EPIDEMIOLOGY The term herpes zoster derives from the Greek words herpein, meaning “to creep or to spread,” and zoster, which means “a girdle, sword, belt, or zone,” applied because
of the belt-like dermatomal distribution of the disease. When
there is involvement of the ophthalmic branch of the trigeminal nerve, the
term herpes zoster ophthalmicus (HZO) is used. In 1818, Mahlis
reported that the cutaneous eruption of herpes zoster followed
the distribution of nerves, but it remained for Hutchinson in 1865 to
describe HZO in detail and to report on several cases.43,44 Herpes zoster causes approximately 1% of all skin disease, and most
commonly affects the dermatomal distributions of the thoracic (56%) and
trigeminal nerves (15%).45 In aggregate, the thoracic dermatomes are the most common areas involved
in herpes zoster, but the trigeminal nerve is the single most common
dermatomal site. Herpes zoster is four to five times more common in
patients who have been immunocompromised by malignancy or immunosuppressive
therapy than in those who have not, however, most patients with
herpes zoster have no underlying systemic disease.46 A retrospective study of 1000 patients with HZO found that only 12 patients (1.2%) presented either with a malignancy or in
an immunocompromised state. No new examples of malignancy or immunosuppression
were found in follow-up evaluations.47 For 15 years, Ragozzino and co-workers48 followed prospectively 590 patients with herpes zoster who did not have
underlying malignancy. They found that: (1) 9.3% had
trigeminal nerve involvement; (2) there was an equal incidence
of herpes zoster between male and female patients; (3) there
was a relatively low incidence of malignancy (1.1% per
year, which is similar to the normal population); and (4) the
average annual incidence rate was 1.3 per 1000 person–years. HZO
does not appear to be more prevalent in a particular race or
gender, but it does seem to have a predilection for elderly patients, who
are also more likely to develop other systemic complications from
the disease. Herpes zoster nearly always occurs in patients who have had previous exposure
to VZV (chickenpox). Two theories have been proposed
to explain the adult form of VZV infection, namely the latency theory
and the theory of altered immunity with reinfection.
The latency theory states that after an initial VZV infection the virus
remains latent in one or more dorsal root ganglia.3,17
Later in life and as a function of T-cell alterations, varicella reappears
from the nerve root ganglion as herpes zoster, in a specific dermatomal
area and not in a diffuse cutaneous distribution.3,17,48
This so-called secondary (or symptomatic) zoster occurs in patients in
whom the host-parasite relationship to latent VZV has been altered by
age, trauma, systemic disease, surgery, or iatrogenic immunosuppression.16
There is a decrease in VZV-neutralizing antibody preceding this event.7,49,15
Herpes zoster may also develop in immunocompetent patients who harbor the
latent virus and who are re-exposed to it, by contact with someone
who has an active varicella or zoster infection (so-called
primary, spontaneous, or infectious zoster).16,50,51 This is the theory of altered immunity with reinfection, but it is not known whether reactivation or fresh exogenous infection
by VZV is the cause.16,51,52 Age is the most common predisposing factor to herpes zoster, probably as
a result of alterations in T cells and a decrease in neutralizing VZV
antibody associated with senescence, allowing reactivation of latent
virus.16 Herpes zoster occurs most commonly in the fifth to seventh decades of
life; however, some patients with zoster are younger, have a history of
recent exposure to someone with active varicella or zoster, and have not had serious predisposing underlying systemic disease.7,8 PATHOGENESIS AND HISTOPATHOLOGY After primary VZV infection, the virus becomes latent within one or more
sensory nerve root ganglia and later reactivates in association with
age, immunocompromised state or trauma to the involved ganglion.7,17,52 Other presumed triggers include tuberculosis, syphilis, radiation therapy, and
systemic corticosteroid use.16 Viral proliferation in the nerve root ganglion causes inflammation and
tissue destruction.
Some of the ophthalmic sequelae of zoster are the result of chronic inflammation
and ischemia secondary to vasculitis.53
The inflammatory reaction can be granulomatous or diffusely lymphocytic.53–55
Granulomatous intracranial arteritis is a specific complication of HZO.53–55
Inflammatory sequelae may also be caused by direct viral infection. VZV
has been identified in sensory neural ganglia and has been recovered
from the skin and corneal surface during the early stages of zoster.53,55,56 The duration of VZV DNA detection on the ocular surface from rash onset
varies from 2 to 34 days.57 Live intraocular VZV has been recovered in a case of acute retinal necrosis
syndrome reported by Culbertson and co-workers.58 Herpes zoster–like particles have been found in the retina and in
the iris as well.59,60 CLINICAL CHARACTERISTICS Dermatomal Considerations The first (ophthalmic) division of the trigeminal nerve is most
frequently involved in HZO, 20 times more often than the second (maxillary) or
third (mandibular) divisions.16 The predilection of VZV for the ophthalmic division may result from trauma (leading
to virus reactivation) or because the localization
of herpes simplex virus in the portions of the ganglion that supply
the maxillary and mandibular branches has an interference effect against
later-acquired VZV.16 HSV preferentially resides in those portions of the ganglion that supply
the upper and lower lid, whereas VZV more commonly resides in the portion
that supplies the upper lid and nasociliary branch of the trigeminal
nerve—usually a sign of dissemination in an immunocompromised
host. The diagnosis of HZO is applied if the area of distribution
of the ophthalmic division of the trigeminal nerve is involved, even if
the globe itself is not inflamed.7 The ophthalmic division of the trigeminal nerve has three parts: the frontal
nerve, the lacrimal nerve, and the nasociliary nerve (Fig. 3). The supraorbital and supratrochlear branches of the frontal nerve
supply the upper lid and forehead and are most often involved in HZO. The
nasociliary nerve provides sensory innervation to the cornea, ciliary
body, iris, and conjunctiva. Its terminal branch is the anterior
ethmoidal nerve, which innervates the sides of the tip of the nose (alae
nasae) via the external nasal nerve. 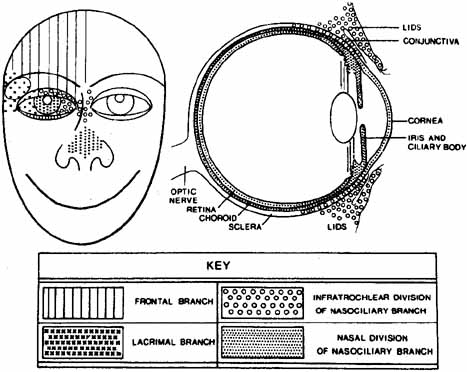 Fig. 3 Distribution of the major branches of first division of trigeminal nerve (cranial
nerve V). (Courtesy of Pavan-Langston
D Herpes Zoster Ophthalmicus: Int Ophthalmol Clin 10(4): 174, 1975) Fig. 3 Distribution of the major branches of first division of trigeminal nerve (cranial
nerve V). (Courtesy of Pavan-Langston
D Herpes Zoster Ophthalmicus: Int Ophthalmol Clin 10(4): 174, 1975)
|
Hutchinson43 was the first to observe that ocular involvement is much more common in
herpes zoster patients who had VZV involvement of the nasociliary branch
of the trigeminal nerve. The classic Hutchinson's sign of cutaneous
VZV involvement on the alae nasae, that is, the side of the tip
of the nose (and not just the tip of the nose), is evidence
of nasociliary nerve involvement. Ocular sequelae occur in 50% to 85% of
such cases.16 It is important to remember, however, that the eye may be seriously affected
in half of all cases, even if Hutchinson's sign is absent (Fig. 4). Thus, Hutchinson's sign is not entirely reliable as a predictor
of ocular involvement; however, if present, ocular inflammation
is more likely to occur. 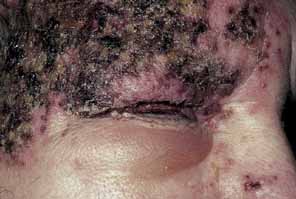 Fig. 4 Severe eruption of herpes zoster ophthalmicus with cutaneous necrosis and
sloughing. Involvement of the upper lid and sparing of the lower lid
is evident. Fig. 4 Severe eruption of herpes zoster ophthalmicus with cutaneous necrosis and
sloughing. Involvement of the upper lid and sparing of the lower lid
is evident.
