ETIOLOGY
Juvenile Rheumatoid Arthritis
Juvenile rheumatoid arthritis (JRA) represents a group of chronic pediatric arthritic disorders with an onset before the age of 16 years. These conditions are sometimes referred to as juvenile chronic arthritis (JCA), especially in the British literature. JRA is the most frequently identifiable etiology of pediatric anterior uveitis.5 The arthritic disease is usually divided into three subtypes: systemic, polyarticular, and pauciarticular (Table 2).9–13 The pauciarticular category is further subdivided into early (type I) and late-onset (type II) forms of the disease.11
TABLE 2. Juvenile Rheumatoid Arthritis Antinuclear
| Subgroup | JRA | Gender | Uveitis | Antinuclear Antibody | Comments |
| Systemic | 15%–20% | M @ F | Rare | Fever, maculopapular rash, hepatosplenome galy, pericarditis, lymphadenopathy | |
| Polyarticular | 30%–50% | F @ M | 5%–7% | 25% | May be associated with malaise and fever |
| Type I | 35% | F @ M | 40%–50% | 50% | Risk of uveitis increases with younger age at onset. |
| Type II | 15% | M @ F | 10%–20% | Late childhood onset; HLA-B27 positive; may develop ankylosing spondylitis |
Pauciarticular arthritis is defined as the involvement of four or fewer joints in the first 6 months after the onset of the disease. It is not uncommon for a patient with the pauciarticular disease to progress to polyarticular involvement after 6 months of the disorder. The risk of uveitis, however, is determined by the nature of the child's arthritis during this initial 6-month time period, even though more joints may later be affected. The characteristics of a patient with the highest risk of developing uveitis is a female ANA-positive individual with the onset of pauciarticular arthritis before the age of 2 years.
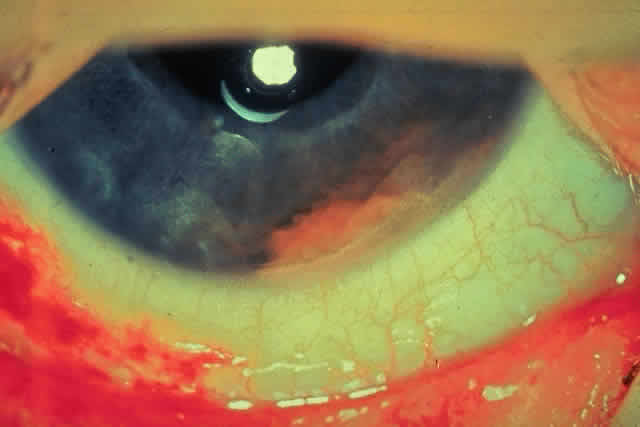
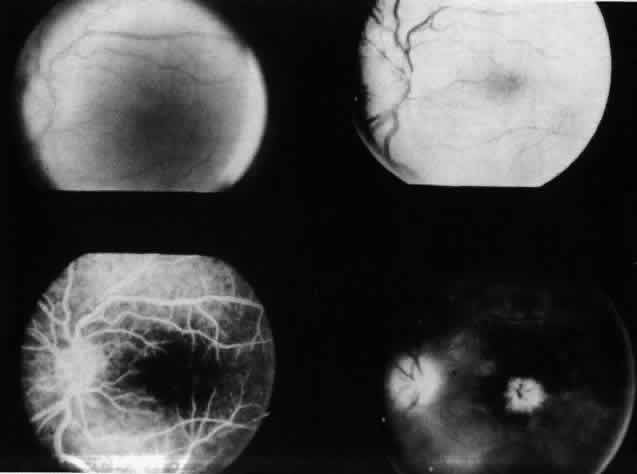
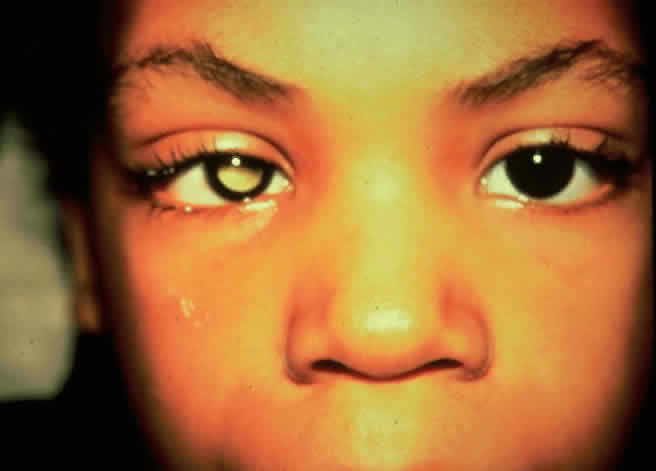
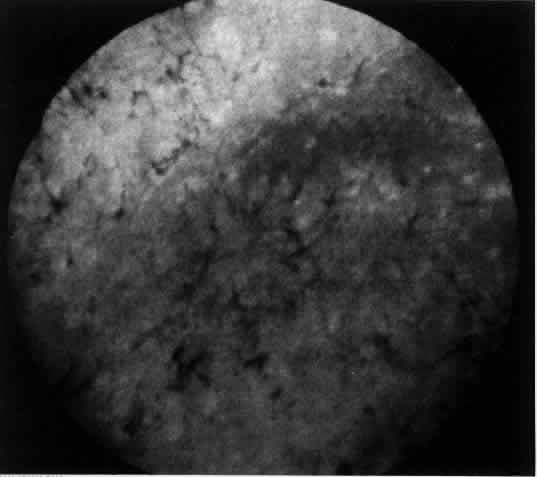
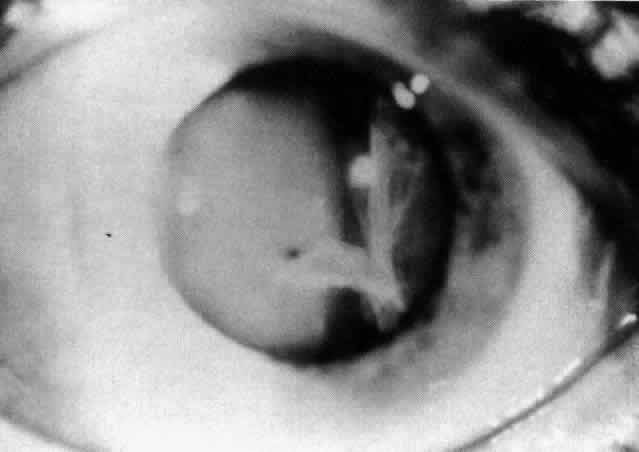
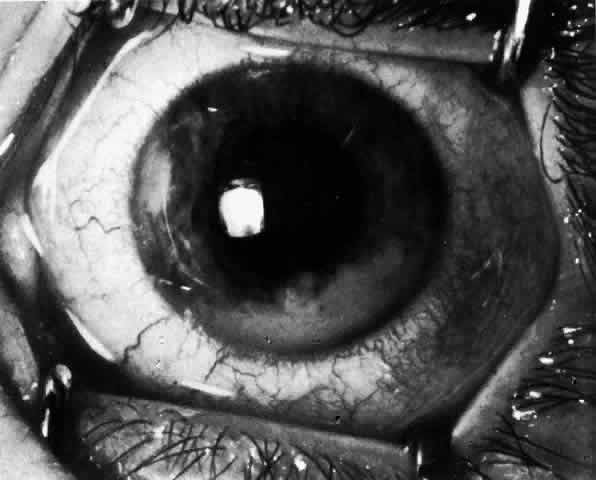
JRA uveitis is most frequently a chronic, nongranulomatous iridocyclitis that is bilateral in 71% of patients.14 Occasional African-American patients exhibit large granulomatous-appearing keratic precipitates. Since the ocular inflammation is usually asymptomatic and the affected eyes are frequently nonerythematous, it is not uncommon for complications to develop before the uveitis is detected.15 The complications include cataract, glaucoma, band keratopathy, posterior synechiae, and phthisis bulbi (Fig. 1; Color Plate 1A).12
|
|
