“Sympathetic ophthalmitis is a specific bilateral inflammation of the entire uveal tract of unknown etiology, characterized clinically by an insidious onset and a progressive course with exacerbation and pathologically by a nodular or diffuse infiltration of the uveal tract with lymphocytes and epithelioid cells. It almost invariably follows a perforating wound involving uveal tissue.”
This disease was known to Hippocrates and was fully described and named by MacKenzie in 1840.2 Schirman, as cited in Duke-Elder and Perkins,1 presented clinical data and supportive rabbit experiments in 1892 and 1905; however, it was Fuchs3 who described the classic histopathology as a distinct ocular disease in a treatise published in 1905. Capitalizing on the theme of SO, Sir Frederick Treves was even able to develop a totally different medical subject into an entertaining horror story in his famous book, The Elephant Man and Other Reminiscences, in 1923.4
PREVALENCE
The prevalence of SO is difficult to measure because it has always been a relatively rare disease; as a result of improvements in modern surgical and medical treatment, it has become even more uncommon. Still, on average, the older literature generally has found the incidence after a perforating injury to be approximately 2%.1 In the American Civil War (1861–1865), 16% of all ocular injuries led to the development of SO. In the Franco-Prussian War (1870–1871), the prevalence of SO after ocular injuries was 55.5% among the Germans and 50% among the French.5 In contrast, only rare cases of SO were reported in World Wars I and II,3,5,6 and none were reported in the Korean, Vietnam, and Persian Gulf conflicts. In surveys unrelated to warfare, Von Holland7 and Allen and Ho8 found SO in 0.7% of 838 and in 0.3% of 348 cases of ocular trauma, respectively.
After trauma, surgical wound is the second most important cause of SO. Allen and Ho8 found one case (0.015%) of SO in a survey of 6,613 cases of ocular surgery (5,325 cataract extraction and 1,288 glaucoma procedures). In 105 cases of SO, Lubin and associates9 found that traumatic perforating wounds accounted for 53.5% of cases, surgical wounds for 40.4%, perforating corneal ulcers for 6%, and malignant melanoma for 4%. Nevertheless, postsurgical SO has been a rare event.
With the development of vitreous surgery and exploratory oculotomy in ocular trauma, however, SO is becoming more prevalent. This may be partly explained by increased awareness and better diagnosis. In 1975, Michaels and Ryan10 reported that SO is an extremely rare complication after vitrectomy. Since then, however, more isolated cases of SO after a number of different intraocular surgeries, including pars plana vitrectomy, have been described.11–16 Other ocular surgeries that may induce SO include iridectomy,9,17,18 iridencleisis,9,19 evisceration,20 retinal detachment repair,21–25 keratectomy,1,9,19 laser photocoagulation,16,26 laser cyclotherapy,27–29 and local irradiation.30,31 Recently, Kilmartin reported a minimum estimated incidence of 0.03/100,000 in the United Kingdom and the Republic of Ireland. The main current risk is surgery, particularly retinal surgery.32