|
HZO begins with a prodrome of severe burning or lancinating pain, dysesthesia
or hyperesthesia over the affected dermatome, mild fever, nausea, and
malaise (Fig. 5). The preeruptive pain may be insidious. At this stage, the cerebrospinal
fluid shows a mild lymphocytic pleocytosis. A cutaneous eruption
usually appears over one dermatomal area 3 to 5 days after the onset
of pain (Figs. 6 and 7). The erythema and swelling may be mistaken for an insect bite or
cellulitis. Initially, the HZO eruption is erythematous and maculopapular, but
soon becomes vesiculopapular, ulcerative, and eventually cicatrizing. Material
in the vesiculopapular eruption is at first clear, but
becomes yellow-brown and purulent as the vesicles erupt and
crust. Because VZV involves the dermis, cutaneous scarring, sloughing, and
subsequent exposure and superinfection can occur (Figs. 8 and 9). Herpes zoster without cutaneous eruption, called zoster sine herpete or zoster sine eruptione, has been observed. Schwab61 reported 16 patients with zoster sine herpete, in whom an anterior uveitis or iridocyclitis was present without ipsilateral
skin lesions; in 9 of these 16 cases there was a documented rise
and fall in VZV titers. 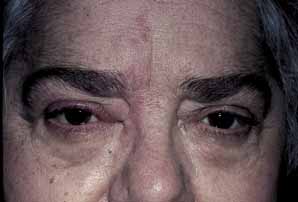 Fig. 5 Herpes zoster ophthalmicus (HZO). Preeruptive phase of HZO, 2 days
before cutaneous vesiculation. Fig. 5 Herpes zoster ophthalmicus (HZO). Preeruptive phase of HZO, 2 days
before cutaneous vesiculation.
|
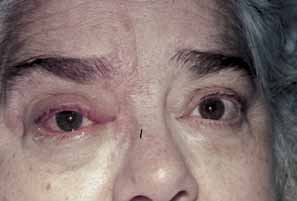 Fig. 6 Herpes zoster ophthalmicus. Same patient as in Figure 5, 1 day before cutaneous eruption. There is mild scleral injection as well
as upper lid vesicles. Fig. 6 Herpes zoster ophthalmicus. Same patient as in Figure 5, 1 day before cutaneous eruption. There is mild scleral injection as well
as upper lid vesicles.
|
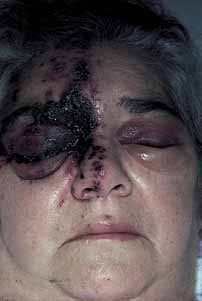 Fig. 7 Herpes zoster ophthalmicus. Same patient
as in Figure 5. Severe
hemorrhagic cutaneous eruption and involvement of the side of the
tip of the nose (positive Hutchinson's sign). Note the presence
of secondary contralateral eyelid edema (this is not dissemination).
Fig. 7 Herpes zoster ophthalmicus. Same patient
as in Figure 5. Severe
hemorrhagic cutaneous eruption and involvement of the side of the
tip of the nose (positive Hutchinson's sign). Note the presence
of secondary contralateral eyelid edema (this is not dissemination).
|
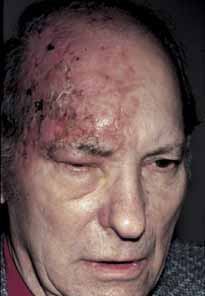 Fig. 8 Herpes zoster ophthalmicus. Initial crusting of vesicles in a 60-year-old
white man, 1 week posteruption. Fig. 8 Herpes zoster ophthalmicus. Initial crusting of vesicles in a 60-year-old
white man, 1 week posteruption.
|
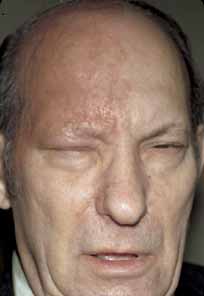 Fig. 9 Herpes zoster ophthalmicus. Same patient as in Figure 8. Prominent scarring 3 months posteruption. Fig. 9 Herpes zoster ophthalmicus. Same patient as in Figure 8. Prominent scarring 3 months posteruption.
|
Edema and vesicular cutaneous eruption of the lower eyelid in HZO do not
necessarily indicate involvement of the maxillary division of the trigeminal
nerve but may just represent local sequela of frontal nerve involvement. The
infratrochlear branch of the nasociliary nerve supplies
the medial aspect of the lower lid and conjunctiva. Therefore, VZV vesicles
can erupt along the medial aspect of the lower lid margin as a
result of involvement of the ophthalmic division of the nerve. Similarly, contralateral
eyelid edema is not usually a sign of dissemination, but
a local consequence of intense inflammation and swelling on the
involved side (see Fig. 7).
The cutaneous eruption of herpes zoster usually follows classic dermatomal
patterns, but vesicular eruption may occur several millimeters outside
a particular dermatomal distribution and still not signify dissemination.7
True herpes zoster dissemination is defined by involvement of two or more
noncontiguous dermatomes. Multiple crops of vesicles in more than one
noncontiguous dermatome are almost always a sign of an immunocompromised
state, and the patient should be appropriately evaluated (see “Systemic
Work-Up” section).
Ocular Involvement A convenient categorization is to define the ophthalmic manifestations
of HZO anatomically. All ocular and adnexal tissues can be affected by
herpes zoster inflammation (Table 1). HZO can cause blepharitis, canaliculitis, conjunctivitis, dacryoadenitis, keratitis, keratouveitis, iridocyclitis, vitritis, retinitis, acute
retinal necrosis, retinal vasculitis, choroiditis, inflammatory
glaucoma, optic neuritis, meningeal encephalitis, inflammatory extraocular
muscle palsies, anterior segment ischemia, episcleritis, scleritis, postherpetic
neuralgia (PHN), and cephalalgia. Secondary
cataract can result from uveitis or prolonged topical or systemic corticosteroid
therapy. Ocular sequelae can occur immediately, a few weeks
later, or seemingly idiopathically months to years after the cutaneous
eruption. It is imperative for the clinician to ask about a prior
history of herpes zoster or “shingles” in any patient with
chronic inflammatory eye disease. TABLE 1. Ocular Involvement in Herpes Zoster Ophthalmicus
| Lid and adnexa |
Anterior chamber angle, ciliary processes |
| Blepharitis—secondary infection with Staphylococcus aureus | Trabeculitis |
| | Glaucoma, secondary to trabeculitis or attendant steroids |
| Lid edema | Hypotonia |
| Vesicular lip eruption | Phthisis bulbi |
| Cicatricial entropion with or without trichiasis | |
| Cicatricial ectropion | |
| Chronic permanent scarring | Vitreous |
| Canaliculitis | Vitritis |
| Ptosis | Vitreous hemorrhage |
| Dacryoadenitis | |
| Conjunctiva | Retina |
| Hyperemic follicular conjunctivitis (rare) | Retinitis or neuroretinitis |
| Papillary conjunctivitis | Thrombophlebitis |
| Petechial hemorrhagic conjunctivitis | Acute retinal necrosis |
| Vesicular conjunctivitis | Retinal detachment, exudative, or rhegmatogenous |
| Conjunctival edema | Perivasculitis and arteritis |
| Cicatricial conjunctival changes | Macular edema |
| Cornea | Pupil |
| Acute epithelial keratitis | Adie's tonic pupil |
| Coarse punctate keratitis | Horner's syndrome |
| Pseudodendritic keratitis (zoster dendrites) | Argyll-Robertson pupils (?) |
| Mucous plaques | |
| Nummular anterior stromal keratitis | Optic nerve |
| Interstitial keratitis | Optic neuritis |
| Fascicular vascularizing keratitis | Retrobulbar neuritis |
| Serpiginous ulceration | Optic atrophy |
| Disciform keratitis | Papillitis and papilledema |
| Corneal hypesthesia or anesthesia | Neuroretinitis (papilledema and macular edema) |
| Neurotrophic keratitis, with or without melting and perforation | |
| Corneal scars | |
| Calcific band keratopathy | Extraocular muscles |
| Lipid keratopathy | Extraocular muscle palsies, myositis |
| Corneal edema | Ptosis |
| Peripheral corneal ulceration | Orbital apex syndrome |
| Epithelial inclusion cysts | Diplopia |
| | Orbit |
| | Orbital apex syndrome |
| Sclera and episclera | Proptosis |
| Scleritis | Exophthalmos |
| Episcleritis | |
| | Brain |
| Iris and uvea | Cephalalgia |
| Iritis | Hypesthesia |
| Sectoral iris atrophy | Anesthesia dolorosa |
| Iridocyclitis, occasionally “plastic” with hypopyon | Postherpetic neuralgia |
| | Contralateral hemiplegia |
| Anterior segment necrosis | Zosteriform temporal arteritis and angiitis |
| Choroiditis | Facial palsy |
| | Cerebrovascular accidents |
| Lens | Guillain-Barré syndrome |
| Cataract, secondary to inflammation or attendant steroids | LIDS AND LASHES.
During the active vesiculoulcerative cutaneous disease, VZV can usually
be cultured from skin lesions (see “Laboratory
Evaluation” section). Zoster dermal involvement can cause cutaneous
sloughing and secondary infection, usually from Staphylococcus aureus
or Streptococcus species.16 The pain
and hyperesthesia of the eyelid skin may be so intense during the first
3 to 5 days of cutaneous eruption that it may be impossible to examine
the conjunctiva, cornea, or internal eye (see Fig.
