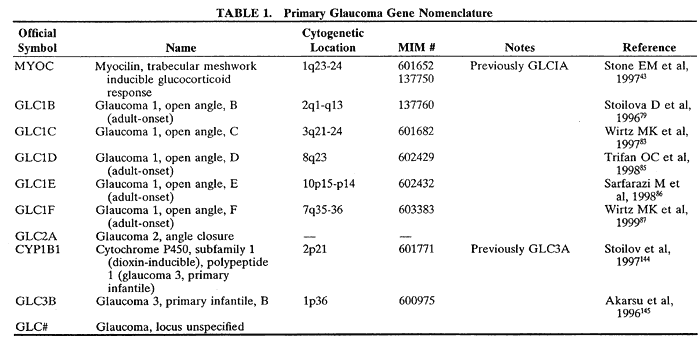
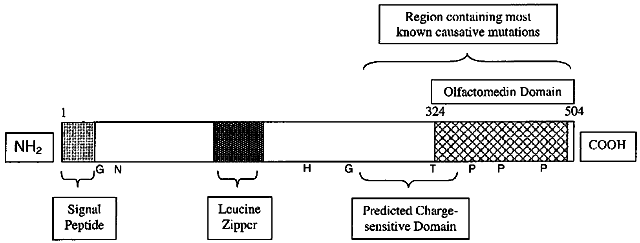
1. Bell J. On some structural anomalies of the eye and on the inheritance
of glaucoma. In Pearson K (ed). The Treasury of Human Inheritance. Cambridge, UK: Cambridge
University Press, 1931:425 2. Benedict TGG. Abhandlungen aus dem Gebiete der Augen-heilkunde. Breslau: L. Freund, 1842:123 3. Mooren A. Ophthalmologische Beobachtungen. Berlin: A. Hirschwald, 1867 4. von Graefe A. Beiträge zur Pathologie und Therapie der Glaucoms. Arch
Ophthalmol 1869;15:108 5. Courthney RH, Hill E. Hereditary juvenile glaucoma simplex. JAMA 1931;97:1602 6. Stokes WH. Hereditary primary glaucoma. Arch Ophthalmol 1940;24:885 7. Allen TD, Ackerman WG. Hereditary glaucoma in a pedigree of three generations. Arch
Ophthalmol 1942;27:139 8. Beiguelman B, Prado D. Recessive juvenile glaucoma.J Genet Hum 1963;12:53 9. Weatherill JR, Hart CT. Familial hypoplasia of the iris stroma associated
with glaucoma. Br J Ophthalmol 1969;53:433 10. Jerndal T. Familial congenital glaucoma with dominant heredity. Acta Ophthalmol 1968;46:459 11. Jerndal T. Dominant goniodysgenesis with late congenital glaucoma: A re-examination
of Berg's pedigree. Am J Ophthalmol 1972;74:28 12. Martin JP, Zorab EC. Familial glaucoma: In nine generations of a South
Hampshire family. Br J Ophthalmol 1974;58:536 13. Tielsch JM, Sommer A, Katz J et al. Racial variations in the prevalence
of primary open-angle glaucoma: The Baltimore Eye Survey. JAMA 1991;266:369 14. Tielsch JM, Katz J, Singh K et al. A population-based evaluation of glaucoma
screening: The Baltimore Eye Survey. Am J Epidemiol 1991;134:1102 15. Klein BE, Klein R, Sponsel WE et al. Prevalence of glaucoma: The Beaver
Dam Eye Study. Ophthalmology 1992;99:1499 16. Othman MI, Sullivan SA, Skuta GL et al. Autosomal dominant nanophthalmos (NNO1) with
high hyperopia and angle-closure glaucoma maps to chromosome 11. Am
J Hum Genet 1998;63:1411 17. Kellerman L, Posner A. The value of heredity in the detection and study
of glaucoma. Am J Ophthalmol 1955;40:681 18. Becker B, Kolker AE, Roth FD. Glaucoma family study. Am J Ophthalmol 1960;50:557 19. Battacharjee H, Chowdury HK. A survey on heredity of open angle glaucoma. Ind
J Ophthalmol 1987;35:138 20. Biro I. Notes on heredity of glaucoma. Ophthalmologica 1939;98:43 21. Francois J, Heintz-deBree. Personal research on the heredity of chronic
simple (open-angle) glaucoma. Am J Ophthalmol 1966;62:1067 22. Tielsch JM, Katz J, Sommer A et al. Family history and risk of primary
open angle glaucoma: The Baltimore Eye Survey. Arch Ophthalmol 1994;112:69 23. Probert LA. A survey of hereditary glaucoma. Can Med Assoc J 1951;66:563 24. McNaught AI, Allen JG, Healey DL et al. Accuracy and implications of a
reported family history of glaucoma: Experience from the Glaucoma Inheritance
Study in Tasmania. Arch Ophthalmol 2000;118:900 25. Goldschmidt E. The heredity of glaucoma. Acta Ophthalmol Suppl 1973;120:27 26. Teikari JM. Genetic factors in open-angle (simple and capsular) glaucoma: A
