, Bray VJ, Enzenauer RW, et al: Acquired Brown syndrome associated with enteropathic arthropathy: A case
report. J Pediatr Ophthalmol Strabismus 31:118, 1994 126. Gladman DD, Rahman P: Psoriatic arthritis. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1071–1079, Philadelphia, WB Saunders, 2001 127. Gladman DD, Shuckett R, Russell ML, et al: Psoriatic arthritis: clinical and laboratory analysis of 220 patients. Q J Med 62:127, 1987 128. Gladman DD, Anhorn KB, Schachter RK, et al: HLA antigens in psoriatic arthritis. J Rheumatol 13:586, 1986 129. Lambert JR, Wright V: Eye inflammation in psoriatic arthritis. Ann Rheum Dis 35:354, 1976 130. Thorne JE, Volpe NJ, Lui GT: Magnetic resonance imaging of acquired Brown syndrome in a patient with
psoriasis. Am J Ophthalmol 127:233, 1999 131. Ansell BM: Chronic arthritis in childhood. Ann Rheum Dis 37:107, 1978 132. Cassidy JT: Juvenile rheumatoid arthritis. In Ruddy S, Harris ED, Sledge CB et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1297–1313. Philadelphia, WB Saunders, 2001 133. Towner SR, Michet CJ, O'Fallon WM, et al: The epidemiology of juvenile arthritis in Rochester, Minnesota, 1960–1979. Arthritis Rheum 26:1208, 1983 134. Pelkenen P: Incidence of arthritis in urban Finnish children. Arthritis Rheum 29:1232, 1986 135. Gare BA: Juvenile chronic arthritis. A population based study on epidemiology, natural history and outcome. Goteborg, Sweden, University of Goteborg, 1994. Lecture. 136. Melin-Aldana H, Giannini EH, Taylor J, et al: Human leukocyte antigen-DR1*1104 in the chronic iridocyclitis
of pauciarticular juvenile rheumatoid arthritis. J Pediatr 121:56, 1992 137. Malagon C, Van Kerckhove C, Giannini EH, et al: The iridocyclitis of early onset pauciarticular juvenile rheumatoid arthritis: Outcome
in immunogenetically characterized patients. J Rheumatol 19:160, 1992 138. Haas JP, Truckenbrodt H, Paul C, et al: Subtypes of HLA-DRB1*03, *08, *11, *12, *13 and *14 in
early onset pauciarticular juvenile chronic arthritis (EOPA) with
and without iridocyclitis. Clin Exp Rheumatol 12(Suppl 10):S7, 1994 139. Schaller J, Kupfer C, Wedgewood RJ: Iridocyclitis in juvenile rheumatoid arthritis. Pediatrics 44:92, 1969 140. Schaller JG, Johnson GD, Holborrow EJ, et al: The association of antinuclear antibodies and the chronic iridocyclitis
of juvenile rheumatoid arthritis (Still's disease). Arthritis Rheum 17:409, 1974 141. Glass D. Litvin D, Wallace K, et al: Early onset pauciarticular juvenile rheumatoid arthritis associated with
human leukocyte antigen DRW5, iritis, and antinuclear antibody. J Clin Invest 66:426, 1980 142. Forre O, Dobloug HJ, Hoyeraal HM, et al: HLA antigens in juvenile arthritis. Arthritis Rheum 26:35, 1983 143. Friis J, Moring N, Pedersen FK, et al: HLA-B27 in juvenile chronic arthritis. J Rheumatol 12:119, 1985 144. Arnett FC, Bias WB, Stevens MD: Juvenile-onset chronic arthritis: Clinical and roentgenographic
features of a unique HLA-B27 subset. Am J Med 69:369, 1980 145. Giannini EH, Brewer EJ, Kuzmina N, et al: Methotrexate in resistant juvenile rheumatoid arthritis: Results of the
USA-USSR double-blind placebo-controlled trial. N Engl J Med 326:1043, 1992 146. Lovell DJ: Ten years of experience with methotrexate. Past, present and future. Rev Rheum Engl Educ 64:186S, 1997 147. Rose CD, Singsen BH, Eichenfield, AH, et al: Safety and efficacy of methotrexate therapy for juvenile rheumatoid arthritis. J Pediatr 117:653, 1990 148. Shetty AK, Zganjar BE, Ellis GS Jr, et al: Low-dose methotrexate in the treatment of severe juvenile rheumatoid
arthritis and sarcoid iritis. J Pediatr Ophthalmol Strabismus 36:125, 1999 149. Weiss AH, Wallace CA, Sherry DD: Methotrexate for resistant chronic uveitis in children with juvenile rheumatoid
arthritis. J Pediatr 133:266, 1998 150. Samson CM, Waheed N, Foster CS: Methotrexate therapy for chronic noninfectious uveitis: Analysis of a case
series of 160 patients. Ophthalmology 108:1134, 2001 151. Lovell DJ, Giannini EH, Reiff A: Etanercept in children with polyarticular juvenile rheumatoid arthritis. N Engl J Med 342:763, 2000 152. Berk AT, Kocak N, Unsal E: Uveitis in juvenile arthritis. Ocul Immunol Inflamm 9:243, 2001 153. Boone MI, Moore TL, Cruz OA: Screening for uveitis in juvenile rheumatoid arthritis. J Pediatr Ophthalmol Strabismus 35:41, 1998 154. Chylack LT: The ocular manifestations of juvenile rheumatoid arthritis. Arthritis Rheum 20:217, 1978 155. Paivonsalo-Hietanen T, Tuominen J, Vaahtoranta-Lehtonen H, et al: Incidence and prevalence of different uveitis entities in Finland. Acta Ophthalmol Scand 75:76, 1997 156. Key SN, Kimura SJ: Iridocyclitis associated with juvenile rheumatoid arthritis. Am J Ophthalmol 80:425, 1975 157. Kanski JJ: Anterior uveitis in juvenile rheumatoid arthritis. Arch Ophthalmol 95:1794, 1977 158. Rosenberg AM, Oen KG: The relationship between ocular and articular disease activity in children
with juvenile rheumatoid arthritis and associated uveitis. Arthritis Rheum 29:797, 1986 159. Gori S, Broglia AM, Ravelli A, et al: Frequency and complications of chronic iridocyclitis in ANA-positive
pauciarticular juvenile chronic arthritis. Int Ophthalmol 18:225, 1994 160. Section on Rheumatology and Section on Ophthalmology: Guidelines for ophthalmologic
examinations in children with juvenile rheumatoid arthritis. Pediatrics 92:295, 1993 161. Merayo-Lloves J, Power WJ, Rodriguez A, et al: Secondary glaucoma in patients with uveitis. Ophthalmologica 213:300, 1999 162. Kanski JJ: Juvenile arthritis and uveitis. Surv Ophthalmol 34:253, 1990 163. Sherry DD, Mellins ED, Wedgwood RJ: Decreasing severity of chronic uveitis in children with pauciarticular
arthritis. Am J Dis Child 145:1026, 1991 164. Cabral DA, Petty RE, Malleson PN, et al: Visual prognosis in children with chronic anterior uveitis and arthritis. J Rheumatol 21:2370, 1994 165. Paikos P, Fotopoulou M, Papathanassiou M, et al: Cataract surgery in children with uveitis. J Pediatr Ophthalmol Strabismus 38:16, 2001 166. Probst LE, Holland EJ: Intraocular lens implantation in patients with juvenile rheumatoid arthritis. Am J Ophthalmol 122:161, 1996 167. Benezra D, Cohen E: Cataract surgery in children with chronic uveitis. Ophthalmology 107:1255, 2000 168. Jabs DA, Houk JL, Bias WB, et al: Familial granulomatous synovitis, uveitis, and cranial neuropathies. Am J Ophthalmol 78:801, 1985 169. Blau EB: Familial granulomatous arthritis, iritis, and rash. J Pediatr 107:689, 1985 170. McKusick VA: Mendelian inheritance in man. Catalogs of Human Genes and Genetic Disorders, 12th ed. Baltimore: The Johns Hopkins University Press, p. 1730, 1998 171. Miller JJ III: Early-onset “sarcoidosis” and “familial granulomatous