7). Eyelid involvement can be lead to permanent scar formation, cicatricial
ectropion or entropion, trichiasis, eyelash whitening (poliosis), eyelash
loss (madarosis), or even frank loss of eyelid tissue. Chronic contracted
eyelid scars may occur. Zoster scarring of the lacrimal punctum can cause
punctal stenosis and epiphora.
The skin vesicles and ulcerations are virologically sterile after 5 to 7 days
and heal within 2 or 3 weeks. Occasionally these lesions require
several months to heal totally as a result of secondary bacterial infection
or an allergic contact dermatitis caused by topical medication.7
Dysesthesia, hyperesthesia, and pain are common throughout the distribution
of the cutaneous eruption, especially in the elderly. PHN may be mild,
moderate, severe, or occasionally incapacitating (see “Neurologic
Involvement” section).
Hemorrhagic complications may occur, especially in patients who have underlying
hematopoietic diseases (e.g., thrombocytopenia, anemia) (see Fig. 7) in association with the cutaneous crisis. Subdural, subarachnoid, and
intracranial hematomas have been described in herpes zoster patients
taking anticoagulant medications concurrent with the onset of the
herpes zoster skin eruption.26 CONJUNCTIVA. Herpes zoster conjunctivitis can manifest as papillary, pseudomembranous, membranous, or
follicular. Transitory nonulcerative vesicles, hemorrhages, and
conjunctival cicatrization may occur.16 Conjunctival cytology reveals mostly a mononuclear and lymphocytic infiltration, but
polymorphonuclear leukocytes can be found in severe cicatrizing
membranous forms of the disease, even in the absence of microbial
superinfection. After the occurrence of pseudomembranous, membranous, or
vesicular types of conjunctivitis, chronic scarring and symblepharon
formation can occasionally ensue.16 CORNEA. Herpes zoster corneal disease is insidious in onset, protean in manifestation, and
potentially visually devastating, even if promptly and appropriately
managed. Herpes zoster keratitis manifests in six basic clinical
forms: (1) an acute or chronic epithelial keratitis; (2) nummular (coin-shaped) stromal keratitis; (3) disciform
keratitis; (4) limbal vascular (fascicular) keratitis; (5) serpiginous ulceration; and (6) neurotrophic keratitis, with or without corneal perforation. Acute Epithelial Keratitis. This coarse, punctate keratitis usually occurs 5 to 10 days after onset
of the dermatomal eruption. It is characterized by multiple fine, raised
intraepithelial lesions located paracentrally or at the limbus.61 There may be a subjacent anterior stromal infiltrate with a ground-glass
appearance. These initially coarse, punctate lesions sometimes
coalesce into elevated, snake-like, dendriform epithelial lesions, which
are called pseudodendrites to distinguish them from the true dendrites of herpes simplex keratitis (Fig. 10).16,62–65 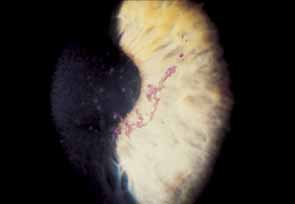 Fig. 10 Herpes zoster pseudodendriform keratitis, with heaped-up epithelial
cells surrounded by coarse punctate keratitis. Note rose bengal staining
of the surface of the pseudodendrite. Fig. 10 Herpes zoster pseudodendriform keratitis, with heaped-up epithelial
cells surrounded by coarse punctate keratitis. Note rose bengal staining
of the surface of the pseudodendrite.
|
Herpes Simplex Keratitis. The dendrite is a true ulceration with terminal end-bulb formations
and represents active viral replication. Corneal sensation is usually
decreased. Because epithelial herpes simplex represents active viral
replication, the edges of these lesions stain vividly with rose bengal
dye and the ulcer base with fluorescein dye. Debridement causes ulceration
of the epithelium, and steroids are contraindicated because they
enhance the growth of these lesions Herpes Zoster Keratitis. Pseudodendrites are typically smaller and snake-like, and they do
not have terminal end-bulb formations. Debridement of these lesions
causes less damage to the epithelium. Corneal sensation is variable, being
either normal or profoundly decreased. These lesions stain
only mildly with fluorescein and variably with rose bengal dye. Steroids
have little or no effect on these lesions.64 Pseudodendriform herpes zoster keratitis lesions are transient and usually
resolve within 2 weeks after the cutaneous eruption. Viral particles
were isolated in one study by Pavan-Langston and McCulley54 but were not identified in another study by Piebenga and Laibson.63 VZV DNA has been recovered from the ocular surface at various time periods
after the cutaneous eruption.56 In evaluating these studies, it is apparent that the timing of retrieval
of live virus is critical. Chronic Epithelial Keratitis. Chronic epithelial keratitis was described by Piebenga and Laibson63 as a chronic form of herpes zoster keratitis characterized by epithelial
mucous plaques that can appear from 1 week to as late as 1 year (typically 3 to 4 months) after the cutaneous eruption. This occurs
in approximately 8% of patients with HZO.64 Corneal mucous plaques may be seen in isolation or in association with
limbal hyperemia and iridocyclitis (Fig. 11A). In areas of mucous-plaque formation, the cornea appears
edematous and desquamated (Fig. 11B). These pleomorphic plaques tend to wander over the surface of the
cornea.61 The chronic coarse mucous plaques of herpes zoster are elevated and associated
with diffuse anterior stromal haze.65 These lesions demonstrate poor fluorescein staining but vivid staining
with rose bengal dye.50 Debridement of these plaques is not associated with any damage to the
underlying epithelium, and steroids have little or no effect.61 Delayed corneal mucous plaques are typically culture-negative and
result from immune, neurotrophic, or abnormal epithelial phenomena.65,68,69 Soft contact lenses, lubrication, steroids, acetylcysteine, and antivirals
have been therapeutically inconsistent.55,62,64,67,68,69 Pavan-Langston and colleagues66 found VZV DNA by polymerase chain reaction (PCR) assay in some
cases of delayed herpes zoster mucous plaques and pseudodendrites, suggesting
an infectious association. Chern and co-workers67 described a chronic form of dendritic herpes zoster epithelial keratitis
in acquired immune deficiency syndrome (AIDS). 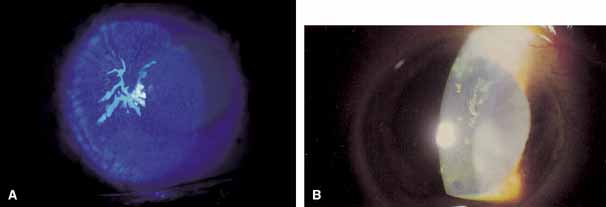 Fig. 11 A. Herpes zoster corneal mucous plaque, stained with fluorescein. B. Chronic epithelial plaque in herpes zoster ophtalmicus. Fig. 11 A. Herpes zoster corneal mucous plaque, stained with fluorescein. B. Chronic epithelial plaque in herpes zoster ophtalmicus.
|
Nummular Keratitis. Superficial, nummular anterior stromal opacities may be found beneath areas
of prior acute epithelial keratitis. Nummular keratitis appears as
granular infiltrates that are located in the anterior stroma.62,65 These lesions are usually evanescent but can lead to nebulous scarring (Fig. 12). 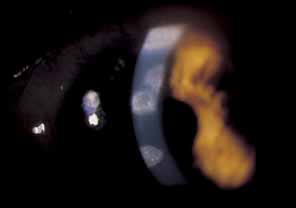 Fig. 12 Herpes zoster nummular stromal keratitis. Several discrete, coin-shaped
opacities are found in the anterior stroma. Fig. 12 Herpes zoster nummular stromal keratitis. Several discrete, coin-shaped
opacities are found in the anterior stroma.
|
Disciform Keratitis. Disciform keratitis may develop weeks to months after the original cutaneous
eruption and often recurs in the vicinity of an area of previous
acute epithelial keratitis. Rarely, it occurs spontaneously, with no
clinical evidence of previous epithelial disease. A central, well-defined, disc-shaped area of diffuse stromal edema without vascularization
is usually seen. If left untreated, disciform edema runs
a chronic course, producing scarring, occasional lipid deposition, and
neovascularization (Fig. 13). Herpes zoster disciform keratitis cannot be distinguished from
disciform keratitis secondary to herpes simplex virus or other viral diseases. Steroids
have some beneficial effect. 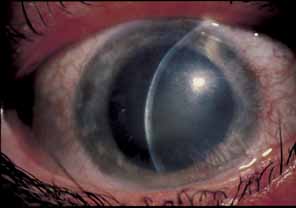 Fig. 13 Herpes zoster disciform keratitis with disc-shaped stromal edema. Fig. 13 Herpes zoster disciform keratitis with disc-shaped stromal edema.