population-based twin study. Acta Ophthalmol 1987;65:715 27. Gottfredsdottir MS, Sverisson T, Musch DC et al. Chronic open-angle glaucoma
and associated ophthalmic findings in monozygotic twins and their
spouses in Iceland. J Glaucoma 1999;8:134 28. Schwartz JT, Reuling FH, Feinleib M. Heritability study on size of the
physiologic cup of the optic nerve head: A summary report. Acta Genet
Med Gemellol 1976;25:181 29. Bengtsson B. The inheritance and development of cup and disc diameters. Acta
Ophthalmol 1980;58:733 30. Knobloch WH, Leavenworth NM, Bouchard TJ et al. Eye findings in twins reared
apart. Ophthalmic Paediatr Genet 1985;5:59 31. Drance SM. Angle closure glaucoma among Canadian Eskimos. Can J Ophthalmol 1973;8:252 32. Arkell SM, Lightman DA, Sommer A et al. The prevalence of glaucoma among
Eskimos of northwest Alaska. Arch Ophthalmol 1987;105:482 33. Clemmesen V, Alsbirk PH. Primary angle-closure glaucoma (a.c.g.) in Greenland. Acta
Ophthalmol 1971;49:47 34. Congdon NG, Quigley NG, Hung PR et al. Screening techniques for angle-closure
glaucoma in rural Taiwan. Acta Ophthalmol Scand 1996;74:113 35. Congdon N, Wang F, Tielsch JM. Issues in the epidemiology and population-based
screening of primary angle-closure glaucoma. Surv Ophthalmol 1992;36:411 36. Salmon JF. Predisposing factors for chronic angle-closure glaucoma. Prog
Retin Eye Res 1999;18:121 37. Alward WL, Fingert JH, Coote MA et al. Clinical features associated with
mutations in the chromosome 1 open-angle glaucoma gene (GLC1A). N Engl
J Med 1998;338:1022 38. Sheffield VC, Stone EM, Alward L et al. Genetic linkage of familial open
angle glaucoma to chromosome 1q21-q31. Nat Genet 1993;4:47 39. Richards JE, Lichter PR, Boehnke M et al. Mapping of a gene for autosomal
dominant juvenile-onset open-angle glaucoma to chromosome Iq. Am J
Hum Genet 1994;54:62 40. Wiggs JL, Haines JL, Paglinauan C et al. Genetic linkage of autosomal dominant
juvenile glaucoma to 1q21-q31 in three affected pedigrees. Genomics 1994;21:299 41. Morissette J, Cote G, Anctil JL et al. A common gene for juvenile and adult-onset
primary open-angle glaucomas confined on chromosome 1q. Am
J Hum Genet 1995;56:1431 42. Lichter PR. Genetic clues to glaucoma's secrets: The L. Edward Jackson
Memorial Lecture. Part 2. Am J Ophthalmol 1994;117:706 43. Stone EM, Fingert JH, Alward WLM et al. Identification of a gene that causes
primary open angle glaucoma. Science 1997;275:668 44. Adam MF, Belmouden A, Binisti P et al. Recurrent mutations in a single
exon encoding the evolutionarily conserved olfactomedin-homology domain
of TIGR in familial open-angle glaucoma. Hum Mol Genet 1997;6:2091 45. Richards JE, Ritch R, Lichter PR et al. Novel trabecular meshwork inducible
glucocorticoid response mutation in an eight-generation juvenile-onset
primary open-angle glaucoma pedigree. Ophthalmology 1998;105:1698 46. Mansergh FC, Kenna PF, Ayuso C et al. Novel mutations in the TIGR gene
in early and late onset open angle glaucoma. Hum Mutat 1998;11:244 47. Rozsa FW, Shimizu S, Lichter PR et al. GLC1A mutations point to regions