The arthritis usually precedes the uveitis. If iridocyclitis does develop, it will occur in approximately half of the affected patients within 2 years of the onset of the arthritis.16 Pauciarticular individuals should be checked every 3 months. If they are ANA positive, the eye examinations are performed every 2 to 3 months. It has been recommended that JRA patients be examined for uveitis during the 7 years following the onset of uveitis.16 Polyarticular patients may be evaluated at 6-month intervals for the development of uveitis, while children with systemic-onset JRA are examined annually. There is no correlation between the degree of arthritis activity and the ocular inflammation. Although the arthritis may diminish and resolve with age, the uveitis often remains chronic and may persist into adulthood.17
Initial treatment of the iridocyclitis involves mydriatics and topical corticosteroids. Periocular corticosteroids, which may require a general anesthetic for administration in this pediatric population, and systemic corticosteroids are used for more severe ocular inflammation. Immunosuppressives are used for unresponsive vision-threatening cases of uveitis.18,19 These medications should only be used with the assistance of qualified pediatric consultants.
Ankylosing Spondylitis
Ten to 15% of patients with ankylosing spondylitis develop their arthritic disease in childhood.20 The onset of juvenile ankylosing spondylitis is usually at 8 to 10 years of age. Males are affected more commonly than females, as is true for the adult disorder form of the disease.20 Unlike the adult disorder, however, juvenile ankylosing spondylitis frequently presents as a peripheral arthropathy before physical or radiographic findings of sacroiliac joint involvement.21 Although both adult and juvenile ankylosing spondylitis manifest an approximately 95% association with HLA-B27,22,23 it is important to note that this histocompatibility antigen is not specific for ankylosing spondylitis and occurs in other forms of anterior uveitis,14 such as Reiter's syndrome and HLA-B27-associated anterior uveitis.
An acute, recurrent nongranulomatous iridocyclitis affects 10% to 20% of patients with juvenile ankylosing spondylitis. This anterior uveitis is typically described as unilateral because, although both eyes may be inflamed, it is unusual to note simultaneous involvement. The uveitis may precede or follow the arthritis, and the levels of activity of the two disorders are unrelated. Treatment of the ocular inflammation requires mydriatics and corticosteroids. Complications of the iridocyclitis include cataract, glaucoma, and posterior synechiae.