arthritis (arteritis)”: The same disease. J Pediatr 109:387, 1986 172. Latkany PA, Jabs DA, Smith JR, et al: Multifocal choroiditis in patients with familial juvenile systemic granulomatosis. Am J Ophthalmol 134:897, 2002 173. Miceli-Richard C, Lesage S, Rybojad M, et al: CARD15 mutations in Blau syndrome. Nat Genet 29:19, 2001 174. Hahn BH: Pathogenesis of systemic lupus erythematosus. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1089–1103. Philadelphia, WB Saunders, 2001 175. Seligman VA, Suarez C, Lum R, et al: The Fcγ receptor IIIA-158F allele is a major risk factor for the development
of lupus nephritis among Caucasians but not non-Caucasians. Arthritis Rheum 44:618, 2001 176. Sato K, Miyasaka N, Yamaska K, et al: Quantitative defect of CD4+2H4+ cells in systemic lupus erythematosus
and Sjogren's syndrome. Arthritis Rheum 30:1407, 1987 177. Kammer GM: High prevalence of T cell type I protein A deficiency in systemic lupus
erythematosus. Arthritis Rheum 42:1458, 1999 178. Deng C, Kaplan MJ, Yang J, et al: Decreased ras-mitogen-activated protein kinase signaling
may cause DNA hypomethylation in T lymphocytes from lupus patients. Arthritis Rheum 44:397, 2001 179. Edworthy SM: Clinical manifestations of systemic lupus erythematosus. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1105–1123. Philadelphia, WB Saunders, 2001 180. Shern MA, Pirofsky B: Disseminated lupus erythematosus: Analysis of thirty-four cases. Arth Intern Med 90:790, 1952 181. Dubois EL, Tuffanelli DL: Clinical manifestations of systemic lupus erythematosus. JAMA 190:104, 1964 182. Estes D, Christian CL: The natural history of systemic lupus erythematosus by prospective analysis. Medicine (Baltimore) 50:85, 1971 183. Hochberg MC, Boyd RE, Aheran JM, et al: Systemic lupus erythematosus: A review of clinico-laboratory features
and immunogenetic markers in 150 patients with emphasis on demographic
subsets. Medicine (Baltimore) 64:285, 1985 184. Feinglass EJ, Arnett FC, Dorsch CA, et al: Neuropsychiatric manifestations of systemic lupus erythematosus: Diagnosis, clinical
spectrum, and relationship to other features of the disease. Medicine (Baltimore) 55:332, 1976 185. Tan, EM, Cohen As, Fries JF, et al: The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25:1271, 1982 186. Harris EN, Boey ML, Mackworth-Young CG, et al: Anti-cardiolipin antibodies: Detection by radioimmunoassay and association
with thrombosis in systemic lupus erythematosus. Lancet 2:1211, 1983 187. Glueck HI, Kant KS, Weiss MA, et al: Thrombosis in systemic lupus erythematosus: Relation to the presence of
circulating anticoagulants. Arch Intern Med 145:1389, 1985 188. Petri M, Rheinschmidt M, Whiting-O'Keefe Q, et al: The frequency of lupus anticoagulant in systemic lupus erythematosus: A
study of sixty consecutive patients by activated partial thromboplastin
time. Russell viper venom time and Anti-cardiolipin antibody level. Ann
Intern Med 106:524, 1987 189. Love PE, Santoro SA: Antiphospholipid antibodies: Anti-cardiolipin and the lupus anticoagulant
in systemic lupus erythematosus (SLE) and in non-SLE
disorders. Ann Intern Med 112:682, 1990 190. Dunn JP, Noorily SW, Petri M, et al: Antiphospholipid antibodies and retinal vascular disease. Lupus 5:313, 1996 191. Cleaner RC, Nigerian LV, Schattten S, et al: Vaso-occlusive retinopathy associated with anti-phospholipid
antibodies (lupus anticoagulant retinopathy). Ophthalmology 96:896, 1989 192. Perez-Vazquez ME, Villa AR, Drenkard C, et al: Influence of disease duration, continued follow-up and further antiphospholipid
testing on the frequency and classification category of
antiphospholipid syndrome in a cohort of patients with SLE. J Rheumatol 20:437, 1993 193. Simioni P, Prandoni P, Zanon E, et al: Deep venous thrombosis and lupus anticoagulant. A case-control study. Thromb Haemost 76:187, 1996 194. Wilson WA, Gharavi EA, Koike T, et al: International consensus statement on preliminary classification criteria
for definite antiphospholipid syndrome. Report of an International Workshop. Arthritis Rheum 42:1309, 1999 195. Durrani OM, Gordon C, Murray PI: Primary anti-phospholipid antibody syndrome (APS): Current
concepts. Surv Ophthalmol 47:215, 2002 196. Hahn BH: Management of systemic lupus erythematosus. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1125–1143. Philadelphia, WB Saunders, 2001 197. Canadian Hydroxychloroquine Study Group: A randomized study of the effect
of withdrawing hydroxychloroquine sulfate in systemic lupus erythematosus. N Engl J Med 324:150, 1991 198. Callen JP: Management of skin disease in lupus. Bull Rheum Dis 46:4, 1997 199. Cathcart ES, Scheinberg MA, Idelson BA, et al: Beneficial effects of methylprednisolone “pulse” therapy in
diffuse proliferative lupus nephritis. Lancet 1:163, 1976 200. Isenberg DA, Morrow WJW, Snaith ML: Methylprednisolone pulse therapy in the treatment of systemic lupus erythematosus. Ann Rheum Dis 41:347, 1982 201. Felson DT, Anderson J: Evidence for the superiority of immunosuppressive drugs and prednisone
alone in lupus nephritis. N Engl J Med 311:1528, 1984 202. Gourley MF, Austin HA III, Scott D, et al: Methylprednisolone and cyclophosphamide, alone or in combination, in patients
with lupus nephritis. Ann Intern Med 125:549, 1996 203. Bansal VK, Beto JA: Treatment of lupus nephritis: A meta-analysis of clinical trials. Am J Kidney Dis 29:193, 1997 204. Brodsky RA, Petri M, Smith BD, et al: Immunoablative high-dose cyclophosphamide without stem-cell
rescue for refractory, severe autoimmune disease. Ann Intern Med 129:1031, 1998 205. Khamashta MA, Cuadrado MJ, Mujic F, et al: The management of thrombosis in the antiphospholipid-antibody syndrome. N Engl J Med 332:993, 1995 206. Rosove MH, Brewer PMC: Antiphospholipid thrombosis: Clinical course after the first thrombotic
event in 70 patients. Ann Intern Med 117:303, 1992 207. Huey C, Jakobiec FA, Iwamoto T, et al: Discoid lupus erythematosus of the eyelids. Ophthalmology 90:1389, 1983 208. Maumenee AE: Retinal lesions in lupus erythematosus. Am J Ophthamol 23:971, 1940 209. Gold DH, Morris DA, Henkind P: Ocular findings in systemic lupus erythematosus. Br J Ophthalmol 56:800, 1972 210. Lanham JG, Barrie T, Kohner EM, et al: SLE retinopathy: Evaluation of fluorescein angiography. Ann Rhuem Dis 41:473, 1982 211. Johnson RT, Richardson EP: The neurological manifestations of systemic lupus erythematosus: A clinical
Pathological study of 24 cases and review of the literature. Medicine (Baltimore) 47:337, 1968 212. Bresnihan B, Hohmeister R, Cutting J, et al: The neuropsychiatric disorder in systemic lupus erythematosus: Evidence