of potential functional importance on the TIGR/MYOC protein. Mol Vis 1998;4:20 48. Fingert JH, Heon E, Liebmann JM et al. Analysis of myocilin mutations in 1703 glaucoma
patients from five different populations. Hum Mol Genet 1999;8:899 49. Shimizu S, Lichter PR, Johnson AT et al. Age-dependent prevalence of mutations
at the GLC1A locus in primary open-angle glaucoma. Am J Ophthalmol 2000;130:165 50. Wiggs JL, Del Bono EA, Schuman JS et al. Clinical features of five pedigrees
genetically linked to the juvenile glaucoma locus on chromosome 1q21-q31. Ophthalmology 1995;102:1782 51. Stoilova D, Child A, Brice G et al. Novel TIGR/MYOC mutations in families
with juvenile onset primary open angle glaucoma. J Med Genet 1998;35:989 52. Polansky JR, Fauss DJ, Chen P et al. Cellular pharmacology and molecular
biology of the trabecular meshwork inducible glucocorticoid response
gene product. Ophthalmologica 1997;211:126 53. Nguyen TD, Chen P, Huang WD et al. Gene structure and properties of TIGR, an
olfactomedin-related glycoprotein cloned from glucocorticoid-induced
trabecular meshwork cells. J Biol Chem 1998;273:6341 54. Kubota R, Noda S, Wang Y et al. A novel Myson-like pretin (Myocilin) expressed
in the connecting cilium of the photoreceptor: Molecular cloning, tissue
expression, and chromosomal mapping. Genomics 1997;41:360 55. Ortego J, Escribano J, Coca-Prados M. Cloning and characterization of subtracted
cDNAs from a human ciliary body library encoding TIGR, a protein
involved in juvenile open angle glaucoma with homology to myosin
and olfactomedin. FEBS Lett 1997;413:349 56. Snyder DA, Rivers AM, Yokoe H et al. Olfactomedin: Purification, characterization, and
localization of a novel olfactory glycoprotein. Biochemistry 1991;30:9143 57. Huang W, Jaroszewski J, Ortega J et al. Expression of the TIGR gene in
the iris, ciliary body, and trabecular meshwork of the human eye. Ophthalmic
Genet 2000;21:155 58. Tomarev SI, Tamm ER, Chang B. Characterization of the mouse Myoc/Tigr gene. Biochem
Biophys Res Commun 1998;245:887 59. Fingert JH, Ying L, Swiderski RE et al. Characterization and comparison
of the human and mouse GLC1A glaucoma genes. Genome Res 1998;8:377 60. Swiderski RE, Ying L, Swiderski RE et al. Expression pattern and in situ
localization of the mouse homologue of the human MYOC (GLC1A) gene in
adult brain. Brain Res Mol Brain Res 1999;68:64 61. Swiderski RE, Ross JL, Fingert JH et al. Localization of MYOC transcripts
in human eye and optic nerve by in situ hybridization. Invest Ophthalmol 2000;41:3420 62. Morissette J, Clepet C, Moisan S et al. Homozygotes carrying an autosomal
dominant TIGR mutation do not manifest glaucoma. Nat Genet 1998;19:319 63. Ueda J, Wentz-Hunter KK, Cheng EL et al. Ultrastructural localization of
myocilin in human trabecular meshwork cells and tissues. J Histochem
Cytochem 2000;48:1321 64. Lütjen-Drecoll E, May CA, Polansky JR et al. Localization of the stress
protein aβ-crystallin and trabecular meshwork inducible glucocorticoid
response protein in normal and glaucomatous trabecular meshwork. Invest