Men have a higher prevalence of trauma-induced SO than women, probably because men have higher overall injury rates.9,33 In postsurgical cases, however, the rates are equal between the sexes.33,34 Trauma-induced disease is more common among children, whereas postsurgical disease is more prevalent among older patients because of their increasing need for ophthalmic surgery.33,35
CLINICAL MANIFESTATIONS
The interval between ocular injury and the onset of SO is important. It has been reported to be as short as 5 days9,36 or as long as 66 years.37,38 In general, 65% of SO cases occur 2 weeks to 2 months after injury, and 90% occur before 1 year.1,9,39
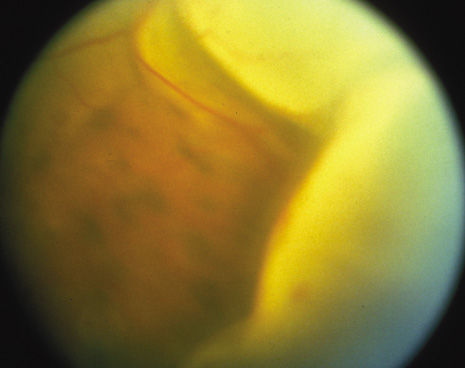
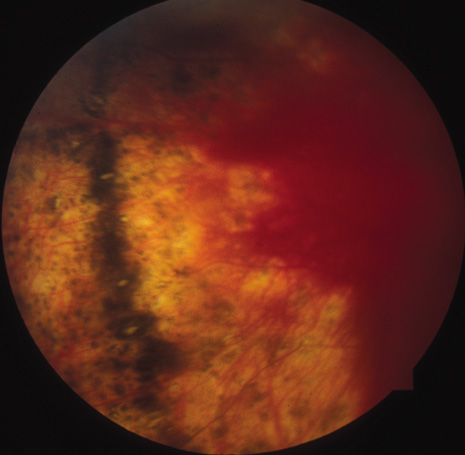
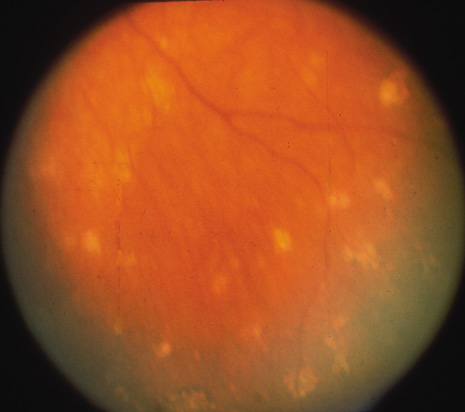
The onset of the disease is insidious. The injured eye becomes painful and photophobic, and visual acuity is diminished. The uninjured (sympathetic) eye then follows a similar course. On ocular examination, the injured eye shows persistent low-grade uveitis despite the healing wound. Characteristic bilateral panuveitis can be recognized by “mutton-fat” keratic precipitates, ciliary flush, aqueous cell and flare, posterior synechiae, vitreous cells and haze, choroidal thickening and infiltration, and optic nerve head edema. The presence of small, yellowish-white inflammatory infiltrates, 60 to 700 μm in diameter, at the level of the retinal pigment epithelium (Dalen-Fuchs nodules), but more commonly located at the midperiphery, is pathognomonic of SO (Fig. 1). In severe cases, rubeosis iridis, cataracts, pupillary membrane formation, and exudative retinal detachment can occur. Chorioretinal scarring may eventually form, and the eyes may become phthisic.1,40–42
|
In some cases of SO, the bilateral uveitis may be so mild or transient that the diagnosis is missed.1,40,43 Sympathetic ophthalmia can present as a mild, nongranulomatous uveitis that is easily misdiagnosed as idiopathic uveitis. This atypical clinical presentation may result from early enucleation of the injured eye before the classic histopathology of SO has appeared. Alternatively, the pathology may be modified by immunosuppressive treatment.43,44 In addition, progressive subretinal fibrosis with multifocal granulomatous chorioretinitis45 can occur in SO.46 This rare entity is characterized by rapid vision loss with progressive subretinal fibrosis involving the macula.45 This form of the disease progresses rapidly and often is unresponsive to systemic immunosuppressive therapy.