|
Limbal Vascular (Fascicular) Keratitis. Limbal vascular (fascicular) keratitis may occur early or late
in relation to the cutaneous eruption. Limbal-vessel ingrowth
and stromal edema are seen. Episcleral or scleral inflammation is often
noted adjacent to the limbal involvement (sclerokeratitis). Limbal
vascular keratitis is thought to represent an immune-complex
vasculitis, resulting in corneal edema, vascularization, and in
some cases, the eventual deposition of lipid and subsequent scarring (Fig. 14). 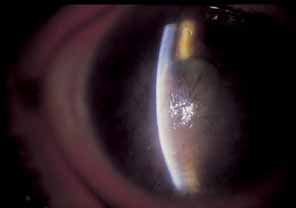 Fig. 14 Herpes zoster limbal vascular (fascicular) keratitis, with lipid
infiltrates and a leash (fascicle) of blood vessels encroaching
from the limbus. Note the intense vascular invasion and acute
interstitial keratitis. Fig. 14 Herpes zoster limbal vascular (fascicular) keratitis, with lipid
infiltrates and a leash (fascicle) of blood vessels encroaching
from the limbus. Note the intense vascular invasion and acute
interstitial keratitis.
|
Serpiginous Ulceration. Large geographic ulcers may form, and the entire epithelium may slough.70 This form of chronic serpiginous ulceration mimics HSV infection, in which
trophic ulceration without secondary pannus or vascularization leads
to corneal melting. Neurotrophic Keratitis. One of the most ominous corneal manifestations of HZO is neurotrophic keratitis; this
can occur because corneal sensation is often chronically
and profoundly decreased or absent. An inferior, horizontally oval, epithelial
defect with rolled-under edges is characteristic. These
indolent ulcers do not readily epithelialize and may lead to trophic
corneal ulceration, corneal melting, descemetocele formation, and ultimately
corneal perforation. Limbal corneal vascularization (pannus) to
fill the epithelial defect is a reparative process and should
be encouraged (Fig. 15).70 The resulting scar is usually in the inferior pupillary axis and is preferable
to corneal melting. Careful, long-term follow-up
for this insidious corneal complication is mandatory. Other corneal
findings in HZO include epithelial inclusion cysts69 and peripheral corneal ulceration.71 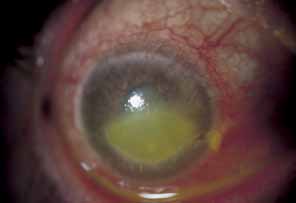 Fig. 15 Herpes zoster neurotrophic keratitis, showing indolent corneal ulceration
with adjacent rolled epithelial edges and stromal vascularization. Fig. 15 Herpes zoster neurotrophic keratitis, showing indolent corneal ulceration
with adjacent rolled epithelial edges and stromal vascularization.
|
IRIS AND UVEA. HZO can cause either a nongranulomatous or granulomatous iridocyclitis (anterior
uveitis) with extensive keratic precipitates and posterior
synechiae.69 The iridocyclitis tends to be chronic and recurrent and may require long-term, carefully
titrated topical corticosteroids for control. Anterior
uveal involvement can occur independent of corneal disease
and is heralded by blurred vision, intense photophobia, ciliary injection, edema
of the iris, hyperemia, miosis, and inflammatory anterior chamber
cells and protein flare.62 A severe “plastic” iridocyclitis, with hypopyon, hyphema, and
intractable secondary glaucoma, can be observed (Fig. 16). As a result of chronic iridocyclitis, corneal edema secondary to
endothelial damage and sectoral iris atrophy can occur.62 Endothelial cell loss is especially common with herpes zoster keratouveitis, even
with normal intraocular pressure.64,65 Histopathologically, herpes zoster iritis is an ischemic, occlusive vasculitis. The
typical sector iris atrophy that accompanies HZO is the
result of focal ischemic necrosis (Fig. 17).70 In contrast, herpes simplex iritis is primarily a lymphocytic infiltration
without ischemia, and causes a more diffuse iris atrophy.16,72,73 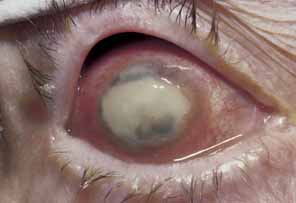 Fig. 16 Herpes zoster ophthalmicus. Corneal edema, abscess formation, and hypopyon
iridocyclitis in an eye that was subsequently enucleated for intractable
glaucoma and pain. Fig. 16 Herpes zoster ophthalmicus. Corneal edema, abscess formation, and hypopyon
iridocyclitis in an eye that was subsequently enucleated for intractable
glaucoma and pain.
|
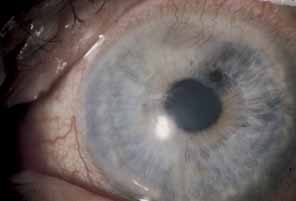 Fig. 17 Sectoral iris atrophy, status post–herpes zoster ophthalmicus. Note
the moth-eaten appearance of iris in sectoral area, underlying
a corresponding area of sclerokeratitis and limbal vascularization. Fig. 17 Sectoral iris atrophy, status post–herpes zoster ophthalmicus. Note
the moth-eaten appearance of iris in sectoral area, underlying
a corresponding area of sclerokeratitis and limbal vascularization.
|
Ciliary-process ischemia can cause hypotonia, cyclitic-membrane
formation, and ultimately atrophy and phthisis bulbi. LENS. HZO can cause cataracts either secondary to the acute anterior uveitis
or as a result of the attendant need for topical or systemic corticosteroids. The
cataracts of HZO are typically posterior subcapsular in location. ANTERIOR CHAMBER ANGLE AND GLAUCOMA. Glaucoma can occur in HZO as a result of several mechanisms: (1) plugging
of the trabecular mesh-work because of the presence
of cellular debris, iris pigment, or hyphema; (2) pupillary-block
glaucoma secondary to posterior synechiae, with resultant
iris bombé; (3) peripheral anterior synechiae; or (4) chronic
open-angle glaucoma long after the acute
infection is resolved, presumably because of damage to the trabecular
meshwork.62 VITREOUS. Vitreous opacities, vitritis, and vitreous hemorrhage have all been seen
in HZO.16 RETINA Retinal hemorrhages, retinal thrombophlebitis, branch or central retinal
artery occlusion, retinal arteritis and necrotizing retinopathy, necrotizing
retinitis, exudative or rhegmatogenous retinal detachment, and
ischemic perivasculitis have been described in HZO.72–75 In addition, neuroretinitis with papillitis and macular edema can be seen.76 Acute retinal necrosis (ARN) is a clinical syndrome with the
following characteristics: anterior chamber reaction, occlusive arteritis
and phlebitis of the retina and choroid, a necrotizing peripheral
circumferential retinitis, and vitritis.77 Anterior segment inflammation and late retinal detachment are prominent
sequelae of this disease. ARN results from viral infection caused by
human herpes viruses, which includes herpes simplex virus types 1 and 2, cytomegalovirus, and
VZV. Of these, varicella zoster virus is thought
to account for the majority of cases of ARN.58,59 ARN is an infrequent complication of HZO.58,78–80 Ganatra et al.60 used PCR to identify both VZV and HSV type I as causative in ARN in patients
over 25 years of age, whereas they found HSV type II caused ARN
in patients younger than 25 years old. Most investigators now accept
that immunosuppression is a condition compatible with ARN. In fact, there
is evidence that there may be an immunogenetic predisposition to the
development of ARN, with an apparent association with the HLA-DQw7 antigen.77 A newer clinical entity known as the progressive outer retinal necrosis (PORN) has
been recognized as a unique and devastating VZV
retinopathy associated with AIDS (Fig 18; also see section below on herpes zoster in AIDS)87. 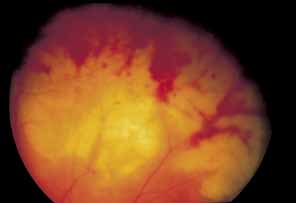 Fig. 18 Progressive outer retinal necrosis in a patient with herpes zoster ophthalmicus (HZO). Fig. 18 Progressive outer retinal necrosis in a patient with herpes zoster ophthalmicus (HZO).