Ophthalmol 1998;39:517 65. Polansky J, Fauss DJ, Zimmerman CC. Regulation of TIGR/MYOC gene expression
in human trabecular meshwork cells. Eye 2000;14:503 66. Tamm ER, Russell P, Epstein DL et al. Modulation of myocilin/TIGR expression
in human trabecular meshwork. Invest Ophthalmol 1999;40:2577 67. Polansky JR, Kurtz RM, Fauss DJ et al. HTM cell culture model for steroid
effects on IOP: overview. In Krieglstein GK (ed). Glaucoma Update IV, Berlin: Springer-Verlag, 1991:20–29 68. Nguyen TD, Huang WD, Bloom E et al. Glucocorticoid effects on HTM cells: Molecular
biology approaches. In Lutjen-Drecoll E (ed). Basic Aspects
of Glaucoma Research III. Stuttgart: Schattauer, 1993:331–343 69. Armaly MF. Effect of corticosteroids on intraocular pressure and fluid
dynamics. I: The effect of dexamethasone in the normal eye. Arch Ophthalmol 1963;70:482 70. Armaly MF. Effect of corticosteroids on intraocular pressure and fluid
dynamics. II: The effect of dexamethasone in the glaucomatous eye. Arch
Ophthalmol 1963;70:492 71. Furuyoshi N, Furuyoshi M, Futa R et al. Ultrastructural changes in the
trabecular meshwork of juvenile glaucoma. Ophthalmologica 1997;211:140 72. Johnson D, Gottanka J, Flügel C et al. Ultrastructural changes in
the trabecular meshwork of human eyes treated with corticosteroids. Arch
Ophthalmol 1997;115:375 73. Raymond V, Rodrigue MA, Gobeil S et al. Heterodimerization between wild-type
and mutant TIGR/myocilin polypeptides is critical for autosomal
dominant open-angle glaucoma. Am J Hum Genet 2000;76:A2220 74. Sarfarazi M, Chaterji R, Kocak Midillioglu I et al. Identification of a
severely affected homozygote TIGR/MYOC mutation in a large family with
juvenile-onset primary open angle glaucoma. Invest Ophthalmol 2000;41:S822 75. Johnson AT, Drack AV, Kwitek AE et al. Clinical features and linkage analysis
of a family with autosomal dominant juvenile glaucoma. Ophthalmology 1993;100:524 76. Johnson AT, Richards JE, Boehnke M et al. Clinical phenotype of juvenile-onset
primary open-angle glaucoma linked to chromosome 1q. Ophthalmology 1996;103:808 77. Suzuki Y, Shirato S, Taniguchi F et al. Mutations in the TIGR gene in familial
primary open-angle glaucoma in Japan letter. Am J Hum Genet 1997;61:1202 78. Michels-Rautenstrauss KG, Mardin CY, Budde WM et al. Juvenile open angle
glaucoma: Fine mapping of the TIGR gene to 1q24.3- q25.2 and mutation
analysis. Hum Genet 1998;102:103 79. Stoilova D, Child A, Trifan OC et al. Localization of a locus (GLC1B) for
adult-onset primary open angle glaucoma to the 2cen-q13 region. Genomics 1996;36:142 80. Faucher M, Dubois S, Cote G et al. An integrated map of the GLC1B locus
for primary open-angle glaucoma at chromosome 2. Invest Ophthalmol 2000;41:A4373 81. Wiggs JL, Allingham RR, Hossain A et al. Genome-wide scan for adult onset
primary open angle glaucoma. Hum Mol Genet 2000;9:1109 82. Desai TD, Child A, Rezaie T et al. Searching for identification of mutations
involved in GLC1B, GLC1D, and GLC1E linked glaucoma loci. Invest