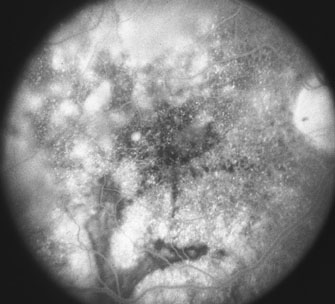
Fluorescein angiography of patients with SO often shows multiple areas of hyperfluorescence and leakage at the level of the retinal pigment epithelium (Dalen-Fuchs nodules) and the choroid (choroidal granuloma).39–41,47,48 If there is a serous retinal detachment, pooling of dye in the late frames of the angiogram can be observed.11,40,48–50 Although early hypofluorescence followed by late hyperfluorescence has been reported, this may indicate obliteration of the choriocapillaris and the presence of numerous Dalen-Fuchs nodules, as well as choroidal granulomas to obscure choroidal fluorescence in the early phase.11,41,50,51 Indocyanine green angiography (ICG) can reveal hypofluorescent patterns that persist throughout angiography or fade in the late phase that represent active or cicatricial lesions in the choroids.29,52,53
There may be a genetic component to disease susceptibility. With racially matched controls, HLA-DR4 and HLA-DRw53 were found to be highly associated with SO in Japanese patients,54 whereas the occurrence of HLA-DR4 and DQw3 was reported significantly elevated in American patients with SO.55 Using HLA serological and polymerase chain reaction amplification (PCR)–based DNA typing in 16 Japanese patients and 50 controls, Shindo and associates56 reported HLA-DRB1*04, DQA1*03, and DQB1 04 were significantly associated with SO. Kilmartin and colleagues57 also confirmed this genotype association (HLA-DRB1*04 and DQA1*03) in Caucasian patients with SO.
The differential diagnosis of SO includes Vogt-Koyanagi-Harada syndrome, phacoanaphylactic uveitis, sarcoidosis, chronic idiopathic uveitis, and other granulomatous uveitis induced by mycobacteria or fungi.1,41,42 A history of ocular injury and a lack of systemic involvement or proven ocular infection, however, are helpful indicators in diagnosing SO. Vitiligo, poliosis, alopecia, dysacusis, and meningeal irritation, findings more commonly reported in Vogt-Koyanagi-Harada syndrome, may be noted rarely in SO.1,40,58 Blodi59 and Lubin and colleagues9 reported SO in 23% of 170 and in 46% of 105 cases of lens-induced endophthalmitis, respectively; in the latter study, of 31 cases of lens-induced endophthalmitis occurring after 1950, there was only one case of SO. In reviewing 100 cases of SO from the Armed Force Institute of Pathology (AFIP) file, Croxatto and associates60 found 14 cases (22% of the 46 eyes enucleated before 1950, in contrast to 7% of the 54 eyes enucleated after 1950) associated with phacoanaphylactic endophthalmitis; the occurrence of SO was unrelated to the severity of the choroidal inflammation.
The more severe the inflammation, the poorer the prognosis of SO. The sooner the diagnosis and more intensive the therapy, the better the outlook.19,34,61,62 In their study of 105 cases, Lubin and associates9 found that 93.3% of the patients with mild inflammation had 20/70 (6/21)* vision or better; and 100% of patients with severely inflamed eyes had 20/70 vision or worse. In a study with long-term follow-up data (mean follow-up of 23 years), Makley and Azar61 demonstrated ocular complication in 70% of patients. In this study, only 65% of the 17 patients treated with corticosteroids attained a visual acuity of 20/60 (6/18) or better. In a study at Moorfield hospital, a visual acuity of between 20/20 and 20/60 was achieved in 14 of 18 patients (77%).62 Chan and colleagues34 reported on 32 SO patients from 1982 to 1992: 16 patients had a final visual acuity of 20/40 or better, and 10 had a visual acuity worse than 20/200. The remaining six patients had visual acuity ranging from 20/40 to 20/200. In this series, prompt and aggressive application of anti-inflammatory therapy was the key to achieving better visual outcome.
HISTOPATHOLOGY
The classic histopathologic features of SO were first described by Fuchs in 1905,3 confirmed by Easom and Zimmerman,63 and summarized by Green.64 Both the injured and the sympathetic eyes show similar histopathologic features. The main feature of SO is a diffuse, non-necrotizing, granulomatous inflammation in the uvea. The choroid is markedly thickened and infiltrated by lymphocytes and nests of macrophages, epithelioid cells, and giant cells. Other classic histopathologic findings include a relative lack of retinal involvement, sparing of the choriocapillaris, and the formation of Dalen-Fuchs nodules, consisting mainly of epithelioid cells located between Bruch's membrane and the retinal pigment epithelium (Fig. 2). Fuchs3 found that approximately 25% of his classic cases of SO had Dalen-Fuchs nodules.