for both vascular and immune mechanisms. Ann Rheum Dis 38:301, 1979 213. Aronson A, Ordinis NG, Diddle KR, et al: Immune-complex deposition in the eye in systemic lupus erythematosus. Arch Intern Med 139:1312, 1979 214. Klinkhoff AV, Beattie CW, Chalmers A: Retinopathy in systemic lupus erythematosus: Relationship to disease activity. Arthritis Rheum 29:1152, 1986 215. Appen RE, Wray SH, Cogan DG: Central retinal artery occlusion. Am J Ophthalmol 79:374, 1975 216. Gold D, Feiner L, Heinkind P: Retinal arterial occlusive disease in systemic lupus erythematosus. Arch Ophthalmol 95:1580, 1977 217. Silverman M, Lubeck MJ, Brimey WG: Central retinal vein occlusion complicating systemic lupus erythematosus. Arthritis Rehem 21:839, 1978 218. Kayazawa F, Honda A: Severe retinal vascular lesions in systemic lupus erythematosus. Ann Ophthalmol 13:1291, 1981 219. Vine AK, Barr CC: Prolifeative lupus retinopathy. Arch Opthalmol 102:852, 1984 220. Coppeto JR, Currie JN, Moneiro MLR, et al: A syndrome of arterial-occlusive retinopathy and encephalopathy. Am J Ophthalmol 98:189, 1984 221. Hall S, Buettner H, Luthra HS: Occlusive retinal vascular disease in systemic lupus erythematosus. J Rheumatol 11:846, 1984 222. Graham EM, Spalton DJ, Barnard RO, et al: Cerebral and retinal vascular changes in systemic lupus erythematosus. Ophthalmology 92:444, 1985 223. Jabs DA, Fine SL, Hochberg MC, et al: Severe retinal vaso-occlusive disease in systemic lupus erythematosus. Arch Ophthalmol 104:558, 1986 224. Diddie KR, Aronson AJ, Ernest JT: Chorioretinopathy in a case of systemic lupus erythematosus. Trans Am Ophthalmol Soc 75:122, 1977 225. Kinyoun JL, Kalina RE: Visual loss from choroidal ischemia. Am J Ophthalmol 101:650, 1986 226. Jabs DA, Hanneken AM, Schachat AP, et al: Choroidopathy in systemic lupus erythematosus. Arch Ophthalmol 106:230, 1988 227. Hackett ER, Martinez RD, Larson PF, et al: Optic neuritis in systemic lupus erythematosus. Arch Neurol 31:9, 1974 228. Shepherd DI, Downie AW, Best PV: Systemic lupus erythematosus and multiple sclerosis. Arch Neurol 30:423, 1974 229. April RS, Vansonnenberg E: A case of neuromyelitis optic (Devic's syndrome) in systemic
lupus erythematosus: Clinicopathologic report and review of the literature. Neuology 26:1066, 1976 230. Cinofro JR, Frenkel M: Systemic lupus erythematousu presenting as optic neuritis. Ann Ophthalmol 10:559, 1978 231. Allen IV, Miller JHD, Kirk J, et al: Systemic lupus erythematosus clinically resembling multiple sclerosis and
with unusual pathological and ultrastructural features. J Jeurol Neurol Neuosurg Psychiatry 42:13, 1979 232. Lessell S: The neuro-ophthalmology of systemic lupus erythematosus. Doc Ophthalmol 47:13, 1979 233. Hayreh SS: Posterior ischemic optic neuropathy. Ophthalmologica 182:29, 1981 234. Smith CA, Pinals RS: Optic neuritis in systemic lupus erythematosus. J Rheumatol 9:963, 1982 235. Mavrikakis M, Anastasiou-Nana M, Kalaitzidou C, et al: Optic neutris and Jaccoud's syndrome in a patient with systemic lupus
erythematosus. Scand J Rheumatol 12:367, 1983 236. Jabs DA, Miller NR, Newman SA, et al: Optic neuropathy in systemic lupus erythematosus. Arch Ophthalmol 104:564, 1986 237. Brandt KD, Lessell S, Cohen AS: Cerebral disorders of vision in systemic lupus erythematosus. Ann Intern Med 83:163, 1975 238. Seibold JR: Scleroderma. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1211–1239. Philadelphia, WB Saunders, 2001 239. Ruffanelli DL, Winkelmann RK: Systemic scleroderma: A clinical study of 727 cases. Arch Dermatol 84:359, 1961 240. Rodnan GP: The natural history of progressive systemic sclerosis (diffuse scleroderma). Bull Rheum Dis 13:301, 1963 241. Rodnan GP, Lipinski E, Luksich J: Skin thickness and collagen content in progressive systemic sclerosis and
localized scleroderma. Arthritis Rheum 22:130, 1979 242. Buckingham RB, Prince RK, Rodnan GP, et al: Increased collagen accumulation in dermal fibroblast cultures from patients
with progressive systemic sclerosis (scleroderma). J Lab Clin Med 92:5, 1978 243. Ungerer RG, Tashkin DP, Furst D, et al: Prevalence and clinical correlates of pulmonary arterial hypertension in
progressive systemic sclerosis. Am J Med 75:65, 1983 244. Kahaleh MB, Sherer GK, LeRoy EC: Endothelial cell injury in scleroderma. J Exp Med 149:1326, 1979 245. Cohen S, Johnson AR, Hurd E: Cytotoxicity of sera from patients with scleroderma. Arthritis Rheum 26:170, 1983 246. Kahaleh MB, Osborn I, LeRoy EC: Elevated levels of circulating platelet aggregates and beta-thromboglobulin
in scleroderma. Ann Intern Med 96:610, 1982 247. Casciola-Rosen L, Wigley F, Rosen A: Scleroderma autoantigens are uniquely fragmented by metal-catalyzed
oxidation reactions: Implications for pathogenesis. J Exp Med 185:71, 1997 248. Haynes DC, Gershwin ME: The immunopathology of progressive systemic sclerosis (PSS). Semin Arthritis Rheum 11:331, 1982 249. Roumm AD, Whiteside TL, Medsger TA, et al: Lymphocytes in the skin of patients with progressive systemic sclerosis. Arthritis Rheum 27:645, 1984 250. Steen VD, Oddis CV, Conte CG, et al: Incidence of systemic sclerosis in Allegheny County, Pennsylvania: A twenty-year
study of hospital-diagnosed cases, 1963–1982. Arthritis Rheum 40:441, 1997 251. Mayes MD, Laing TJ, Gillespie BW, et al: Prevalence, incidence and survival rates of systemic sclerosis in the Detroit
metropolitan area. Arthritis Rheum 39:S150, 1996 252. Maricq HR: Widefield capillary microscopy. Technique and rating scale for abnormalities seen in scleroderma and related
disorders. Arthritis Rheum 24:1159, 1981 253. Ellis WW, Baer AN, Robertson RM, et al: Left ventricular dysfunction induced by cold exposure in patients with
systemic sclerosis. Am J Med 80:385, 1986 254. Furst DE, Davisn JA, Clements PJ, et al: Abnormalities of pulmonary vascular dynamics and inflammation in early
progressive systemic sclerosis. Arthritis Rheum 24:1403, 1981 255. Steen VD, Owens GR, Fino GJ, et al: Pulmonary involvement in systemic sclerosis (scleroderma). Arthritis Rheum 28:759, 1985 256. Roberts NK, Cabeen WR, Moss J, et al: The prevalence of conductive defects and cardiac arrhythmias in progressive
systemic sclerosis. Ann Intern Med 94:38, 1981 257. Cannon PJ, Hassar M, Case DB, et al: The relationship of hypertension and renal failure in scleroderma (progressive
systemic sclerosis) to structural and functional abnormalties
of renal cortical circulation. Medicine (Baltimore) 53:1, 1974 258. Kovalchik MT, Guggenheim SJ, Silverman MH, et al: The kidney in progressive systemic sclerosis: A prospective study. Ann Intern Med 89:881, 1978 259. Mitnick PD, Feig PU: Control of hypertension and reversal of renal failure in scleroderma. N Engl J Med 299:871, 1978 260. Lopez-Overjero JA, Saal D, D'Angelo WA, et al: Reversal of vascular and renal crisis of scleroderma by oral angiotensin-converting-enzyme blockage. N Engl J Med 300:1417, 1979 261. Thurm RH, Alexander JC: Captopril in the treatment of scleroderma renal crisis. Arch Intern Med 144:733, 1984 262. Teasdall RD, Frayha RA, Shulman LE: Cranial nerve involvement in systemic sclerosis (scleroderma): A