|
PUPIL. Involvement of the sympathetic nervous system can result in Horner's
syndrome, which involves pupillary miosis, ptosis, and anhidrosis of
the involved side. A pseudo-Argyll-Robertson or a tonic (Adie's) pupil, secondary to herpes zoster ciliary ganglionitis, can
also occur.16 OPTIC NERVE. Herpes zoster optic neuritis can appearing as an isolated finding, in association
with macular edema in neuroretinitis, as a retrobulbar neuritis, or
as an ischemic optic neuropathy.76 A finding of herpes zoster optic neuritis does not necessarily indicate
central nervous system involvement by herpes zoster because local transmission
of the virus within the orbit can occur from the fifth to the
second cranial nerve.50,72 Optic neuritis associated with cerebrospinal fluid pleocytosis and increased
protein, however, indicates meningeal involvement.16 EXTRAOCULAR MUSCLES. Ophthalmoplegia is a well-known complication of HZO, various studies
have showin a rate of 11% to 31%.81 Marsh and colleagues81 found extraocular motor palsies in 31% of 77 consecutive patients
with HZO; however, only 28% of these patients were symptomatic.81 A highly significant association was observed between the occurrence of
oculomotor palsies and the clinical severity of the herpes zoster infection, as
well as the presence of iritis and iris atrophy.81 Typically, the third cranial nerve is involved, usually within the first 2 weeks
of the initial cutaneous eruptions. The fourth and sixth cranial
nerves can also be involved, either in isolation or with third-nerve paresis. Most
cases of herpes zoster extraocular muscle palsies resolve within 1 year.81 Contralateral or bilateral involvement occurs in approximately 12% of
these patients 81 ORBIT. HZO orbital myositis resulting proptosis and extraocular muscle palsies
has been described.82 HZO can produce ptosis from a secondary Horner's syndrome. Kattah
and Kennerdell83 reported on two patients with orbital apex syndrome (optic neuropathy
with complete ophthalmoplegia and anesthesia), associated with
meningoencephalitis. A patient with HZO with proptosis, optic neuropathy, and
restriction of abduction and up-gaze showed marked enlargement
of the inferior and medial rectus muscles on computed tomography (CT) scanning, which resolved with systemic corticosteroid
therapy (Fig. 19A and 19B).84 Orbital myositis is a cause of HZO-associated ophthalmoplegia, an
entity most often attributed to primary or secondary cranial nerve
VZV involvement. 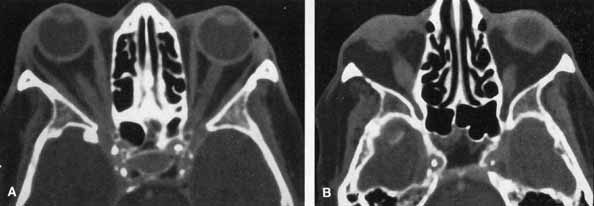 Fig. 19 Orbital myositis in herpes zoster ophthalmicus (HZO). Axial contrasted
orbital computed tomographic scan, demonstrating enlargement
of right medial rectus (A) and right inferior rectus (B) muscles. Fig. 19 Orbital myositis in herpes zoster ophthalmicus (HZO). Axial contrasted
orbital computed tomographic scan, demonstrating enlargement
of right medial rectus (A) and right inferior rectus (B) muscles.
|
Neurologic Involvement One of the most dreaded complications of HZO is PHN and cephalgia. The
Scandinavian word for herpes zoster, helvedesild (“hellfire”) is descriptive of this intense, incapacitating
neuralgia and cephalgia.16 By definition, zoster pain is divided into acute pain (so-called
zoster-associated pain or ZAP), and chronic PHN. ZAP
is pain occurring any time within the first 4 weeks of the initial cutaneous
crisis. In contradistinction, PHN is pain that begins after the
fourth week of HZO. PHN can occur in any patient with HZO; however, it
is not usually seen in patients younger than 50 years of age, and
its frequency and severity increase with age. PHN affects approximately
one-half of all patients with herpes zoster who are older than 70. In
some instances, the pain may be so extreme and persistent that
the patient is driven to consider suicide.16 Children in whom HZO develops, usually secondary to immunosuppressive
disease, do not have much PHN and in fact, may have a paradoxical sensation
of numbness in the involved dermatome (so-called anesthesia
dolorosa).7,16 Contralateral hemiplegia, facial palsies, and cerebral angiitis also have
been described.85 These neurologic sequelae occur months after the original episode, and
their association with HZO may be overlooked. Zoster pseudotemporal arteritis has also been reported.55 Intense pain in the temporal area can occur in the preeruptive phase of
HZO and can be mistaken for temporal arteritis. We have seen three elderly
patients who presented with intense unilateral temporal area pain, in
whom ipsilateral HZO subsequently developed. Biopsy of the temporal
artery was performed in each case and showed evidence of granulomatous
arteritis without breaks in the tunica intima. Therefore, in addition
to the possibility of temporal arteritis, the differential diagnosis
of periorbital pain out of proportion to clinical findings should
include the preeruptive phase of HZO. HERPES ZOSTER OPHTHALMICUS IN AIDS HZO is an important early clinical marker for AIDS, especially in young
patients in high-risk groups.86–91 In a prospective study, Sandor and associates85 followed 54 patients with HZO for a 2-year period. Of the 23 patients
younger than 45 years of age, 14 were at high risk for contracting
human immunodeficiency virus (HIV), and 3 of these patients
subsequently were diagnosed with AIDS. Kestelyn and co-workers91 examined 19 young Africans who presented with HZO and found that they
were all HIV positive. These patients had a higher proportion of keratitis, uveitis, and
PHN compared with immunocompetent patients with HZO. Therefore, a
physician who finds HZO in a young person should be alerted
to the strong possibility of coexistent HIV infection. A more prolonged and severe disease course may characterize HZO in AIDS. Recurrence
of HZO may be more frequent than in immunocompetent patients, in
whom recurrence is very uncommon.92 Successful treatment with intravenous acyclovir has been reported in numerous
cases, and this has become the recommended treatment for patients
at high risk for acquiring HIV.89 Because cessation of treatment may result in subsequent recurrence, repeated
and prolonged treatment may be required. ARN is a retinopathy not unique to patients with AIDS but is more commonly
seen in that population. The causative agent in ARN is usually a herpes
virus, either VZV, HSV type I or II, or CMV.59 In contrast, PORN syndrome is a distinct form of acyclovir-resistant
necrotizing herpetic retinopathy seen in patients with AIDS.87 VZV is considered the most likely etiology in most cases.92,93 PORN is characterized by multifocal, deep retinal lesions that progress
rapidly to confluence, minimal or no intraocular inflammation, absence
of vascular inflammation, and perivenular clearing of retinal opacification (Fig. 20). It is an extremely rapid and visually devastating form of retinal
necrosis. After CMV retinopathy, PORN is the second most frequent opportunistic
retinal infection in patients with AIDS in North America.87 In a retrospective study of 38 patients with PORN, Engstrom and colleagues87 reported that 67% had a history of cutaneous herpes zoster and 41% had
HZO. VZV retinopathy in patients with AIDS may present
a particularly difficult therapeutic challenge with regard to recurrence
and resistance. 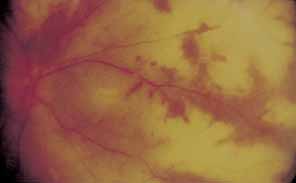 Fig. 20 Progressive outer retinal necrosis. (Courtesy of Lawrence S. Morse, MD, PhD) Fig. 20 Progressive outer retinal necrosis. (Courtesy of Lawrence S. Morse, MD, PhD)
|
LABORATORY EVALUATION VZV can be isolated from cutaneous vesicles at the time of acute dermatologic
crisis. The collected material should be placed in viral transport
medium for immediate shipment to the virology laboratory.50 The virus can be propagated in embryonic lung tissue culture. Cells may
also be examined by fluorescent antibody techniques.50 There are several serologic tests that can also detect herpes zoster, including
complement-fixation and neutralizing antibodies. Antibody
titers rise for 2 weeks after infection and then fall to a lower
level, where they may remain for many years. Because many VZV antigens
are shared by HSV, there may be false-positives with herpes simplex
serologies. Cytology of cutaneous vesicular scrapings with Papanicolaou's stain
displays multiple eosinophilic intranuclear inclusions, the so-called
Lipschütz type II bodies (Fig. 21). In addition, multinucleated giant cells identical to those of herpes
simplex virus infection can be seen. Although the inclusions cannot
be seen with either Giemsa or Gram's stain, the giant cells are
readily visible with Giemsa stain (Tzanck preparation).50 The spherical icosahedral proteinaceous coat (capsid) of the
VZV can be seen with electron microscopy, although it is impossible to
distinguish VZV from other members of the herpesvirus group unless it
is previously tagged with specific peroxidase-labeled antibody.50 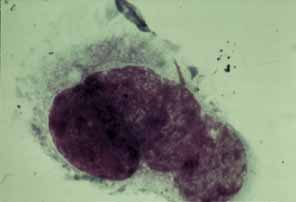 Fig. 21 Herpes zoster ophthalmicus (HZO): scraping of vesicle. Multinucleated
giant epithelial cell with intracytoplasmic basophilic inclusions, so-called
Lipschutz type II bodies (Giemsa stain, Tzanck
preparation). Fig. 21 Herpes zoster ophthalmicus (HZO): scraping of vesicle. Multinucleated
giant epithelial cell with intracytoplasmic basophilic inclusions, so-called
Lipschutz type II bodies (Giemsa stain, Tzanck
preparation).
|
SYSTEMIC WORK-UP
A careful history should elucidate identifiable factors predisposing
the patient to HZO, such as childhood varicella, older age, stress, trauma,
radiation, surgery (especially on the trigeminal nerve), heavy-metal poisoning,
pharmacologic immunosuppression,7 or systemic
disease. Predisposing systemic diseases include leukemia, lymphoma (especially
Hodgkin's disease), other malignant neoplastic diseases, tuberculosis,
syphilis, and (congenital or acquired immunodeficiency syndromes.6,7,51,92,93
Patients with HZO who have cutaneous dissemination, immune compromise, or
a history of chronic illness should undergo a general medical examination.94 In HZO patients who are otherwise healthy, medical examination is usually
not necessary, although screening tests such as liver function studies, chest
x-ray film, and complete blood count occasionally reveal
an underlying chronic illness.