|
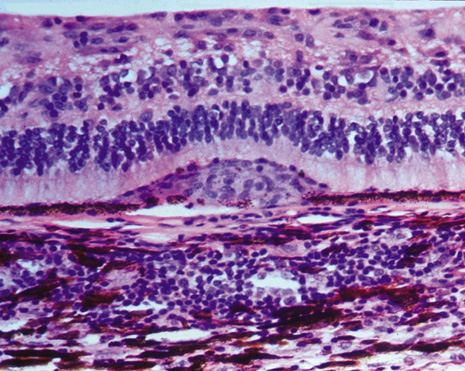
Lubin and associates9 reported that more than one nodule was present in 33 of 93 cases (35.5%). Kuo and co-workers65 observed Dalen-Fuchs nodules in all 50 Chinese patients with severe SO. Nine of 29 patients (31%) had Dalen-Fuchs nodules in an AFIP series.66 Breaks in Bruch's membrane at the site of Dalen-Fuchs nodules also have been identified.50,67
Many other histopathologic features of SO have been reported in the literature. Retinal involvement (e.g., retinal perivasculitis, retinitis, retinal gliosis) has been documented in 30% of cases of SO by Winter,19 in 42.2% by Lubin and associates,9 and in 55% by Croxatto and colleagues.60 Other features include choriocapillaris obliteration, reported in 40% of cases by Croxatto and colleagues60 and in 52% by Kuo and co-workers;65 chorioretinal scarring was reported in 7% of cases by Croxatto and colleagues60 and in 25% by Makley and Azar.61 Lubin and associates9 found optic atrophy in 54.4% of SO cases. Nongranulomatous inflammation and focal inflammation also can be observed in SO. In the 29 AFIP cases of SO, 11 did not have granulomatous inflammation and 13 presented only with focal choroidal inflammation.66 Eosinophils and plasma cells were more commonly seen in the non–corticosteroid-treated and more severely inflamed cases.
Marak and co-workers68 observed histologic variations related to race in SO. They found thickened choroid, an increase in the number and size of choroidal granulomas, and eosinophilia more commonly among African-American patients than among white American patients. Kuo and associates65 did not find any significant histopathologic differences between Chinese patients and white American patients. The finding of Marak and co-workers may reflect the fact that the cases in the African-American group were more severe at the time of diagnosis.
As stated, there is a rare form of SO characterized by a progressive subretinal fibrosis with multifocal granulomatous chorioretinitis leading to rapid vision loss.45 Histopathologic findings include multifocal granulomatous inflammation involving the outer retina and choroid with giant cells distributed along the Bruch's membrane.46 In this variant serum antibodies against photoreceptor and retinal pigmented epithelium can be detected.
Immunohistopathologic studies of SO show that choroidal infiltrates are predominantly composed of T lymphocytes.16,67,69–72 We have demonstrated a greater number of CD4+ T cells (T helper/inducer cells) in the early stages of the disease and CD8+ T cells (T suppressor/cytotoxic cells) in the late stages of the disease.67 In contrast, B cells are found in less than 5%16,69,70,72 to 15%16,67 of choroidal infiltrates; however, higher percentages of B cells were found in four of the 29 AFIP eyes at the end stage of the disease.66 Formation of pseudogerminal center with aggregate of B cells surrounding by T cells is found in the variant of SO with progressive subretinal fibrosis and blindness associated with multifocal granulomatous chorioretinitis.73
The main cellular component of both choroidal granuloma and Dalen-Fuchs nodules are bone marrow–derived monocytic (histocytic) cells. The granulomas of SO have immunohistochemical staining profiles that are similar to the granulomas of sarcoidosis.74 In the late stage of SO, however, degenerated retinal pigment epithelium can be an important component of Dalen-Fuchs nodules.75 Despite the accumulation of T lymphocytes in eyes with SO, there is no reported difference in the lymphocytic subpopulations in the peripheral blood of patients with SO, with the exception of one atypical case reported by Kaplan and co-workers.72 Expressions of cell adhesion molecules and major histocompatibility (MHC) class II antigens on the ocular resident cells have been observed in eyes with SO and may be important in the pathogenesis of the disease.16,76,77
PATHOGENESIS