Ophthalmol 2000;41:S526 83. Wirtz MK, Child A, Rezaie T et al. Mapping a gene for adult-onset primary
open-angle glaucoma to chromosome 3q. Am J Hum Genet 1997;60:296 84. Xu H, Acott TS, Wirtz MK. Identification and expression of a novel type
I procollagen C-proteinase enhancer protein gene from the glaucoma candidate
region on 3q21-24. Genomics 2000;66:264 85. Trifan OC, Traboulsi EI, Stoilova D et al. A third locus (GLC1D) for adult-onset
primary open-angle glaucoma maps to the 8q23 region. Am J Ophthalmol 1998;126:17 86. Sarfarazi M, Child A, Stoilova D et al. Localization of the fourth locus (GLC1E) for
adult-onset primary open-angle glaucoma to the 10p15-p14 region. Am
J Hum Genet 1998;62:641 87. Wirtz MK, Samples JR, Rust K et al. GLC1F, a new primary open-angle glaucoma
locus, maps to 7q35-q36. Arch Ophthalmol 1999;117:237 88. Sverrisson, TM, Rust K, Huynh L et al. Refining the GLC1F physical map
and analysis of candidate genes. Invest Ophthalmol Vis Sci 2000;41:A4373 89. Andersen JS, Pralea AM, DelBono EA et al. A gene responsible for the pigment
dispersion syndrome maps to chromosome 7q35-q36. Arch Ophthalmol 1997;115:384 90. Yokoyama A, Nao-i N, Date Y et al. Detection of a new TIGR gene mutation
in a Japanese family with primary open angle glaucoma. Jpn J Ophthalmol 1999;43:85 91. Yoon SJ, Kim HS, Moon JI et al. Mutations of the TIGR/MYOC gene in primary
open-angle glaucoma in Korea [letter]. Am J Hum Genet 1999;64:1775 92. Kee C, Ahn BH. TIGR gene in primary open-angle glaucoma and steroid-induced
glaucoma. Korean J Ophthalmol 1997;11:75 93. Lam DS, Leung YF, Chua JK et al. Truncations in the TIGR gene in individuals
with and without primary open-angle glaucoma. Invest Ophthalmol
Vis Sci 2000;41:1386 94. Zhou Y, Ge J, Guo Y. To screen, clone and sequence TIGR gene mutation in
Chinese patients with primary open-angle glaucoma. Chin J Ophthalmol 2000;36:416 95. Schwartz M, Yoles E. Self-destructive and self-protective processes in
the damaged optic nerve: implications for glaucoma. Invest Ophthalmol
Vis Sci 2000;41:349 96. Alsbirk PH. Anterior chamber depth and primary angle-closure glaucoma. II: A
genetic study. Acta Ophthalmol 1975;53:436 97. Drance SM, Morgan RW, Bryett J et al. Anterior chamber depth and gonioscopic
findings among the Eskimos and Indians in the Canadian Arctic. Can
J Ophthalmol 1973;8:255 98. Lowe RF. Primary angle-closure glaucoma: Inheritance and environment. Br
J Ophthalmol 1972;56:13 99. Spaeth GL. Gonioscopy: Uses old and new—The inheritance of occludable
angles. Ophthalmology 1978;85:222 100. Tomlinson A, Leighton DA. Ocular dimensions in the heredity of angle-closure
glaucoma. Br J Ophthalmol 1973;57:475 101. Lichter PR and Anderson DR. Discussions on Glaucoma. New York: Grune & Stratton, 1977 102. Gieser DK, Wilensky JT. HLA antigens and acute angle-closure glaucoma. Am
J Ophthalmol 1979;88:232 103. Brooks AM, Gillies WE. Blood groups as genetic markers in glaucoma. Br
J Ophthalmol 1988;72:270 104. Brockhurst RJ. Nanophthalmos with uveal effusion: A new clinical entity. Trans