The epidemiologic data (see “Epidemiology”
section earlier in this chapter) support the notion that medical work-ups
to detect underlying immunosuppression or malignancy in patients with
HZO are usually not contributory. Nevertheless, it is usually prudent
to have an internist or dermatologist follow a patient with HZO, both
to confirm the usual absence of underlying disease and to assist in the
medical management of HZO, which may require the use of systemic corticosteroids
or other immunosuppressive agents (see “Management
Strategies” section). If a patient with HZO has no previous
history of varicella and no recent exposure to the virus, the possibility
of underlying systemic disease should be considered and the patient medically
evaluated. In a young patient with HZO, HIV should be suspected, especially
if any of the usual risk factors are present. An appropriate history should
be obtained, and testing for HIV should be considered if it is at all
suspected.
MANAGEMENT STRATEGIES General Considerations The multitude of therapeutic strategies for HZO underscores the lack of
any one successful prophylactic or curative treatment for this disease. Human leukocytic interferon, intravenous vidarabine, and cytarabine have
been shown to accelerate healing of cutaneous vesicles in HZO and to
lessen visceral complications in immunocompromised patients if they are
treated within the first 3 days of onset of the cutaneous eruption.7 However, the data on the efficacy of these agents have been mixed, principally
because of drug toxicity or systemic immunocompromise. Cytarabine
can actually enhance the dissemination of VZV because of its immune-suppressing
properties.95 ACYCLOVIR. Acyclovir (acycloguanosine, Zovirax®) has in vitro and in vivo efficacy against VZV. Balfour and co-workers22 and Bean and associates98 documented improvement in the cutaneous eruption and acceleration of VZV
virus clearance from vesicles in immunocompromised patients with herpes
zoster. Compared with placebo, intravenous acyclovir also reduced
PHN-associated pain. In the initial, prospective, longitudinal, randomized, double-blind, placebo-controlled trial using
oral acyclovir, Cobo and co-workers80,96 prospectively evaluated 71 immunocompetent patients with HZO, who presented
within 7 days of onset of characteristic skin eruptions. They found
that a 10-day course of treatment with 600 mg of oral acyclovir, 5 times
per day, significantly reduced the incidence and severity
of the most common complications of HZO, including dendritic keratopathy, stromal
keratitis, and uveitis. The drug is well tolerated, significantly
decreases cutaneous herpes zoster dissemination, and accelerates
clearance of VZV virus from vesicles. It also reduces the pain, especially
if initiated within 72 hours of the cutaneous eruption. The
current standard suprathreshold dosage of acyclovir is 800 mg, 5 times
per day for 10 days (not the original 600-mg dosage in the
study by Cobo and associates). This higher dosing is based on acceptable
tolerance, adequate MICs-minimum inhibitory concentration and
drug pharmacokinetics.99 For immunocompromised hosts acyclovir should be administered intravenously
at 30 g/kg per day.100 NEWER ANTIVIRAL DRUGS. A number of promising newer and more effective antiviral agents are now
available.100 These drugs have been developed with improved activity against VZV, as
well as more favorable pharmacokinetic properties. They include famciclovir, valacyclovir, and
sorivudine. The precise role of each of these
in the treatment of HZO has yet to be elucidated by comparative trials. Famciclovir™ (Famvir™) is the diacetyl ester prodrug
of penciclovir. It is hepatically deesterified to pencyclovir after
oral administration.102 After oral administration, famciclovir has a much higher bioavailability (77%) than acyclovir (18%). The recommended
oral dosage is 750 mg of famciclovir, 3 times per day for 7 days; This
dosing schedule (fewer doses per day for a fewer number
of days than with acyclovir) creates better compliance. A recent
randomized, double-blind, placebo-controlled, multicenter
trial demonstrated that famciclovir was well tolerated and safe.102 Significantly, patients who took famciclovir had faster resolution of
PHN (by approximately twofold) than placebo recipients. Valaciclovir (Valtrex™) is the L-valine ester prodrug of acyclovir.100,103 The bioavailability after oral adminis-tration is also high (80%) compared to acyclovir (18%). Valaciclovir is converted to acyclovir by hepatic first-pass
metabolism.100,103 The dosage is 1000 mg three times per day for 7 days. This is a simpler
dosing regimen with a similar mechanism of action as acyclovir in the
treatment of HZO and the prevention of its sequelae. Sorivudine (5-bromo-vinyl-arabinosyluracil) is
one of two new potent uracil derivatives that are active against
VZV.100,104 The other is BW882C87, a nucleoside analogue that is approximately seven
times as potent as acyclovir, with a bioavailability of 21% to 25%.103 Sorivudine's mechanism of action is the inhibition of DNA polymerase.100,104 The dosage of sorivudine is 40 mg once daily for 7 days.104 Sorivudine 40 mg per day is equivalent to 800 mg of acyclovir 5 times
per day. A recent report has shown sorivudine to be effective in the treatment
of an AIDS patient who had PORN syndrome, presumably caused by
VZV.106 Sorivudine is inactive against HSV, and is toxic if taken in combination
with 5-fluorouracil.100 Sorivudine is not available in the United States. Foscarnate (trisodium phosphonoformate) is an inorganic phosphate
analogue.100 The mechanism of antiviral action of foscarnate is a reversible inhibition
of virus-induced RNA and DNA polymerases.100 Foscarnate does not require phosphoryation to be active, and is thus an
option for acyclovir-resistant strains of VZV, and is available
only in intravenous and intravitreal forms.99 Other antivirals used for acyclovir-resistant VZV strains include
ganciclovir, cidofovir, and lubocavir.100,101 Herpes Zoster Ophthalmicus: Treatment and Management Dividing the treatment of HZO into the management of acute herpes zoster
dermal eruption, corneal sequelae, other ocular involvement, and PHN
provides a framework for the clinician when attempting to manage this
protean disease. ACUTE HERPES ZOSTER DERMAL ERUPTION. Local treatment of cutaneous zoster is mostly palliative. Topical idoxuridine 40% in
dimethyl sulfoxide (DMSO) has some benefit, but
is not approved for use in the United States.107 The astringent Domboro's (Burow's) solution (acetic
acid/aluminum acetate), cool compresses, and mechanical
cleansing of the involved skin are helpful. Topical antibiotic ointment
may inhibit scarring and decrease the possibility of bacterial superinfection. Sensitizing
antibiotics with neomycin should not be used. Topical
corticosteroid ointment should be avoided, at least during the
initial few days after the acute dermatologic crisis, during which active
replicating virus is present in the epidermis and dermis.50 Drying preparations should also be eschewed, because they may prolong healing, produce
confluence and necrosis of lesions, and enhance cicatrization
and immobilization of the upper eyelid, resulting in exposure.100 The use of 5% acyclovir ointment topically on the zoster skin rash may have some value. However, the
usual natural history of the acute dermatologic eruption in HZO
is vesiculation and crusting with eventual scarring, regardless of the
use of any of the above strategies. CORNEAL SEQUELAE. The punctate and pseudodendritic forms of herpes zoster epithelial keratitis
can represent active viral replication in the corneal epithelium.56,57 Whether this live VZV is at the terminal corneal nerve ending or deposited
on the corneal surface from the lid margin or conjunctiva is debated.64,65 The antivirals idoxuridine, vidarabine, and trifluridine have in vitro activity against VZV, but little or no in vivo effect. Theoretically, these agents might be of benefit during the first
few days of HZO by inhibiting viral replication in the epithelium and
reducing the residual amount of viral antigen.7 However, because the MICs for VZV are so high, these drugs are of no clinical
value for the treatment of HZO keratitis. The apparent simultaneous
occurrence of herpes simplex and herpes zoster may actually represent
herpes zoster pseudodendrites, rather than simultaneous infection
with herpes simplex. Therefore, the management of HZO keratitis is not