The etiology of SO is still an enigma. Two hypotheses dominate the older literature: (a) autoimmunity against uveal melanin, uveal melanocytes, retinal pigment epithelium, or retinal antigens; or (b) a viral or bacterial infection.1 Later studies have emphasized immune system involvement in patients with ocular inflammation.41 In 1971, investigators reported that lymphocytes from patients with SO might respond to heterogeneous or homogeneous retinal and uveal tissues.78–80 Using lymphocyte transformation and a leukocyte migration inhibition assay, Rahi and associates43 reported that cells from five of six patients with SO showed positive reactions to uveoretinal tissues, whereas seven of ten cases of posttraumatic nongranulomatous uveitis showed negative responses to uveoretinal antigen. These observations suggested that cell-mediated hypersensitivity could be an important pathogenic mechanism. The immunohistopathologic finding of the predominantly T-lymphocytic infiltration in ocular tissue supports a cell-mediated immunologic response (delayed hypersensitivity) to ocular antigens in both typical and atypical cases of SO.16,70–72,81,82
Although the immune nature of SO appears to be established, the inciting antigen is still in question.40,41,43,44,69 Retinal extracts are highly antigenic and easily produce retinouveitis in experimental animals. Four main retinal antigens are rhodopsin,83 retinal soluble antigen,84,85 interphotoreceptor retinoid binding protein,86 and recoverin.87 Using a single injection of purified rhodopsin with adjuvant in monkeys, Wong and co-workers79 observed choroidal granulomatous inflammation, loss of outer segment photoreceptor, perivascular retinitis, and optic disc edema. Extensive investigations on experimental autoimmune uveoretinitis models have been reported since the isolation of retinal soluble antigen in 1977 by Wacker and colleagues,84 who found it to be located on the membrane of the outer segment of the photoreceptor. Like retinal soluble antigen, interphotoreceptor retinoid binding protein (located in the retinal interphotoreceptor space) and recoverin (a retinal calcium-binding protein) are highly uveitogenic and can induce T-cell–mediated autoimmune uveoretinitis.86,87 Similar clinical and histopathologic pictures of SO can be produced in experimental models by alternating the immunizing dose, route of administration, adjuvants, and animal species.88,89 These features include Dalen-Fuchs nodules, nongranulomatous and granulomatous uveitis, retinal vasculitis, retinal detachment, corneal keratic precipitates, iridocyclitis, and vitreitis.
In 1992, Broekhuyse and co-workers90 described an insoluble uveal melanin preparation that can produce an inflammation limited to the uvea in immunized animals. Chan and associates91 reported that spontaneous recurrence occurred in this experimental model; however, Bora and colleagues92,93 emphasized the anterior uveal involvement with immunization of melanin protein. Such a role of a uveal antigen is more consistent with clinical data because SO usually follows uveal tissue trauma. Recently Bora's group reported that type I collagen is the autoantigen in their experimental autoimmune anterior uveitis model.94
The role of an ocular penetrating wound in the development of SO has been proposed.95,96 Barker and Billingham proposed that absence of lymphatics within the eye lets intraocular antigens circulate to the blood and spleen and may induce blocking antibodies or suppressor cells; when the antigens go to the regional lymph nodes, a cell-mediated immune response can be induced. Thus, in SO cases, penetrating trauma may result in exposure of uveoretinal antigens to the conjunctival lymphatics, as confirmed by subconjunctival injection of retinal soluble antigen in rabbits.88 Exposure to bacteria (e.g., Propionibacterium acne), viruses, and other infectious agents through the wound may serve as an adjuvant to induce the disease.41,97
TREATMENT
Enucleation