Reiter's Syndrome
Reiter's syndrome is infrequent in children, although it has been described in this patient age group.14,20 It is classically described as a triad of noninfectious urethritis, arthritis, and conjunctivitis.20 Males are more often affected than females. Although patients with Reiter's syndrome may have sacroiliitis and HLA-B27-positivity, the presence of urethritis, keratoderma blennorrhagica of the palms and soles, and ulcerations of the mouth and genitals differentiates this disorder from ankylosing spondylitis. Unlike Behçet's disease, the mucosal lesions are usually not painful.
A nongranulomatous anterior uveitis affects 3% to 12% of patients.22 This recurrent anterior uveitis is treated with topical and periocular corticosteroids.
Fuchs' Heterochromic Iridocyclitis
Although this disorder usually occurs in adults, it has been reported in the pediatric population.24 It is frequently asymptomatic with a chronic low-grade iridocyclitis, which is almost always unilateral. Hypochromia of the involved eye, as a result of iris atrophy, is typical but not invariable. The hypochromia may be difficult to detect in patients with blue irides. The keratic precipitates that accompany this anterior uveitis have a characteristic small, stellate appearance.25 Cataracts and open-angle glaucoma are the major complications of Fuchs' heterochromic iridocyclitis. Diagnosis is determined by the history and clinical manifestations, since there are no laboratory tests available for this disorder. Treatment with topical corticosteroids is usually ineffective. Mydriatics are often not required because posterior synechiae are uncommon.
Kawasaki Disease
Kawasaki disease is an acute multisystemic vasculitis characterized by fever, cervical lymphadenopathy, erythematous mucocutaneous lesions, and desquamation of the skin of the hands and feet.26–28 It is also referred to as mucocutaneous lymph node syndrome. This idiopathic disorder primarily affects infants and young children. It may be associated with arthritis and cardiac abnormalities, including coronary aneurysms, arrhythmias, and myocarditis. These heart complications produce sudden death in 1% to 2% of patients with Kawasaki disease.28
Bilateral bulbar conjunctival erythema has been reported in 96% of these children.26,29 Anterior uveitis is also common, occurring in 66% of individuals. If examined within 1 week of the onset of this disease, 83% of patients were reported to have an anterior uveitis.26 The ocular inflammation was mild and resolved without sequelae.26 No therapy is indicated for this uveitis.
Psoriatic Arthritis
Pauciarticular or polyarticular arthritis is an uncommon complication of psoriasis in childhood.20 The joint inflammation may precede or follow the development of the psoriasis. Patients with psoriatic arthritis may develop a bilateral, chronic anterior uveitis.14 Topical corticosteroids are the treatment for this ocular inflammation.
Acute Interstitial Nephritis
Acute interstitial nephritis is an uncommon renal disorder that may be the result of an immune reaction to antibiotics, nonsteroidal anti-inflammatory drugs, or infection.30,31 It is characterized by the nonspecific systemic complaints of low-grade fever, pallor, fatigue, and weight loss.30,31 Acute interstitial nephritis may be associated with an increased erythrocyte sedimentation rate, elevated serum creatinine value,31 proteinuria, glucosuria, microhematuria, leukocyturia, and excretion of casts.30,31 The diagnosis is established by renal biopsy. The prognosis in childhood is good.
A bilateral anterior uveitis may be associated with acute interstitial nephritis.30–33 The iridocyclitis may precede, follow, or occur concomitantly with this renal disorder. The ocular inflammation is treated with topical corticosteroids and responds well to therapy.33
Orbital Pseudotumor
Orbital pseudotumor is an idiopathic inflammatory lesion within the orbit that simulates a neoplasm.34 It is primarily a disorder of adults, although it may also occur in the first or second decade of life.34 This inflammatory disorder may present with multiple nonspecific signs and symptoms.34 Systemic complaints include headache, sore throat, lethargy, anorexia, weight loss, abdominal pain, and vomiting. Ocular findings include eyelid swelling, ptosis, pain, motility disorders, and diplopia. Proptosis occurs in 80% of patients.35
Unlike the adult form of the disease, uveitis may be associated with pediatric orbital pseudotumor.34 The ocular inflammation may involve the anterior, the posterior, or both segments of the eye. Patients with uveitis have an increased incidence of recurrence of orbital pseudotumor, as well as bilateral involvement.34
The possibility of an orbital pseudotumor should be considered in those pediatric patients with a persistent or recurrent uveitis and a previous negative diagnostic workup.36 These children may present without proptosis. Diagnostic evaluation includes ultrasonography, computed tomography, or magnetic resonance imaging. Treatment requires the use of systemic corticosteroids.
Lens-Induced Uveitis
Rupture of the lens capsule liberates lens material into the eye cavities. This results in one of two different types of uveitis that subside when the lens substance is removed. The uveitis is often associated with glaucoma. The phacotoxic reaction usually occurs in the presence of a hypermature cataract. The lens material acts as a chemical irritant, probably acting directly on the iris and ciliary body. Macrophages enter to engulf the liberated material. No polymorphonuclear cells are seen.