favorably influenced by the use of first-generation antivirals. Second-generation antiviral agents such as acyclovir do have in vivo efficacy against VZV, and intravenous acyclovir has been shown to be of
value in the management of herpes zoster keratitis. Oral acyclovir in
HZO keratitis also has shown efficacy.80,96 McGill and Chapman109,110 have successfully used topical ophthalmic acyclovir in HZO keratitis and
keratouveitis, however, topical ophthalmic acyclovir is not available
for corneal use in the United States. Maudgal and associates111,112 and DeClerq113 used topical or oral 5-2-bromovinyl-2'-deoxyuridine (BVDU) in the management of HZO keratitis. No
placebo-controlled studies of BVDU have been performed to date. BVDU
is not available in the United States. Topical corticosteroids are of little benefit in the treatment of acute
epithelial herpes zoster keratitis. In the past, corticosteroids were
considered deleterious in HZO because of the possibility of superinfection
or dissemination, but more recent evidence shows that herpes zoster
pseudodendrites can respond to topical steroids.56,57,63 Because most cases of herpes zoster epithelial keratitis usually have
a self-limited and visually asymptomatic course even without treatment, the
most appropriate management strategy is to use ocular lubrication
with nonpreserved artificial tears and possible debridement and
to avoid topical antivirals and steroids.7 Topical steroids do have a place in the management of the components of
zoster corneal disease associated that are inflammatory in etiology, namely, sclerokeratitis, keratouveitis, interstitial keratitis as well
as immune-related sequelae such as anterior stromal infiltrates, disciform
keratitis, and delayed corneal mucous plaques.65 Herpes zoster interstitial keratitis is probably an antigen-antibody-complement-mediated reaction, whereas disciform keratitis
is a manifestation of delayed hypersensitivity.65 The use of topical steroids in these settings diminishes inflammation
and vascularization, but the patient must be evaluated frequently and
carefully because of the rapidity of changes in herpes zoster keratitis
and because of the potential for corneal melting. Patients should be
forewarned of the potential for chronic topical corticosteroid treatment, even
at low, seemingly homeopathic doses, with the potential for
side effects such as cataract or elevated intraocular pressure. Steroids
should not be used in cases of exposure or neurotrophic keratitis because
of the possibility of keratolysis.65 Indolent epithelial defects associated with neurotrophic keratitis (Fig. 22) that are unresponsive to nonpreserved artificial tears, bland ointments, pressure
patching, or therapeutic soft contact lenses should
be considered for lateral, central, or total tarsorrhaphy, bridge or full
Gundersen conjunctival flap, or amniotic membrane transplantation. A
lateral or central tarsorrhaphy is one of the simplest and most effective
means of reducing the sight-threatening sequelae of HZO
neurotrophic keratitis (Fig. 23). Waring and Ekins114 have delineated the various factors that can cause corneal perforation
in HZO (Fig. 24) and underscored the prophylactic benefit of early, large tarsorrhaphy
to prevent initial epithelial erosion and subsequent corneal melting
and perforation.115 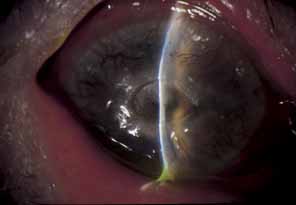 Fig. 22 Herpes zoster ophthalmicus (HZO) neurotrophic keratitis, demonstrating
the leathery appearance of the corneal surface rolled epithelial
edges and indolent ulceration with reparative limbal vascularization. Fig. 22 Herpes zoster ophthalmicus (HZO) neurotrophic keratitis, demonstrating
the leathery appearance of the corneal surface rolled epithelial
edges and indolent ulceration with reparative limbal vascularization.
|
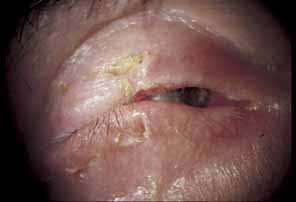 Fig. 23 Lateral tarsorrhaphy for herpes zoster ophthalmicus (HZO) neurotrophic
keratitis. Fig. 23 Lateral tarsorrhaphy for herpes zoster ophthalmicus (HZO) neurotrophic
keratitis.
|
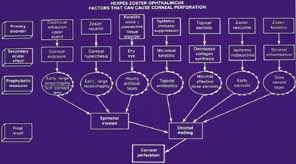 Fig. 24 Algorithm of factors that can cascade to corneal perforation in herpes
zoster ophthalmicus (HZO). (Waring GO, Ekins MB: Corneal perforations secondary to exposure keratitis. Ophthalmic
Surg 15:153, 1984) Fig. 24 Algorithm of factors that can cascade to corneal perforation in herpes
zoster ophthalmicus (HZO). (Waring GO, Ekins MB: Corneal perforations secondary to exposure keratitis. Ophthalmic
Surg 15:153, 1984)
|
Therapeutic soft contact lenses may be a reasonable short-term strategy
in herpes zoster neurotrophic keratitis, but they must be used
with caution and with frequent follow-up to guard against lens
spoilage, microbial superinfection, and hypoxic iridocyclitis. The development of cicatricial entropion or ectropion with subsequent trichiasis
further exacerbates the corneal surface.115 Timely repair of ectropion or entropion from HZO protects the corneal
epithelium and makes subsequent tarsorrhaphy easier to perform. Superior
lid cicatrization can make subsequent tarsorrhaphy in HZO quite difficult
to perform, resulting in immediate retraction or breakage of the
tarsal scar. When there is severe cicatricial ectropion, a simple tarsorrhaphy (performed
by scraping the superior and inferior tarsal
plates and placing 6-0 silk sutures on bolsters) may not
be effective in closing the lids permanently; in these cases a tongue-in-groove
tarsorrhaphy or Hughes-type procedure
may be required. If less than 1.0 mm in diameter, impending or actual corneal perforation
can be managed with 2-butyl-cyanoacrylate (Histacryl™) adhesive
and a thick therapeutic soft contact lens (e.g., Bausch & Lomb piano T, or Wessley-Jensen CSI) (Figs. 25 and 26). Larger perforations can be managed acutely with scleral grafting
or corneal patch grafting. These grafts should in turn be covered with
conjunctiva to prevent the initial melting process from destroying
the scleral or corneal graft (Figs. 27 and 28). Steroid therapy should be tapered and then stopped, and cycloplegics
added. Some patients with herpes zoster keratitis end up with vascularized, lipid-containing scars and may have some residual return
of corneal sensation. In this particular subset of patients, corneal
transplantation can be considered (Figs. 29 and 30). Tanure and associates117 presented positive follow-up data on patients who had keratoplasty
for VZV keratopathy. Simultaneous lateral tarsorraphy was helpful
in these patients. In patients with HZO keratitis who have profound corneal
anesthesia, however, corneal transplantation could lead to a greater
risk of corneal wound dehiscence, healing problems, and graft rejection, and
is not recommended.117 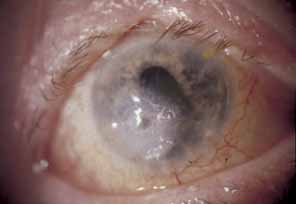 Fig. 25 Corneal perforation secondary to herpes zoster ophthalmicus (HZO) neurotrophic
keratitis. Fig. 25 Corneal perforation secondary to herpes zoster ophthalmicus (HZO) neurotrophic
keratitis.
|
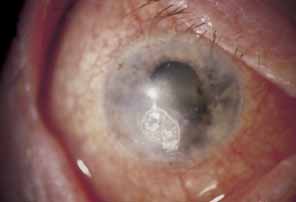 Fig. 26 Same patient as in Figure 25. Cyanoacrylate adhesive and therapeutic contact lens applied for corneal
perforation secondary to herpes zoster neurotrophic keratitis. Fig. 26 Same patient as in Figure 25. Cyanoacrylate adhesive and therapeutic contact lens applied for corneal
perforation secondary to herpes zoster neurotrophic keratitis.
|
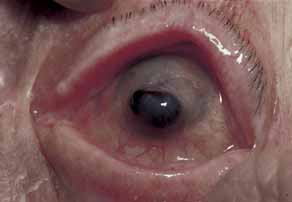 Fig. 27 Herpes zoster ophthalmicus. Large spontaneous corneal melt, perforation, and
iris prolapse in a 92-year-old woman, 1 year postcutaneous
herpes zoster ophthalmicus (HZO) eruption. Fig. 27 Herpes zoster ophthalmicus. Large spontaneous corneal melt, perforation, and
iris prolapse in a 92-year-old woman, 1 year postcutaneous
herpes zoster ophthalmicus (HZO) eruption.
|
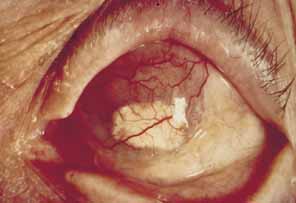 Fig. 28 Same patient as in Figure 27. Scleral patch graft and overlying conjunctival flap applied for corneal
melt, perforation, and iris prolapse in herpes zoster ophthalmicus (HZO). Fig. 28 Same patient as in Figure 27. Scleral patch graft and overlying conjunctival flap applied for corneal
melt, perforation, and iris prolapse in herpes zoster ophthalmicus (HZO).
|
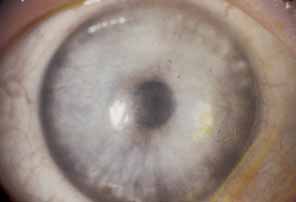 Fig. 29 Herpes zoster ophthalmicus (HZO) keratopathy. Lipoidal scarring, vascularization
and intact corneal sensation. Best correctable vision 20/400. Fig. 29 Herpes zoster ophthalmicus (HZO) keratopathy. Lipoidal scarring, vascularization
and intact corneal sensation. Best correctable vision 20/400.
|
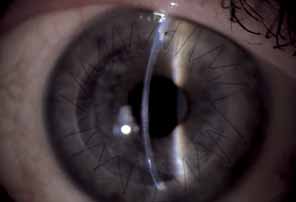 Fig. 30 Herpes zoster ophthalmicus (HZO) keratopathy.