The classic and only known prevention against SO is enucleation of the injured eye before the other eye develops disease.1,40 However, because this is an extremely rare disease and techniques for the surgical repair of injured eyes have greatly improved, one should always make every attempt to save the injured eye if it has useful vision. Once SO develops, enucleation of the injured eye usually cannot improve the symptoms and final visual acuity of the sympathizing eye.19,40 Lubin and associates9 did suggest, however, that enucleation within 2 weeks after symptoms of SO occurred may improve the visual prognosis. Kuo and co-workers65 reported significantly fewer recurrences of inflammatory disease in patients who underwent early enucleation but a lack of improvement in ultimate visual acuity. In a retrospective clinicopathologic study of 30 cases of SO, Reynard and colleagues98 found that early enucleation of the existing eye was associated with a benign clinical course: visual acuity better than 20/50 (6/15) and fewer and milder relapses than eyes that underwent late enucleation (p = 0.008). Even when corticosteroid-treated patients were excluded in the study, the association between early enucleation and a benign clinical course remained significant (p = 0.018). There still is controversial whether enucleation is helpful to prevent development of SO in the uninjured eye.32,99
Sympathetic ophthalmia occasionally develops after enucleation of the injured eye. Of the 18 cases of SO from the Moorfield Eye Hospital, there was one case of SO that occurred in a patient whose injured eye was enucleated before the onset of disease;62 similarly, two of the 29 AFIP cases occurred after enucleation of the traumatized eye.66
Pharmaceutical Agents
CORTICOSTEROIDS.
Intensive systemic and topical corticosteroid treatment can improve visual outcome and decrease relapse of SO.1,9,34,40,61,65,98 Adequate high-dose corticosteroids should be given for the first few days until the disease is controlled. Makley and Azar61 found that 9 of 14 treated patients attained 20/60 vision or better, Lubin and associates9 noted that 13 of 18 treated patients achieved 20/50 vision or better, and Reynard and co-workers98 reported that 18 of 22 treated patients had 20/50 vision or better. It is known, however, that prophylactic corticosteroids do not prevent the development of SO. This is based on the repeated observation in the 1950s that SO developed in patients who were on high-dose steroids.1 Recently intravitreal steroids have been successfully used in the management of SO.10
Cytotoxic Agents
Despite the advantages of corticosteroids, their side effects should be taken into consideration. Other immunosuppressive therapies such as azathioprine, chlorambucil, or cyclophosphamide may be used when patients are unresponsive to corticosteroids or are having severe corticosteroid-related side effects.35,101 Corticosteroids also may be used in combination with other immunosuppressive agents. When these cytotoxic agents are used, it is essential to monitor the status of the patient's bone marrow; it is also advisable to coordinate the patient's care with an internist.
CYCLOSPORINE.
Because eyes with SO usually are infiltrated with numerous activated T cells, cyclosporine, a potent inhibitor of T-cell function, can be a very effective therapeutic agent in the treatment of SO. Nussenblatt and colleagues34,97,102 have used cyclosporine as a corticosteroid-sparing agent in patients with SO, and the results have been encouraging. However, because cyclosporine can cause hepatic and renal toxicities, patients need to be monitored closely. The recommended dosages for a combination of cyclosporine and steroids are cyclosporin A, 3 to 5 mg/kg per day; and prednisone, 15 to 20 mg/day. An optimal therapy for SO depends on improved understanding of the disease mechanism
Biologics
Recently several biologics agents are available as therapeutic target for various autoimmune diseases such as Crohn's disease, Behçet's syndrome, arthritis, psoriasis, and uveitis.103–105 A number of these agents are available and effective for the treatment of uveitis.106,107 Nussenblatt and his associates108,109 have reported the success of humanized anti–interleukin-2 receptor alpha antibody in the treatment of noninfectious uveitis. Several investigators have reported the success of anti–tumor necrosis factor-alpha in the treatment of uveitis associated with Behçet's syndrome and other autoimmune uveitis.110,111 Although these biologics are relatively expensive, they could be considered as an alternative therapeutic agents for sympathetic ophthalmia.