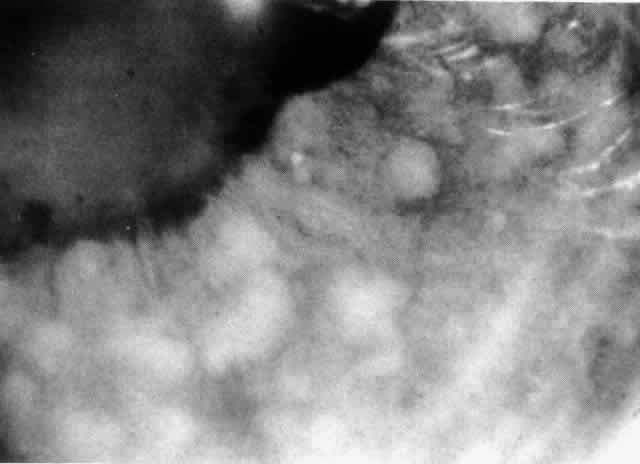
Endophthalmitis phacoanaphylactica results from an underlying sensitivity to lens protein that is probably amplified by bacteria or their toxins. Typically, a break in the lens capsule occurs in one eye as a result of surgery or injury. After inflammation has subsided in the first eye, the second eye develops a severe anterior granulomatous uveitis after surgery or trauma. Polymorphonuclear cells and macrophages are found in the aqueous iris and lens. Treatment involves removal of the lens, in addition to the general therapy for uveitis discussed in the section on treatment.
PROPOSED EVALUATION
Although it is traditional to present a flow sheet and table suggesting any etiologic evaluation in a patient with uveitis, Table 3 is offered with some hesitation. The “shotgun” approach to an etiologic evaluation should be abandoned in favor of evaluation on the basis of history and cost effectiveness. Table 3 is relevant only for the patient with severe disease in whom no localizing clue to etiology can be elicited.
TABLE 3. Diagnostic Evaluation of AnteriorUveitis in Children
History
Ocular examination
Pediatric evaluation
Complete blood cell count
Serologic
Antinuclear antibody
Serum lysozyme
Serum protein electrophoresis
Fluorescent treponemal antibody absorption
HLA typing
Enzyme-linked immunosorbent assay, indirect fluorescent antibody for Lyme
disease
Skin tests
Tuberculin
Radiographic
Chest
Sacroiliac joint
Gastrointestinal series
Aqueous tap
Fluorescein angiography
Lumbar puncture
Routine blood studies should include a complete blood cell count to rule out the presence of leukemia as well as the fluorescent treponemal antibody absorption test for syphilis, which is most specific in cases of syphilitic ophthalmologic involvement. A number of serologic determinations can be performed to determine the etiology of anterior uveitis. These include determination of angiotensin converting enzyme levels, serum lysozyme elevation, and serum protein electrophoresis, antinuclear antibody, and HLA-B27 studies. The angiotensin converting enzyme is of great value in the presence of active sarcoid disease in adults, but results in children must be interpreted with caution (see Sarcoidosis). Serum lysozyme elevation and serum protein electrophoresis with an increased α2-globulin fraction are highly suggestive of sarcoid. Because of its association with JRA, antinuclear antibody determination is a most important serologic examination. Finally, while the presence of HLA-B27 is an important marker in patients with uveitis, it is not helpful for management and therefore is not obtained routinely except in a research setting. Until a clearer picture regarding the ocular manifestations of Lyme disease is available, immunologic testing for this spirochete (by enzyme-linked immunosorbent assay [ELISA] or indirect fluorescent antibody testing) is indicated in patients with idiopathic chronic uveitis.
Skin testing with the Kveim test has been used in some centers. Blind conjunctival biopsy has proved generally ineffective in attempts to diagnose sarcoid. When a significant conjunctival nodule is present, the yield is high. Biopsy of the lacrimal gland and surrounding conjunctiva has also proved helpful in establishing a diagnosis of sarcoid. The tuberculin skin test (intermediate-strength purified protein derivative), which is used to diagnose the rare case of tuberculous-induced uveitis, identifies patients in whom antituberculous therapy is indicated when systemic corticosteroids are used.
Radiographic studies include chest roentgenograms to rule out sarcoid and tuberculosis, a sacroiliac joint study to exclude rheumatoid spondylitis, and a gastrointestinal series in patients in whom ulcerative colitis or regional enteritis is suspected. Additional studies using fluorescein angiography, vitreous and aqueous aspiration, and lumbar puncture are only occasionally helpful.
TREATMENT
Over the past 35 years, corticosteroids have been the cornerstone of therapy for anterior uveitis. Of currently available local corticosteroids, prednisolone acetate 1% is the preferred solution because of its ability to penetrate the intact corneal epithelium. When this agent is used as frequently as every 30 minutes, most children do not need periocular and systemic corticosteroids.
Treatment requirements of the identified etiologic entities vary widely. Some, such as traumatic anterior uveitis and Fuchs' heterochromic iridocyclitis, require minimal amounts of local corticosteroids to control inflammation. On the other hand, acute fibrinous uveitis in patients with spondylitis may require maximal application of corticosteroids. With the onset of chronic anterior segment inflammation, careful monitoring is required to regulate drug dosage. Generally, the presence of flare without significant anterior chamber cells does not warrant treatment. We prefer to use chronic corticosteroid therapy to maintain the anterior segment with slightly less than a 1+ response present (scale = trace to 4+). Periocular corticosteroids have been widely employed for 2 decades, but their use in children younger than 14 years of age is somewhat limited because general anesthesia is usually needed for their delivery. Systemic corticosteroids are seldom required to treat anterior segment inflammatory disease.