Same patient as in Figure
29. Successful penetrating keratoplasty, 12 weeks after surgery,
with running suture and two remaining interrupted sutures. Best
correctable vision 20/30.
Fig. 30 Herpes zoster ophthalmicus (HZO) keratopathy.
Same patient as in Figure
29. Successful penetrating keratoplasty, 12 weeks after surgery,
with running suture and two remaining interrupted sutures. Best
correctable vision 20/30.
|
OTHER OCULAR INVOLVEMENT. The use of topical cycloplegics is quite helpful in decreasing the ciliary
spasm associated with herpes zoster inflammatory disease and in breaking
posterior synechiae. For many patients with HZO, cycloplegia is
the only form of treatment necessary for ocular comfort. Focal corneal infiltrates, stromal edema, stromal keratitis, and disciform
keratitis may be treated with topical corticosteroids (the equivalent
of prednisolone acetate 12.5% four times per day). Pulsing
steroids is sometimes preferable to chronic treatment to decrease
the associated complications of steroid therapy. Again, steroids
should be reserved for patients who have intact epithelium with herpes
zoster disciform keratitis, endotheliitis, or keratouveitis. Glaucoma in HZO is typically secondary to plugging of the trabecular meshwork
as a consequence of inflammatory anterior chamber reactivation, or
it may result from peripheral anterior synechiae formation. The use
of topical β-blockers and carbonic anhydrase inhibitors (e.g., timolol-dorzolamide combination) and α-adrenergic
medications (e.g., brimonidine) along with topical
corticosteroids usually results in a significant reduction in intraocular
pressure. Herpes zoster vitritis, vitreous hemorrhage, or vitreous
debris is usually secondary to severe inflammation and may respond
to topical, periocular, or systemic steroids. HZO retinitis, HZO optic neuritis, HZO chorioretinitis, and the ARN syndrome
are most appropriately treated with a combination of systemic steroids
and intravenous acyclovir.77 PORN syndrome (acyclovir-resistant VZV retinopathy in AIDS) management
requires the intravenous administration of various combinations
of antivirals, including foscarnet, ganciclovir, and vidarabine.93,100 Another strategy for PORN syndrome is the intravitreal injection of ganciclovir
and foscarnet.101 VZV retinopathy in patients with AIDS requires long-term suppressive
antiviral therapy to prevent recurrences. POSTHERPETIC NEURALGIA. Most therapeutic strategies for PHN have been of only modest value. These
include analgesics, with or without narcotics; the tricyclic antidepressants
chlorprothixene, desipramine, nortriptyline and amitriptyline; and
the neurotonic agents carbamazepine and phenytoin. The tricyclic
antidepressants should be used in low doses in the elderly to avoid
anticholinergic effects and drowsiness. Sklar and co-workers118 performed a randomized, placebo-controlled, double-blind
trial of intramuscular adenosine monophosphate (AMP) for PHN
and found that herpes zoster skin ulcers healed faster and virus shedding
was reduced compared with placebo. AMP, a naturally occurring antiviral
agent, is converted intracellularly to adenosine and is then taken
up by the actively replicating VZV. After a 4-week trial, 88% of
the AMP-treated patients were free of pain as opposed
to only 43% in the placebo group. In the placebo group, patients
who still had pain after 4 weeks were then given AMP treatment, and
all of these patients recovered within 3 weeks after initiation.118 Cimetidine has been used successfully in PHN management in an uncontrolled
trial.119 However, a controlled study reported no significant benefit from cimetidine
over placebo.120 Cimetidine prevents itching, redness, swelling, and neuronal irritation. In
addition, cimetidine is an H2 receptor antagonist, and thymus-dependent suppressor T lymphocytes
have H2 histamine receptors on their surface.119 Cimetidine may therefore work by attaching to the H2 receptor and serving as an immune-modulating agent, thereby enhancing
the immune response by preferentially blocking H2 receptor sites on suppressor T cells and favorably modulating helper T-cell
activity.120 Substance P is one of the principal mediators of pain impulses from the
peripheral nervous system to the central nervous system.121 Capsaicin (trans-8-methyl-N-vanillyl-6-nonemaide, Zostrix™) is a vanilyl alkaloid
extract of the plant Solanaceae family Capsicum (a genus that also includes the red, chili, and jalapeno peppers). Capsaicin
depletes and prevents the reaccumulation of substance
P from small peripheral sensory neurons. Capsaicin is available in a 0.025% cream, and
has been found to be effective in some patients
with PHN. The reported failures with capsaicin are attributed to inadequate
usage. When used appropriately, capsaicin can cause improvement
in up to 75% of patients.121,122 Capsaicin cream needs to be applied three to four times per day and may
require several weeks before pain relief occurs.121 The main side effect of capsaicin application is stinging. If no improvement
in neuralgia is noted after 6 weeks, it is unlikely that further
usage will be effective. Topical forehead administration of a eutectic mixture of local anesthetics (EMLA) patch, which is a 5% cream made of lidocaine 2.5% and
procaine 2.5%, can be an effective strategy
for mild PHN.100 The anticonvulsant gabapentin (1-aminoethyl cyclohexaneacetic
acid, Neurontin™) has been used successfully in the management
of PHN.123 Gabapentin is structurally similar to the inhibitory neurotransmitter
gamma-aminobutyric acid (GABA), but it does not act by
mimicking GABA. The precise mechanism action of gabapentin in the management
of PHN is unknown. Gabapentin is begun at 100 mg orally at bedtime, for
several days. Side effects include somnolence, dizziness, and
ataxia.123 The dosage can be increased as tolerated to 300 mg orally three times
daily and higher, up to a maximum dosing of 3600 mg per day.123 Despite favorable reports on the use of single categories of medication
in the management of PHN—the antivirals acyclovir or famcyclovir
or valacyclovir or oral corticosteroids—there are few controlled
studies comparing the relative benefits of one, all, or combinations
of these medications. A large prospective, placebo-controlled
trial would have to be initiated, and considering the pleomorphic nature
of PHN in HZO, it would be difficult to compare patients and outcomes.124 In the original placebo-controlled study of oral acyclovir in the
management of acute HZO, Cobo and associates80,96 concluded that the drug had no effect on the incidence, severity, or duration
of PHN. A more recent study by McGill and White,125 however, showed a significant decrease in the occurrence of PHN with oral
acyclovir versus placebo. The role of systemic corticosteroids in
the treatment of PHN is moot. Scheie45 initially reported on the successful use of oral corticosteroids in treating
PHN and in reducing the severity of keratiti, uveitis, and glaucoma, without
dissemination of herpes zoster. Epstein126 reported favorable responses to subcutaneous triamcinolone in HZO patients
with PHN. The initial caution expressed by Merselis and co-workers127 on the possibility of HZO dissemination with oral corticosteroid use is
tempered by the fact that 11 of their 17 patients had preexisting immunologic
compromise, including lymphoma, Hodgkin's disease, or leukemia. Of the 6 otherwise immunocompetent patients, only 2 actually received steroids
and neither had dissemination of disease.127 A more recent prospective, double-blind study showed no difference
between oral prednisolone and placebo in the occurrence of PHN.128 Two large, controlled clinical trials examined the role of acyclovir with
or without oral prednisone in the management of PHN.129,130 In both of these studies, there was a statistically significant reduction
in ZAP (the acute pain in the first 4 weeks after the zoster
cutaneous eruption), and in the rate of healing of vesicles, but
no effect on the incidence or duration of PHN.129,130 If oral corticosteroids are to be used in acute HZO, wait a few days before
instituting them, to allow humoral immunity to arise and to let the
oral antiviral (acyclovir or famcyclovir or valacyclovir) kill
circulating VZV. An oral corticosteroid such as prednisone can then
be initiated and tapered during a period of two weeks. The use of
oral corticosteroids without concomitant antiviral therapy is not recommended.4 Oral corticosteroids should not be used in high-risk patients such
as those with diabetes or those with gastritis.4 Olson and Ivy131 successfully treated 27 patients with PHN refractory to medical management
with sympathetic (stellate ganglion) block. There is evidence
that the early use of stellate ganglion block may increase the
rate of relief from PHN.132 Other strategies for PHN include transcutaneous electrical nerve stimulation, acupuncture, psychotherapy, hypnosis, biofeedback, and in extreme
cases, trigeminal rhizotomy. It is strongly recommended that a neurologist
or anesthesiologist specializing in pain management closely follow
HZO patients with PHN. |