report of ten cases. Medicine (Baltimore) 59:149, 1980 263. Fritzler MJ, Kinsella TD, Garbutt E: The CREST syndrome: A distinct serologic entity with anticentromere antibodies. Am J Med 69:520, 1980 264. McCarty GA, Rice JR, Bembe ML, et al: Anticentromere antibody: Clinical correlations and association with favorable
prognosis in patients with scleroderma variants. Arthritis Rheum 26:1, 1983 265. Powell FC, Winkelmann RK, Venecie-Lemarchand F, et al: The anticentromere antibody: Disease specificity and clinical significance. Mayo Clin Proc 59:700, 1984 266. Fritzler MJ, Kinsella TD, Garbutt E: The CREST syndrome: A distinct serologic entity with anticentromere antibodies. Am J Med 69:520, 1980 267. Sharp GC, Irvin WS, Tan EM, et al: Mixed connective tissue disease: An apparently distinct rheumatic disease
syndrome associated with a specific antibody to an extractable nuclear
antigen (ENA). Am J Med 52:148, 1972 268. Nimelstein SH, Brody S, McShane D, et al: Mixed connective tissue disease: A subsequent evaluation of the original 25 patients. Medicine (Baltimore) 59:239, 1980 269. Silver RM, Miller KS, Kinsella MB, et al: Evaluation and management of scleroderma lung disease using bronchoalveolar
lavage. Am J Med 88:470, 1990 270. Rodeheffer RJ, Rommer JA, Wigley F, et al: Controlled double-blind trial of nifedipine in the treatment of
Raynaud's phenomenon. N Engl J Med 308:880, 1983 271. White CJ, Phillips WA, Abrahams LA, et al: Objective benefit of nifedipine in the treatment of Raynaud's phenomenon. Am J Med 80:623, 1986 272. Horna EC: Ophthalmic manifestations of progressive systemic sclerosis. Br J Ophthalmol 53:388, 1969 273. West RH, Barnett AJ: Ocular involvement in scleroderma. Br J Ophthalmol 63:845, 1979 274. Cipoletti JF, Buckingham RB, Barnes EL, et al: Sjogren's syndrome in progressive systemic sclerosis. Ann Intern Med 87:535, 1977 275. Osial TA, Whiteside TL, Buckingham RB, et al: Clinical and serologic study of Sjogren's syndrome in patients with
progressive systemic sclerosis. Arthritis Rheum 26:500, 1983 276. Dorwart BB: Periorbital edema in progressive systemic sclerosis. Ann Intern Med 80:273, 1974 277. Rush JA: Isolated superior oblique paralysis in progressive systemic sclerosis. Ann Ophthalmol 13:217, 1981 278. Mabon M, Whitcher JP, Anderson R: Bilateral scleral pit associated with systemic sclerosis. Am J Ophthalmol 128:521, 1999 279. Campbell WW, Bajandas FJ: Restrictive ophthalmopathy associated with linear scleroderma. J Neuroophthalmol 15:95, 1995 280. Arnett FC, Michels RG: Inflammatory ocular myopathy in systemic sclerosis (scleroderma). Arch Intern Med 132:740, 1973 281. Saari KM, Rudenburg HA, Laitinen O: Bilateral central retinal vein occlusion in a patient with scleroderma. Ophthalmologica 182:7, 1981 282. Pollack IP, Becker B: Cytoid bodies of the retina. Am J Ophthalmol 54:655, 1962 283. Klien BA: Comments on the cotton wool lesions of the retina. Am J Ophthalmol 59:17, 1965 284. Ashton N, Coomes EN, Garner A, et al: Retinopathy due to progressive systemic sclerosis. J Pathol Bacteriol 96:259, 1968 285. MaClean H, Guthrie W: Retinopathy in scleroderma. Trans Ophthalmol Soc UK 139:209, 1969 286. Farkas TG, Sylvester V, Archer D: The choroidopathy of progressive systemic sclerosis (scleroderma). Am J Ophthalmol 74:875, 1972 287. Grennan DM, Forrester J: Involvement of the eye in SLE and scleroderma. Ann Rheum Dis 36:152, 1977 288. Heese RJ, Slagle DF: Scleroderma choroidopathy: Report of an unusual case. Ann Ophthalmol 14:524, 1982 289. Serup L, Serup J, Hagdrup H: Fundus fluorescein angiography in generalized scleroderma. Ophthalmic Res 19:303, 1987 290. Wortmann RL: Inflammatory diseases of muscle and other myopathies. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1273–1296. Philadelphia, WB Saunders, 2001 291. Bohan A, Peter JB, Bowman RL, et al: A computer assisted analysis of 153 patients with polymyositis and dermatomyositis. Medicine (Baltimore) 56:225, 1977 292. Benbassat J, Gefel D, Larholt K, et al: Prognostic factors in polymyositis/dermtommyositis: A computer assisted
analysis of ninety-two cases. Arthritis Rheum 28:249, 1985 293. Whitaker JN, Engle WK: Vascular deposits of immunoglobulin and complement in idiopathic inflammatory
myopathy. N Engl J Med 286:333, 1972 294. Crowe WE, Bove KE, Levinson JE, et al: Clinical and pathogenic implications of histopathology in childhood polydermatomyositis. Arthritis Rheum 25:126, 1982 295. Currie S, Saunders M, Knowles M, et al: Immunologic aspects of polymyositis: The in vitro activity of lymphocytes
on incubation with muscle antigen and with muscle cultures. Q J Med 40:63, 1971 296. Love LA, Leff RL, Fraser DD, et al: A new approach to the classification of idiopathic inflammatory myopathy: Myositis
specific autoantibodies define useful homogeneous patient
groups. Medicine 70:360, 1991 297. Garlepp MJ: Genetics of idiopathic inflammatory myopathies. Curr Opin Rheum 8:514, 1996 298. Arnett FC, Hirsch TJ, Bias WB, et al: The Jo-1 antibody system in myositis: Relationships to clinical
features and HLA . J Rheumatol. 8:925, 1981. 299. Yoshida S, Akizuki M, Mimori T, et al. The precipitating antibody to an acidic nuclear protein antigen, the Jo-1, in
connective tissue diseases. Arthritis Rheum 26:604, 1983 300. Cronin ME, Plotz PH: Idiopathic inflammatory myopathies. Rheum Dis Clin North Am 16:655, 1990 301. Bohan A, Peter JB: Polymyositis and dermatomyositis (first of two parts). N Engl J Med 292:403, 1975 302. Bohan A, Peter JB: Polymyositis and dermatomyositis (second of two parts). N Engl J Med 292:403, 1975 303. Barnes BE: Dermatomyositis and maligancy. Ann Intern Med 84:68, 1976 304. Callen JP, Hyla JF, Bole GG, et al: The relationship of dermatomyositis and polymyositis to internal malignancy. Arch Dermatol 116:295, 1980 305. Oddis C: Idiopathic inflammatory myopathy. In Wortmann RL (ed): Diseases of Skeletal Muscle, pp 111–128. Philadelphia, Lippincott Williams & Wilkins, 2000 306. Metzger AL, Bohan A, Goldberg LS, et al: Polymyositis and dermatomyositis: Combined methotrexate and corticosteroid
therapy. Ann Intern Med 81:182, 1974 307. Bunch TW, Worthington JW, Combs JJ, et al: Azathioprine with prednisone for polymyositis: A controlled, clinical trial. Ann Intern Med 92:365, 1980 308. Joffe MM, Love LA, Leff RL, et al: Drug therapy of the idiopathic inflammatory myopathies: Predictors of response
to prednisone, azathioprine, and methotrexate and a comparison