Cycloplegic-mydriatic agents are important in the treatment of childhood uveitis because of the tendency for posterior synechiae to form. In the early phases of inflammation, atropine 1% may be used as often as two to four times daily. Some ophthalmologists are concerned that long-term use of mydriatic agents may result in a chronically dilated pupil. Most of the complications associated with the pupil are not related to dilatation, however, but to relative miosis and the formation of posterior synechiae. As soon as the inflammation has diminished, homatropine 5% four times daily may be substituted for atropine. In chronic anterior uveitis with minimal anterior chamber reaction, the pupil is dilated once daily. The parent is instructed to administer homatropine 5% one hour before the child's sleeping hour, to inspect the pupil carefully for change in size and regularity, and to report any change in the pupil to the ophthalmologist the next morning. Refraction and prescription of bifocals are important in these patients because of the potential long-term use of these cycloplegic agents. Chronic unilateral cycloplegia may produce amblyopia in children unless the refractive error is corrected.
Specific therapy for anterior uveitis in children is limited to the use of specific antiviral agents (trifluridine for herpes simplex) and antibacterial agents in infectious uveitis (penicillin for syphilis). Nonsteroidal anti-inflammatory agents such as indomethacin (Indocin), phenylbutazone (Butazolidin), piroxicam (Feldene), and aspirin are only occasionally effective in anterior segment inflammatory disease. Whenever high-dosage corticosteroids have failed, nonsteroidal anti-inflammatory agents have also failed.
COURSE AND COMPLICATIONS
With early diagnosis and prompt treatment, inflammation generally subsides within 2 to 6 weeks. The poor response reported in childhood anterior uveitis is directly related to the delay in diagnosis. It is this delay that makes complications of uveitis more common in children than those associated with adult-onset disease. keratopathy.Band keratopathy is the most consistent of all complications associated with chronic anterior chamber inflammation in children.Although hypercalcemia and phthisis bulbi also cause band keratopathy, most band formation is secondary to uveitis. Unless the band results in a cosmetic defect or reduces acuity, treatment is not indicated. When treatment is required, the band is easily removed. After the administration of general anesthesia, the cornea is de-epithelialized and Gelfoam, moistened with a 0.1 molar edentate disodium (sodium versenate) solution, is placed over it; 5- to 10-minute applications produce chelation of the band. If vision is present in the other eye, patching the affected eye after instilling a local antibiotic or sulfa medication for 24 hours aids in the re-epithelialization process. When there is little or no vision in the untreated eye, no patch is used. This procedure may be repeated as often as necessary and is almost uniformly successful.
The mechanism for the production of glaucoma is as varied in childhood uveitis as it is in the adult form of the disorder.Unfortunately, the prognosis for children with inflammatory glaucoma is far worse than for adults. Medical management, when successful, may require the long-term use of carbonic anhydrase inhibitors, since ß-adrenergic blocking agents (timolol, betaxolol) and epinephrine derivatives are not always successful. Medical therapy controls elevated intraocular pressure in 50% of patients who develop it.
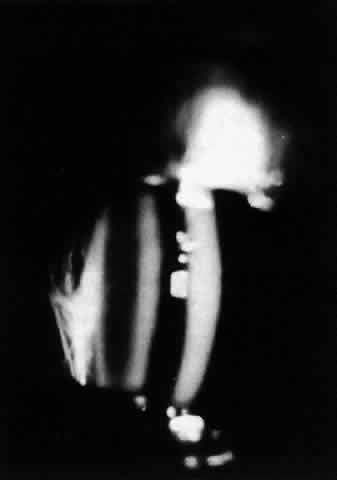
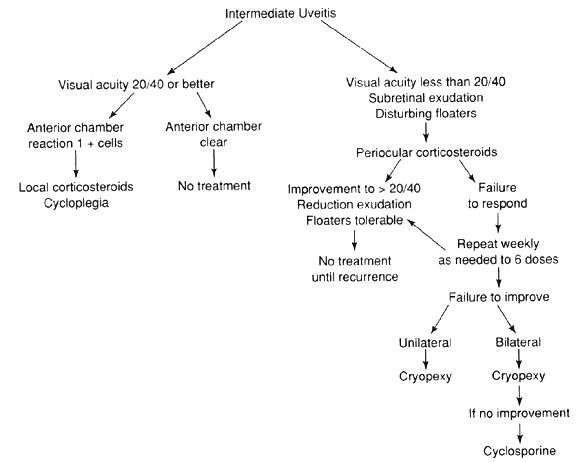
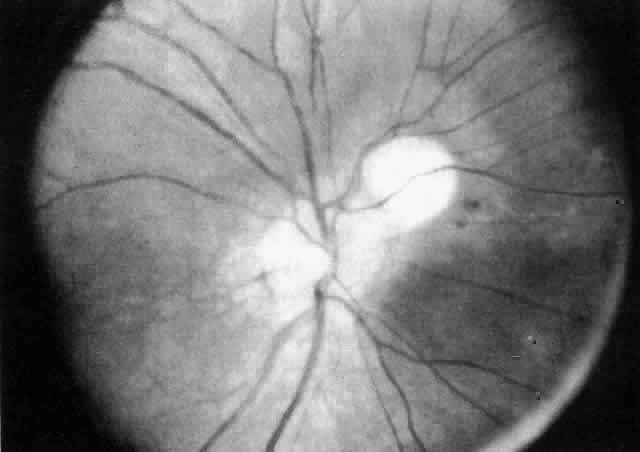
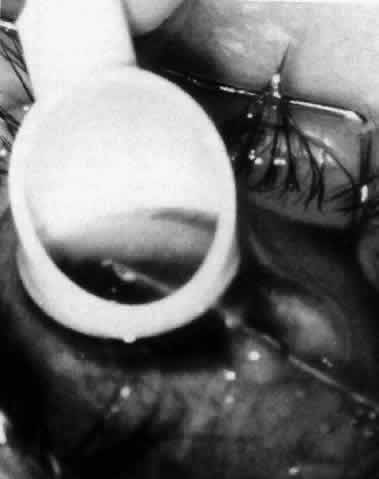
The variety of surgical procedures suggested by glaucoma specialists is adequate testimony to their lack of success. Filtering procedures have always been fraught with difficulty in all patients with inflammatory disease, but in younger patients the success rate is less than 50%. Trabeculectomy, peripheral iridectomy with thermal sclerostomy (advocated by Preziosa and Scheie), trabeculodialysis (advocated by Kanski; Fig. 2), and trabeculectomy with mitomycin have all failed to appreciably improve the prognosis of this complication.
|
Although the prognosis remains guarded in the management of cataract associated with inflammation, improved techniques offer significant hope for the young patient who presents with advanced cataract formation.Elimination of the posterior capsule prevents the later development of secondary membranes, thereby reducing the incidence of hypotony from ciliary body shutdown. A period of 6 months of relative inactivity of the inflammation is desirable before surgery. Periocular corticosteroids are administered at the time of surgery, and most of these eyes do not show marked additional inflammation. Orally administered postoperative corticosteroids are given only when indicated on a case-by-case basis. When extensive band keratopathy suggests that aphakic corneal contact lens fitting will prove difficult, the cornea should be chelated at the time of cataract surgery. Because of the character of the uveitis causing cataracts in children, intraocular lenses are seldom indicated. However, they may be used in carefully selected patients bulbi.Phthisis bulbi represents the end state of severe anterior uveitis in children. On occasion, however, these eyes respond well to a lensectomy-vitrectomy procedure even in the presence of a clear lens. The success of this operation probably depends on the removal of the cyclitic membrane, with restoration of aqueous formation and the elimination of hypotony.
PROGNOSIS
The prognosis for patients with anterior uveitis varies directly with the ability of the clinician to intervene and treat the disease in its early stages. The single largest group of patients who require careful monitoring are those with pauciarticular JRA.14,16 It is prudent to follow all patients who have a presumptive diagnosis of JRA in the previously described manner for 7 years after the diagnosis. For a few patients, early intervention will still fail to control many of the complications, and some of these patients may require immunosuppressive agents (e.g., cyclosporine). Although controlled evaluations of these agents in children are not available, results of preliminary studies are encouraging.
DIFFERENTIAL DIAGNOSIS
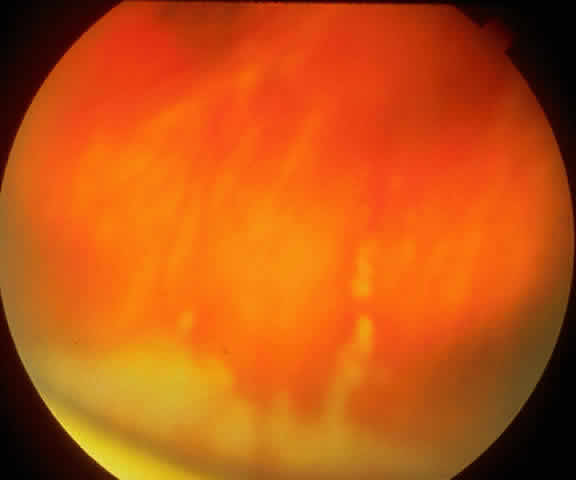
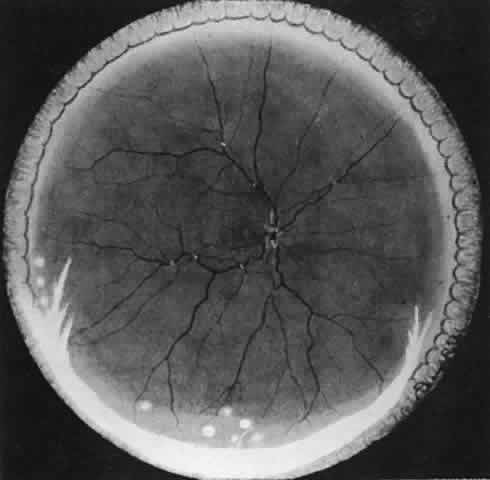
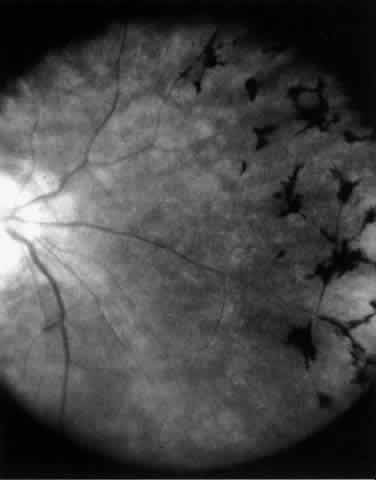
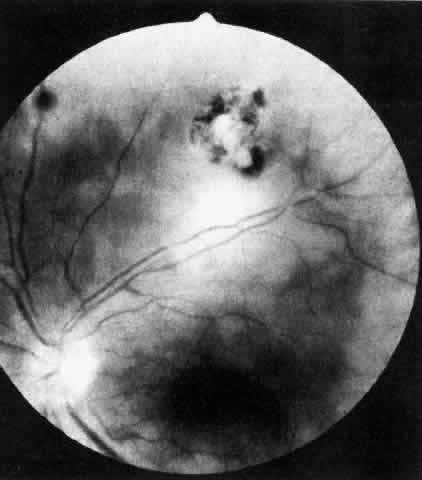
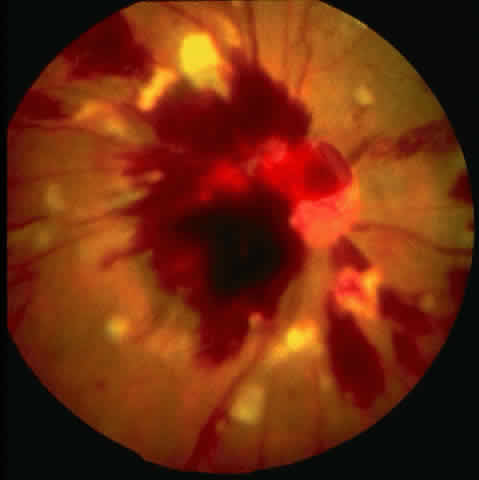
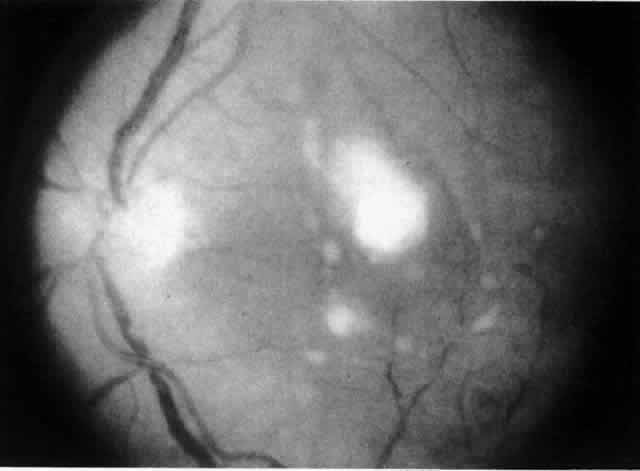
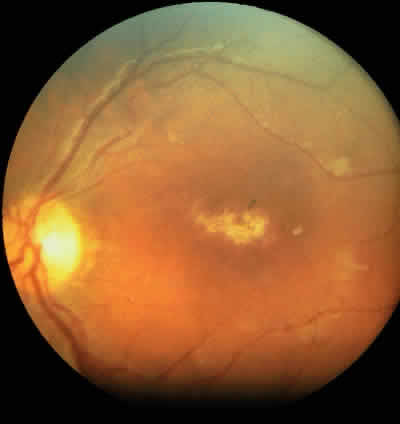
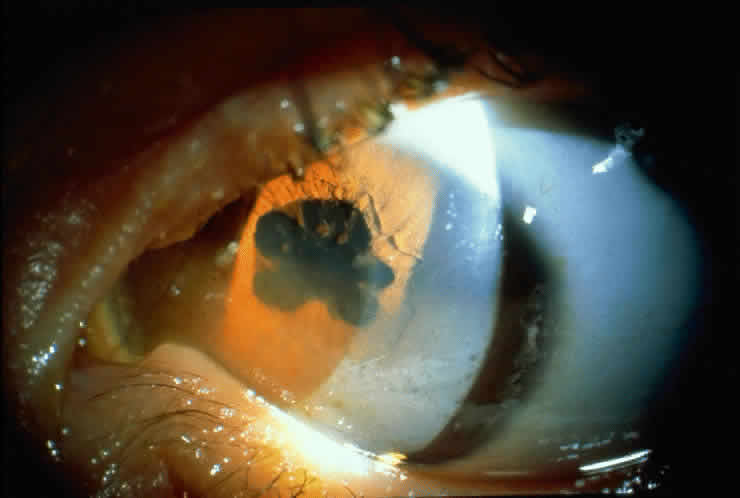
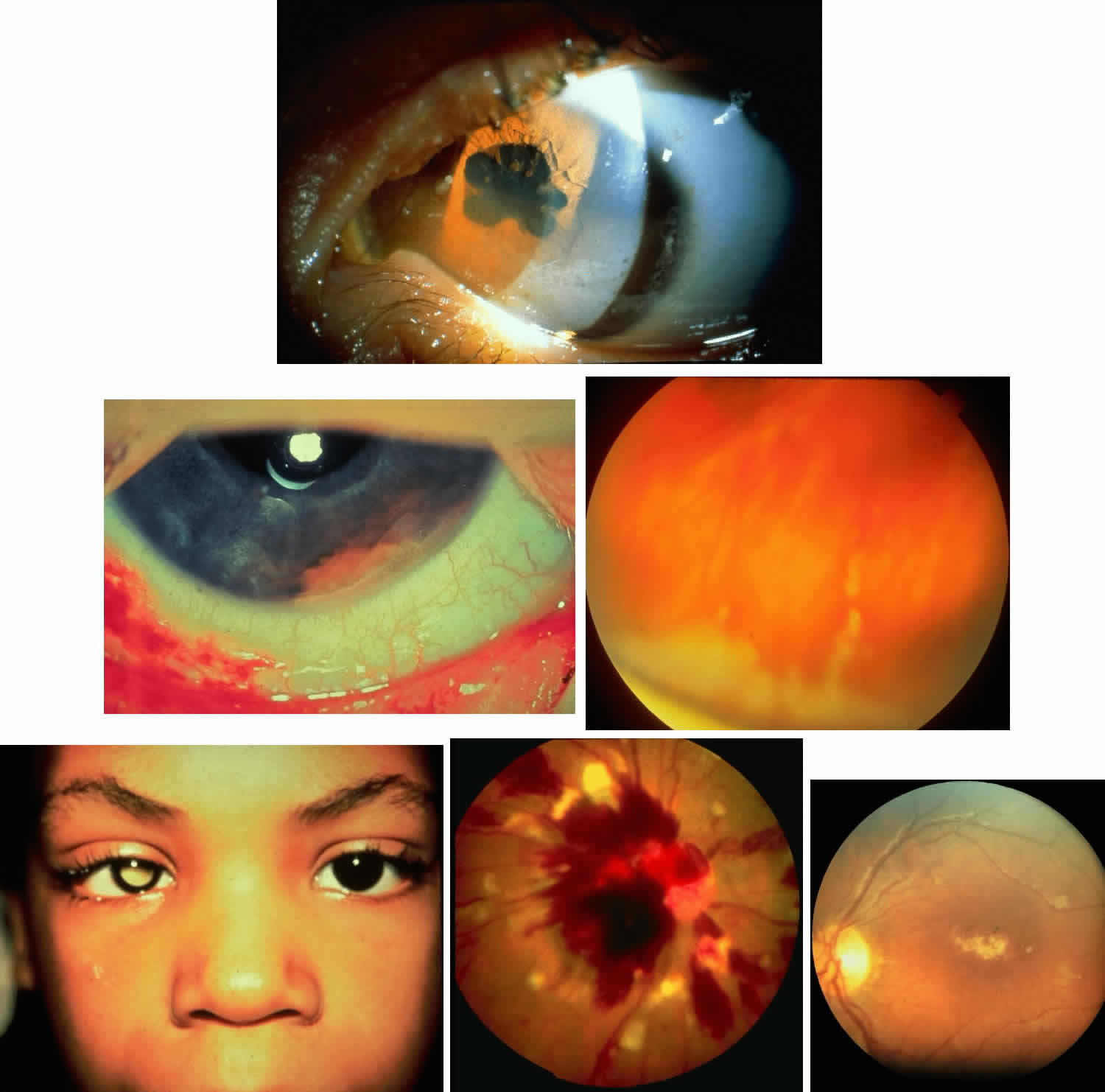
Table 4 lists the anterior segment masquerade syndromes. A pseudohypopyon in a patient with retinoblastoma (Fig. 3), although a diagnostic consideration, can easily be ruled out with a biomicroscopic or fundus examination. Juvenile xanthogranuloma characteristically presents as hyphema and a fleshy