of their efficacy. Am J Med 94:379, 1993 309. Bunch TW, Worthington JW, Combs JJ, et al: Azathioprine with prednisone for polymyositis. A controlled, clinical trial. Ann Intern Med 92:365, 1980 310. Adams EM, Plotz PH: The treatment of myositis. How to approach resistant disease. Rheum Dis Clin North Am 21:179, 1995 311. Susac JO, Garcia-Mullin R, Glaser JS: Ophthalmoplegia in dermatomyositis. Neurology 23:305, 1973 312. Bruce GM: Retinitis in deramatomyositis. Trans Am Ophthalmol Soc 36:282, 1938 313. Lisman JV: Dermatomyositis with retinopathy: Report of a case. Arch Ophthalmol 37:155, 1947 314. Devries S: Retinopathy in dermatomyositis. Arch Ophthalmol 46:432, 1951 315. Munro S: Fundus appearances in a case of acute dermatomyositis. Br J Ophthalmol 43:548, 1958 316. Liebman S, Cook C: Retinopathy with dermatomyositis. Arch Ophthalmol 74:704, 1965 317. Zamora J, Pariser K, Hedges T, et al: Retinal vasculitis in polymyositis-dermatomyositis. Arthritis Rheum 30:S106, 1987. 318. Bloch KJ, Buchanan WW, Wohl MJ, et al: Sjögren's syndrome: A clinical, pathological, and serological
study of sixty-two cases. Medicine (Baltimore) 44:187, 1965 319. Shearn MA: Sjogren's Syndrome. Philadelphia, WB Saunders, 1971 320. Whaley K, Williamson J, Chisholm DM, et al: Sjögren's syndrome. I. Sicca components. Q J Med 42:249, 1973 321. Whaley K, Webb J, McAvoy BA, et al: Sjögren's syndrome.2. Clinical associations and immunological phenomenon. Q J Med 42:513, 1973 322. Fox RI, Michelson P, Törnwall J: Approaches to the treatment of Sjögren's syndrome. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1027–1038. Philadelphia, WB Saunders, 2001 323. Greenspan JS, Daniels TE, Talal N, et al: The histopathology of Sjögren's syndrome in labial salivary gland
biopsies. Oral Surgery 37:217, 1974 324. Daniels TE: Labial salivary gland biopsy in Sjögren's syndrome: Assessment
as a diagnostic criterion in 362 suspected cases. Arthritis Rheum 27:147, 1984 325. Fox RI, Carstens SA, Fong S, et al: Use of monoclonal antibodies to analyze peripheral blood and salivary gland
lymphocyte subsets in Sjögren's syndrome. Arthritis Rheum 25:419, 1982 326. Adamson TC, Fox RI, Frisman DM, et al: Immunohistologic analysis of lymphoid infiltrates in primary Sjögren's
syndrome using monoclonal antibodies. J Immunol 130:203, 1983 327. Akata F, Pflugfelder SC, Lee SF, et al: Immunocytologic features of lacrimal gland biopsies in Sjögren's
syndrome. ARVO Abstracts. Invest Ophthalmol Vis Sci 30(suppl):386, 1989 328. Jabs DA, Prendergast RA: Murine models of Sjögren's syndrome: Evolution of the lacrimal
gland inlfammatory lesions. Invest Ophthalmol Vis Sci 32:371, 1991 329. Alexander GE, Provost TT, Stevens MB, et al: Sjögren's syndrome: Central nervous system manifestations. Neurology 31:1391, 1981 330. Alexander EL, Provost TT, Stevens MB, et al: Neurologic complications in primary Sjögren's syndrome. Medicine (Baltimore) 61:247, 1982 331. Alexander EL, Malinow K, Lejewski JE, et al: Primary Sjögren's syndrome with central nervous system disease
mimicking multiple sclerosis. Ann Intern Med 104:323, 1986 332. Harley JB, Alexander EL, Bias WB, et al: Anti-Ro (SSA) and anti-La (SSB) in patients
with Sjögren's syndrome. Arthritis Rheum 29:196, 1986 333. Gunduz K, Ozdemir on: Topical cyclosporin treatment of keratoconjunctivitis sicca in secondary
Sjögren's syndrome. Acta Ophthalmol (Copenh) 72:438, 1994 334. Rosenbaum JT, Bennett RM: Chronic anterior and posterior uveitis and primary Sjögren's
syndrome. Am J Ophthalmol 104:346, 1987 335. Hochberg MC: Relapsing polychondritis. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1463–1467. Philadelphia, WB Saunders, 2001 336. Foidart JM, Abe S, Martin GR, et al: Antibodies to type II collagen in relapsing polychondritis. N Engl J Med 299:1203, 1978 337. Pearson CM, Kline HM, Newcomer VD: Relapsing polychondritis. N Engl J Med 263:51, 1960 338. Kaye RL, Sones DA: Relapsing polychondritis: Clinical and pathological features in fourteen
cases. Ann Intern Med 60:653, 1964 339. Dolan DL, Lemmon GB, Teitelbaum SL: Relapsing polychondritis: Analytical literature review and studies on pathogenesis. Am J Med 41:285, 1966 340. McAdam LP, O'Hanlan MA, Bluestone R, et al: Relapsing polychondritis: Prospective study of 23 patients and a review
of the literature. Medicine (Baltimore) 55:193, 1976 341. Trentham DE, Le CH: Relapsing polychondritis. Ann Intern Med 129:114, 1998 342. Yang CL, Brinckmann J, Rui HF, et al: Autoantibodies to cartilage collagens in relapsing polychondritis. Arch Dermatol Res 285:245, 1993 343. Martin J, Roenigk HH, Lynch W, et al: Relapsing polychondritis treated with dapsone. Arch Dermatol 112:1272, 1976 344. Barranco VP, Minor DB, Solomon H: Treatment of relapsing polychondritis with dapsone. Arch Dermatol 112:1286, 1976 345. Michel CJ, McKenna CH, Luthra HS, et al: Relapsing polychondritis: Survival and predictive role of early disease
manifestations. Ann Intern Med 104:74, 1986 346. Svenson KLG, Holmdahl R, Klareskog L, et al: Cyclosporin A treatment in a case of relapsing polychondritis. Scand J Rheumatol 13:329, 1984 347. Priori R, Paroli MP, Lua FL, et al: Cyclosporin A in the treatment of relapsing polychondritis with severe
eye involvement [letter]. Br J Rheumatol 32:352, 1992 348. Park J, Gowin KM, Schumacher HR Jr: Steroid sparing of methotrexate in relapsing polychondritis. J Rheumatol 23:937, 1996 349. Hoang-Xuan T, Foster CS, Rice BA: Scleritis in relapsing polychondritis: Response to therapy. Ophthalmology 97:892, 1990 350. Zeuner M, Straub RH, Rauh G, et al: Relapsing polychondritis: Clinical and immunogenetic analysis of 62 patients. J Rheumatol 24:96, 1997 351. Isaak BL, Liesegang TJ, Michel CJ: Ocular and systemic findings in relapsing polychondritis. Ophthalmology 93:681, 1986 352. Hilding AC: Syndrome of cartilage pathology, destructive iridocyclitis, multiple joint
dislocations. Arch Ophthalmol 48:420, 1952 353. Anderson B: Ocular lesions in relapsing polychondritis and other rheumatoid syndromes. Trans Am Acad Ophthalmol Otol 71:227, 1967 354. McDay DAR, Watson PG, Lyne AJ: Relapsing polychondritis and eye disease. Br J Ophthalmol 58:600, 1974 355. Zion VM, Brackup AH, Weingeist S: Relapsing polychondritis, erythema nodosum and sclerouveitis: A case report
with anterior segment angiography. Surv Ophthalmol 19:107, 1974 356. Magargal LE, Donoso LA, Goldberg RE, et al: Ocular manifestations of relapsing polychondritis. Retina 1:96, 1981 357. Fauci AS, Haynes BF, Katz P: The spectrum of vasculitis: Clinical, pathologic, immunologic, and theraputic
considerations. Ann Intern Med 89:660, 1978 358. DeRemee RA, Weiland LH, McDonald TJ: Respiratory vasculitis. Mayo Clin Proc 55:492, 1980 359. Sergent JS: Classification of vasculitis. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1153–1154. Philadelphia: WB Saunders, 2001 360. Lie JT: Illustrated histopathologic classification criteria for selected vasculitis
syndromes. Arthritis Rheum 33:1074, 1990 361. Jennette JC, Falk RP, Andrassy K, et al: Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum 37:187, 1994 362. Weyand CM, Hunder NN, Hicok KC, et al: The HLA-DRB1 locus as a genetic component in giant cell arteritis: Mapping
of a disease-linked sequence motif to the antigen binding
site of the HLA-DR molecule. J Clin Invest 90:2355, 1992 363. Hirose H, Takagi M, Miyagawa N, et al: Genetic risk factor for abdominal aortic aneurysm: HLA-DR2. A Japanese study. J Vasc Surg 27:500, 1998 364. Yazici H, Chamberlain MA, Schreuder I, et al: HLA antigens in Behçet's disease: A reappraisal by a comparative
study of Turkish and British patients. Ann Rheum Dis 39:344, 1980 365. Ohno S, Ohguchi M, Hirose S, et al: Close association of HLA-Bw51 with Behçet's disease. Arch Ophthalmol 100:1455, 1982 366. Behar S, Porcelli SA: Review: Mechanisms of autoimmune disease induction: The role of the immune
response to microbial pathogens. Arthritis Rheum 38:458, 1995 367. Cuchacovich R: Immunopathogenesis of vasculitis. Curr Rheum Rep 4:9, 2002 368. Citron BP, Halpern M, McCarron M, et al: Necrotizing angiitis associated with drug abuse. N Engl J Med 283:1003, 1970 369. Duffy J, Lidsky MD, Sharp JT, et al: Polyarthritis, polyarteritis and hepatitis B. Medicine (Baltimore) 55:19, 1976 370. Sergent JS, Lockshin MD, Chrisuian CI, et al: Vasculitis with hepatitis B antigenemia: Long-term observations
in nine patients. Medicine (Baltimore) 55:1, 1976 371. Gocke DJ, Hsu K, Morgan C, et al: Association between polyarteritis and Australian antigen. Lancet 2:1149, 1970 372. Misiani R, Bellavita P, Fenili D, et al: Hepatitis C virus infection in patients with essential mixed cryoglobulinemia. Ann Intern Med 117:573, 1992 373. Agnello V, Chung RT, Kaplan LM: A role for hepatitis C virus infection in type II cryoglobulinemia. N Engl J Med 327:1490, 1992 374. Lunel F, Musset L, Cacoub P, et al: Cryoglobulinemia in chronic liver disease: Role of HCV and liver damage. Gastroenterology 106:1291, 1994 375. Hunder GG: Giant cell arteritis and polymyalgia rheumatica. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology 6th ed, pp. 1155–1164. Philadelphia, WB Saunders 2001 376. Bengtsson BA, Malmvall BE: The epidemiology of giant cell arteritis including temporal arteritis and
polymyalgia rheumatica. Arthritis Rheum 24:899, 1981 377. Gonzalez-Gay MA, Garcia-Porrua C: Systemic vasculitis in the adults in Northwestern Spain, 1988–97. Clinical and epidemiological aspects. Medicine (Baltimore) 78:292, 1999 378. Watts RA, Scott DG: Classification and epidemiology of the vasculitides. Baillieres Clin Rheumatol 11:191, 1997 379. Ostberg G: Temporal arteritis in a large necropsy series. Ann Rheum Dis 30:224, 1971 380. Hunder GG, Bloch DA, Michel BA, et al: The American College of Rheumatology 1990 criteria for the classification
of giant cell arteritis. Arthritis Rheum 33:122, 1990 381. Fauchald P, Rygvold O, Oystese B: Temporal arteritis and polymyalgia rheumatica. Ann Intern Med 77:845, 1972 382. Huston KA, Hunder GG, Lie JT, et al: Temporal arteritis: A 25-year epidemiologic, clinical, and pathologic
study. Ann Intern Med 88:162, 1978 383. Good BW, Jr: Temporal arteritis. Am J Med 67:839, 1979 384. Chuang TY, Hunder GG, Ilstrup DM, et al: Polymyalgia rheumatica: A 10-year epidemiologic and clinical study. Ann Intern Med 97:672, 1982 385. Gonzalez-Gay MA, Garcia-Porrua C, Salvarani C, et al: Diagnostic approach in a patient presenting with polymyalgia. Clin Exp Rheumatol 17:276, 1999 386. Keltner JL: Giant cell arteritis: Signs and symptoms. Ophthalmology 89:1101, 1982 387. Barilla-LaBarca ML, Lenschow DJ, Brasington RD: Polymyalgia rheumatica/temporal arteritis: Recent advances. Curr Rheum Rep 4:39, 2002 388. Cantini F, Salvarani C, Olivieri I, et al: Erythrocyte sedimentation rate and C-reactive protein in the evaluation
of disease activity and severity in polymyalgia rheumatica: A
prospective follow-up study. Semin Arthritis Rheum 30:17, 2000 389. Boyd RV, Hoffbrand BI: Erythrocyte sedimentation rate in elderly hospital in-patients. BMJ 1:901, 1966 390. Bottiger LE, Svedberg CA: Normal erythrocyte sedimentation rate and age. BMJ 2:85, 1967 391. Klein SM: Erythrocyte sedimentation rate in the elderly. Arch Ophthalmol 88:617, 1972 392. Hayes GS, Stinson IN: Erythrocyte sedimentation rate and age. Arch Ophthalmol 94:939, 1976 393. Kansu T, Corbett JJ, Savino P, et al: Giant cell arteritis with normal sedimentation rate. Arch Neurol 34:624, 1977 394. Biller J, Asconape J, Weinblatt ME, et al: Temporal arteritis associated with normal sedimentation rate. JAMA 274, 486, 1982 395. Gonzalez-Gay MA, Garcia-Porrua C, Llorca J, et al: Biopsy-negative giant cell arteritis: Clinical spectrum and predictive
factors for positive temporal artery biopsy. Semin Arthritis Rheum 30:249, 2001 396. Albert DM, Ruchman MC, Keltner JL: Skip areas in temporal arteritis. Arch Ophthalmol 94:2072, 1976 397. Klein RG, Campbell RJ, Carney JA: Skip lesions in temporal arteritis. Mayo Clin Proc 51:504, 1976 398. Hedges TR, Geiger GL, Albert DM: The clinical value of negative temporal artery biopsy specimens. Arch Ophthalmol 101:1251, 1983 399. Hall S, Lie JT, Kurland LT, et al: The therapeutic impact of temporal artery biopsy. Lancet 26:1217, 1983 400. Roth AM, Milsow L, Keltner JL: The ultimate diagnoses of patients undergoing temporal artery biopsies. Arch Ophthalmol 102:901, 1984 401. McDonnell PJ, Moore GW, Miller NR, et al: Temporal arteritis: A clinicopathologic study. Ophthalmology 93:518, 1986 402. Hunder GG, Sheps SG, Allen GL, et al: Daily and alternate-day corticosteroid regimens in treatment of
giant cell arteritis. Ann Intern Med 82:613, 1975 403. Rosenfeld SI, Kosmorsky GS, Klingele TG, et al: Treatment of temporal arteritis with ocular involvement. Am J Med 80:413, 1986 404. Hoffman G, Cid M, Hellmann D, et al: A multicenter placebo controlled study of methotrexate in giant cell arteritis. Arthritis Rheum 43:S115, 2000 405. Jover JA, Hernandez-Garcia C, Morado IC, et al: Combined treatment of giant-cell arteritis with methotrexate and
prednisone. Ann Intern Med 134:106, 2001 406. Hunder GG: Clinical features of GCA/PMR. Clin Exp Rheumatol 18:S6, 2000 407. Cullen JF: Temporal arteritis: Occurrence of ocular complications 7 years after diagnosis. Br J Ophthalmol 56:584, 1972 408. Wagener HP, Hollenhorst RW: The ocular lesions of temporal arteritis. Am J Ophthalmol 45:617, 1958 409. Cullen JF, Coleiro JA: Ophthalmic complications of giant cell arteritis. Surv Ophthalmol 20:247, 1976 410. Wang FM, Henkind P: Visual system involvement in giant cell (temporal) arteritis. Surv Ophthalmol 23:264, 1979 411. Gaudric A, Coscas G, Margerie JD: Acute sectorial choroidal ischemia. Am J Ophthalmol 98:707, 1984 412. Ellis CJ, Hamer DB, Hunt RW, et al: Medical investigation of retinal vascular occlusion. BMJ 2:1093, 1984 413. Tang RA, Kaldis LC: Retinopathy in temporal arteritis. Ann Ophthalmol 14:652, 1982 414. Lockskin MD: Diplopia as early sign of temporal arteritis: Report of two cases. Arthritis Rheum 13:419, 1970 415. Verdick M, Nielsen NV: Acute transient ophthalmoplegia in giant-cell arteritis: Report