tumor on the iris (Fig. 4; Color Plate 1B).37 This rare condition commonly has associated skin lesions and is responsive to the use of local, periocular, or oral corticosteroids as well as to irradiation. Rarely, leukemia in childhood may present a significant anterior chamber reaction and hypopyon, although a general physical examination and associated laboratory signs will help identify this masquerade syndrome. Peripheral retinal detachment may have flare and cells in the anterior chamber, but careful ophthalmoscopy will reveal the presence of the detachment. An intraocular foreign body should be suspected in patients with unilateral uveitis who have a history of trauma and fail to respond to treatment.
TABLE 4. Masquerade Syndromes: Anterior Segment
| Disease or Disorder | Age | Signs of Inflammation | Diagnostic Studies |
| Retinoblastoma | <15 | Flare, cells, pseudohypopyon | Aqueous tap for lactic dehydrogenase levels and cytology |
| Leukemia | <15 | Flare, cells, heterochromia | Bone marrow, peripheral blood smear, aqueous cytology |
| Intraocular foreign body | Any | Flare, cells | Radiography, ultrasonography |
| Malignant melanoma | Any | Flare, cells | 32P test, fluorescein angiography, ultrasonography |
| Juvenile xanthogranuloma | <15 | Flare, cells, hyphema | Examination of skin, iris biopsy |
| Peripheral retinal detachment | Any | Flare, cells | Careful ophthalmoscopy |
|
|