of a case. Acta Ophthalmol (Copenh) 53:875, 1975 416. Barricks ME, Traviesa DB, Glaser JS, et al: Ophthalmplegia in cranial arteritis. Brain 100:209, 1977 417. Dimant J, Grob D, Brunner NG: Ophthalmoplegia ptosis, and miosis in temporal arteritis. Neurology 30:1054, 1980 418. Zion VM, Goodside V: Anterior segment ischemia with ischemic optic neuropathy. Surv Ophthalmol 19:19, 1974 419. Radda TM, Bardach H, Riss B: Acute ocular hypotony. Ophthalmologica 182:148, 1981 420. Miller NR: Visual manifestations of temporal arteritis. Rheum Dis Clin North Am 27:781, 2001 421. Wilkinson IMS, Russell RWR: Arteries of the head and neck in giant cell arteritis. Arch Neurol 27:378, 1972 422. Model DG: Reversal of blindness in temporal arteritis with methylprednisolone. Lancet 1:340, 1978 423. Schnieder HA, Weber AA, Ballen PH: The visual prognosis in temporal arteritis. Ann Ophthalmol 3:1215, 1971 424. Fraga A, Mintz G, Valle L, et al: Takayasu's arteritis: Frequency of systemic manifestations (study
of 22 patients) and favorable response to maintenance steroid
therapy with adrenocorticosteroids (12 patients). Arthritis Rheum 15:617, 1972 425. Lupi-Herrera E, Sanchez-Torres G, Marcushamer J: Takayasu's arteritis: Clinical study of 107 cases. Am Heart J 93:94, 1977 426. Ishikawa K: Natural history and classification of occlusive thromboaortopathy (Takayasu's
disease). Medicine (Baltimore) 57:27, 1978 427. Sergent JS: Polyarteritis and related disorders. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1185–1195. Philadelphia, WB Saunders, 2001 428. Sharma BK, Jain S, Radotra BD: An autopsy study of Takayasu arteritis in India. Int J Cardiol 66:S85, 1998 429. Fraga A, Medina F: Takayasu's arteritis. Curr Rheum Rep 4:30, 2002 430. Chun YS, Park SJ, Park IK, et al: The clinical and ocular manifestations of Takayasu arteritis. Retina 21:132, 2001 431. Tanaka T, Shimizu K: Retinal arteriovenous shunts in Takayasu's disease. Ophthalmology 94:1380, 1987 432. Lightfoot RW Jr, Michel BA, Bloch DA, et al: The American College of Rheumatology 1990 criteria for the classification
of polyarteritis nodosa. Arthritis Rheum 33:1088, 1990 433. Cohen RD, Conn DI, Ilstrup DM: Clinical features, prognosis, and response to treatment in polyarteritis. Mayo Clin Proc 55:146, 1980 434. Moore PM, Fauci AS: Neurologic manifestations of systemic vasculitis: A retrospective and prospective
study of the clinicopathologic features and responses to therapy
in 25 patients. Am J Med 71:517, 1981 435. Medical Research Council: Treatment of polyarteritis nodosa with cortisone: Results
after three years. BMJ 1:1399, 1960 436. Fauci AS, Katz P, Haynes BF, et al: Cyclophosphamide therapy of severe systemic necrotizing vasculitis. N Engl J Med 301:235, 1979 437. Fauci AS, Doppmann JL, Wolff SM: Cyclophosphamide induced remissions in advanced polyarteritis nodosa. Am J Med 64:890, 1978 438. Gayraud M, Guillevin L, Cohen P, et al: Treatment of good-prognosis polyarteritis nodosa and Churg-Strauss
syndrome: Comparison of steroids and oral or pulse cyclophosphamide
in 25 patients. Br J Rheumatol 36:1290, 1997 439. Maumenee AE: Ocular manifestations of collagen diseases. Arch Ophthalmol 56:557, 1956 440. Gaynon IE, Asbury MK: Ocular findings in a case of periarteritis nodosa. Am J Ophthalmol 26:1072, 1943 441. Goldsmith J: Periarteritis nodosa with involvement of the choroidal and retinal arteries. Am J Opthtalmol 29:435, 1946 442. Ingalls RG: Bilateral uveitis and keratitis accompanying periarteritis nodosa. Trans Am Acad Ophthalmol 55:630, 1951 443. Goar EL, Smith LS: Polyarteritis nodosa of the eye. Am J Ophthalmol 35:1619, 1952 444. Wise GN: Ocular periarteritis nodosa: Report of two cases. Arch Ophthalmol 48:1, 1952 445. Boeck J: Ocular changes in periarteritis nodosa. Am J Ophthalmol 42:567, 1956 446. Sheehan B, Harriman DGF, Bradshaw JPP: Polyarteritis nodosa with ophthalmic and neurological complications. Arch Ophthalmol 60:537, 1958 447. Blodi FC, Sullivan PB: Involvement of the eyes in periarteritis nodosa. Trans Am Acad Sci 63:161, 1959 448. Moore JG, Sevel D: Corneo-scleral ulceration in periarteritis nodosa. Br J Ophthalmol 50:651, 1966 449. Kimbrell OC, Wheliss JA: Polyarteritis nodosa complicated by bilateral optic neuropathy. JAMA 201:139, 1967 450. Rosen ES: The retinopathy in polyarteritis nodosa. Br J Ophthalmol 52:903, 1968 451. Kielar RA: Exudative retinal detachment and scleritis in polyarteritis. Am J Ophthalmol 82:694, 1976 452. Stefani FH, Brandt F, Pielsticker : Periarteritis nodosa and thrombotic thrombocytopenic pupura with serous
retinal detachment in siblings. Br J Ophthalmol 62:402, 1978 453. Appen RE, deVenecia G, Ferwerda J: Optic disc vasculitis. Am J Ophthalmol 90:353, 1980 454. Purcell JJ, Birkenkamp R, Tsai CC: Conjunctival lesions in periarteritis nodosa. Arch Ophthalmol 102:736, 1984 455. Kinyoun JL, Kalina RE, Klein ML: Choroidal involvement in systemic necrotizing vasculitis. Arch Ophthalmol 105:939, 1987 456. Akova YA, Jabbur NS, Foster CS: Ocular presentation of polyarteritis nodosa. Clinical course and management with steroid and cytotoxic therapy. Ophthalmology 100:1775, 1993 457. Churg J, Strauss L: Allergic granulomatosis, allergic angiitis, and periarteritis nodosa. Am J Pathol 27:277, 1951 458. Chumbley LC, Harrison EG, DeRemee RA: Allergic granulomatosis and angiitis (Churg-Strauss syndrome): Report
and analysis of 30 cases. Mayo Clin Proc 52:477, 1977 459. Cooper BJ, Bacal E, Patterson R: Allergic angiitis and granulomatosis. Arch Intern Med 138:367, 1978 460. Finan MC, Winkelmann RK: The cutaneous extravascular necrotizing granuloma (Churg-Strauss
granuloma) and systemic disease: A review of 27 cases. Medicine (Baltimore) 62:142, 1983 461. Masi AT, Hunder GG, Lie JT, et al: The American College of Rheumatology 1990 criteria for the classification
of Churg-Strauss syndrome (allergic granulomatosis and
angiitis). Arthritis Rheum 33:1094, 1990 462. Calabrese LH, Duna G: Vasculitis associated with antineutrophil cytoplasmic antibody. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1165–1184. Philadelphia, WB Saunders, 2001 463. Cury D, Breakey AS, Payne BF: Allergic granulomatosis angiitis associated with uveoscleritis and papilledema. Arch Ophthalmol 55:261, 1956 464. Miesler DM, Stock EL, Wertz RD, et al: Conjunctival inflammation and amyloidosis in allergic granulomatosis and
angiitis (Churg-Strauss syndrome). Am J Ophthalmol 91:216, 1981 465. Weinstein JM, Chui H, Lane S, et al: Churg-Strauss syndrome (allergic granulomatosis and angiitis): Neuro-ophthalmologic manifestations. Arch Ophthalmol 101:1217, 1983 466. Robin JB, Schanzlin DJ, Meisler DM, et al: Ocular involvement in the respiratory vasculitides. Surv Ophthalmol 30:127, 1985 467. Fauci AS, Wolff SM: Wegener's granulomatosis: Studies in eighteen patients and a review
of the literature. Medicine (Baltimore) 52:535, 1973 468. Wolff SM, Fauci AS, Horn RG, et al: Wegener's granulomatosis. Ann Intern Med 81:513, 1974 469. Brandwein S, Esdaile J, Danoff D, et al: Wegener's granulomatosis: Clinical features and outcome in 13 patients. Arch Intern Med 143:476, 1983 470. Fauci AS, Haynes BF, Katz P, et al: Wegener's granulomatosis: Prospective clinical and therapeutic experience
with 85 patients for 21 years. Ann Intern Med 98:76, 1983 471. Leavitt RY, Fauci AS, Bloch DA, et al: The American College of Rheumatology 1990 criteria for the classification
of Wegener's granulomatosis. Arthritis Rheum 33:1101, 1990 472. Cassan SM, Coles DT, Harrison EG: The concept of limited forms of Wegener's granulomatosis. Am J Med 49:366, 1970 473. Coutu RE, Klein M, Lessel S, et al: Limited form of Wegener's granulomatosis: Eye involvement as a major
sign. JAMA 233; 868, 1975 474. Harman LE, Margo CE: Wegener's granulomatosis. Surv Ophthalmol 42:458, 1998 475. Hollander D, Manning RT: The use of alkylating agents in the treatment of Wegener's granulomatosis. Ann Intern Med 67:393, 1967 476. Hoffman GS, Kerr GS, Leavitt RY, et al: Wegener's granulomatosis: An analysis of 158 patients. Ann Intern Med 116:488, 1992 477. Lacki JK, Schochat T, Sobieska M, et al: Immunological studies in patients with rheumatoid arthritis treated with
methotrexate or cyclophosphamide. Z Rheumatol 53:76, 1994 478. Straatsma BR: Ocular manifestations of Wegener's granulomatosis. Ann J Ophthalmol 44:789, 1957 479. Greenberger MH: Central retinal artery closure in Wegener's granulomatosis. Am J Ophthalmol 63:515, 1967 480. Haynes BF, Fishman ML, Fauci AS, et al: The ocular manifestations of Wegener's granulomatosis: Fifteen years
of experience and review of the literature. Am J Med 63:131, 1977 481. Jaben SL, Norton EWD: Exudative retinal detachment in Wegener's granulomatosis: Case report. Ann Ophthalmol 14:717, 1982 482. Bullen CL, Liesegang TJ, McDonald TJ, et al: Ocular complications of Wegener's granulomatosis. Ophthalmology 90:279, 1983 483. Blodi FC, Gass JDM: Inflammatory pseudotumor of the orbit. Br J Ophthalmol 52:579, 1968 484. Sainz de la Maza M, Foster CS, Jabbur NS: Scleritis associated with systemic vasculitic diseases. Ophthalmology 102:687, 1995 485. Ghate JV, Jorizzo JL: Behçet's disease. In Ruddy S, Harris ED, Sledge CB, et al: (eds): Kelly's Textbook of Rheumatology, 6th ed, pp. 1205–1209. Philadelphia, WB Saunders, 2001 486. Yazici H, Chamberlain MA, Schreuder I, et al: HLA antigens in Behçet's disease: A reappraisal by a comparative
study of Turkish and British patients. Ann Rheum Dis 39:344, 1980 487. Ohno S Ohguchi M, Hirose S, et al: Close association of HLA-BW51 with Behçet's disease. Arch Ophthalmol 100:1455, 1982 488. Mason RM, Barnes CG: Behçet's syndrome with arthritis. Ann Rheum Dis 28:95, 1969 489. Chajek T, Fainaru M: Behçet's disease: Report of 41 cases and a review of the literature. Medicine (Baltimore) 54:179, 1975 490. Chamberlain MA: Behçet's syndrome in 32 patients in Yorkshire. Ann Rheum Dis 36:491, 1977 491. Sakane T, Takeno M, Suzuki N, et al: Behçet's disease. N Engl J Med 341:1284, 1999 492. Zizic TM, Stevens MB: The arthropathy of Behçet's disease. Johns Hopkins Med J 136:243, 1975 493. Schotland DL, Wolf SM, White HH, et al: Neurologic aspects of Behçet's disease. Am J Med 34:544, 1963 494. Kalbain VV, Challis MT: Behçet's disease: Report of twelve cases with three manifesting
as papilledema. Am J Med 49:823, 1970 495. O'Duffy JD, Golstein NP: Neurologic involvement in seven patients with Behçet's disease. Am J Med 61:170, 1976 496. Shimizu T, Mishima S, Miyoshi K, et al: Behçet's disease. Jpn J Opthalmol 18:93, 1974 497. International Study Group for Behçet's Disease: Criteria for
diagnosis of Behçet's disease. Lancet 335:1078, 1990 498. O'Duffy JD, Robertson DM, Goldstein NP: Chlorambucil in the treatment of uveitis and meningoencephalitis of Behçet's
disease. Am J Med 7675, 1984 499. Abdalla MI, Baghat NED: Long-lasting remission of Behçet's disease after chlorambucil
therapy. Br J Ophthalmol 57:706, 1973 500. Tessler HH, Jennings T: High-dose short-term chlorambucil for intractable sympathetic
ophthalmia and Behçet's disease. Br J Ophthalmol 74:353, 1990 501. Dinning WJ, Perkins ES: Immunosuppressive agents in uveitis: A preliminary report of experience
with chlorambucil. Br J Ophthalmol 59:397, 1975 502. Mamo JG, Azzam SA: Treatment of Behçet's disease with chlorambucil. Arch Ophthalmol 84:446, 1970 503. Mamo JG: Treatment of Behçet's disease with chlorambucil. Arch Ophthalmol 94:580, 1976 504. Tricoulis D: Treatment of Behçet's disease with chlorambucil. Br J Ophthalmol 60:55, 1976 505. Tabbara KF: Chlorambucil in Behçet's disease: A reappraisal. Ophthalmology 90:906, 1983 506. Jabs DA, Rosenbaum JT, Foster CS, et al: Guidelines for the use of immunosuppressive drugs in patients with ocular
inflammatory disorders: Recommendations of an expert panel. Am J Ophthalmol 130:492, 2000 507. Nussenblatt RB, Palestine AG, Chan CC, et al: Effectiveness of cyclosporin therapy for Behçet's disease. Arthritis Rheum 28:671, 1985 508. Masuda K, Nakajima A, Urayama A, et al: Double-masked trial of cyclosporine versus colchicine and long-term
open study of cyclosporine in Behçet's disease. Lancet 1:1093, 1989 509. Whitcup SM, Salvo EC, Nussenblatt RB: Combined cyclosporine and corticosteroid therapy for sight-threatening
uveitis in Behçet's disease. Am J Ophthalmol 118:39, 1994 510. Palestine AG, Austin HA, Balow JE, et al: Renal histopathologic alterations in patients treated with cyclosporin
for uveitis. N Engl J Med 314:1293, 1986 511. Yazici J, Pazarli H, Barnes CG, et al: A controlled trial of azathioprine in Behçet's syndrome. N Engl J Med 322:281, 1990 512. Mamo JG, Baghdassarian A: Behçet's disease: A report of 28 cases. Arch Ophthalmol 71:4, 1964 513. Colvard DM, Robertson DM, O'Duffy JD: The ocular manifestations of Behçet's disease. Arch Ophthalmol 95:1813, 1977 514. Michelson JB, Chisari FV: Behçet's disease. Surv Ophthalmol 26:190, 1982 515. Scouras J, Koutroumanos J: Ischemic optic neuropathy in Behçet's syndrome. Ophthalmologica 173:11, 1976 516. Michelson JB, Michelson PE, Chisari FV: Subretinal neovascular membrane and disciform scar in Behçet's
disease. Am J Ophthalmol 90:182, 1980 517. Mamo JG: The rate of visual loss in Behçet's disease. Arch Ophthalmol 84:451, 1970 |