Am Ophthalmol Soc 1974;72:371 105. Kimbrough RL, Trempe CS, Brockhurst RJ et al. Angle-closure glaucoma in
nanophthalmos. Am J Ophthalmol 1979;88:572 106. Cross HE, Yoder F. Familial nanophthalmos. Am J Ophthalmol 1976;81:300 107. Martorina M. Familial nanophthalmos. J Fr Ophthalmol 1988;11:357 108. Neelakantan A, Venkataramakrishnan P, Rao BS et al. Familial nanophthalmos: Management
and complications. Ind J Ophthalmol 1994;42:139 109. Altintas AK, Acar MA, Yalvac IS et al. Autosomal recessive nanophthalmos. Acta
Ophthalmol Scand 1997;75:325 110. Vingolo EM, Steindl K, Forte R et al. Autosomal dominant simple microphthalmos. J
Med Genet 1994;31:721 111. Malta RF, Povoa CA, Castro EFS et al. Nanophthalmos (NNo1): Study of a
Brazilian family. Invest Ophthalmol Vis Sci 2000;41:A4370 112. Bessant DA, Khaliq S, Hameed A et al. A locus for autosomal recessive congenital
microphthalmia maps to chromosome 14q32. Am J Hum Genet 1998;62:1113 113. deLuise VP, Anderson DR. Primary infantile glaucoma (congenital glaucoma). Surv
Ophthalmol 1983;28:1 114. Lehrfeld L, Reber J. Glaucoma at the Wills Eye Hospital (1925-1935). Arch
Ophthalmol 1937;18:712 115. Gencik A. Epidemiology and genetics of primary congenital glaucoma in Slovakia. Description
of a form of primary congenital glaucoma in gypsies
with autosomal-recessive inheritance and complete penetrance. Dev Ophthalmol 1989;16:76 116. Jaafar MS. Care of infantile glaucoma patients. In Reinecke RD (ed). Ophthalmology
Annual. New York: Raven Press, 1988:15 117. Westerlund E. On the heredity of congenital hydrophthalmus. Acta Ophthalmol 1943;21:330 118. Shaffer RN. Genetics and the congenital glaucomas. Am J Ophthalmol 1965;60:981 119. Bejjani BA, Lewis RA, Tomey KF et al. Mutations in CYP1B1, the gene for
cytochrome P4501B1, are the predominant cause of primary congenital glaucoma
in Saudi Arabia. Am J Hum Genet 1998;62:325 120. Merin S, Morin D. Heredity of congenital glaucoma. Br J Ophthalmol 1972;56:414 121. Demenais F, Elston RC, Bonaiti C et al. Congenital glaucoma: genetic models. Hum
Genet 1979;46:305 122. Gencik A, Gencikova A, Gerinec A. Genetic heterogeneity of congenital glaucoma. Clin
Genet 1980;17:241 123. Sarfarazi M, Stoilov I. Molecular genetics of primary congenital glaucoma. Eye 2000;14:422 124. Sarfarazi M, Akarsu AN, Hossain A et al. Assignment of a locus (GLC3A) for
primary congenital glaucoma (buphthalmos) to 2p21 and evidence for
genetic heterogeneity. Genomics 1995;30:171 125. Stoilov I, Akarsu AN, Sarfarazi M. Identification of three different truncating
mutations in cytochrome P4501B1 (CYP1B1) as the principal cause
of primary congenital glaucoma (buphthalmos) in families linked to
the GLC3A locus on chromosome 2p21. Hum Mol Genet 1997;6:641 126. Stoilov I, Akarsi AN, Alonzie I et al. Sequence analysis and homology modeling
suggest that primary congenital glaucoma on 2p21 results from
mutations disrupting either the hinge region or the conserved core structures
of cytochrome P4501B1. Am J Hum Genet 1998;62:573 127. Plasilova M, Stoilov I, Sarfarazi M et al. Identification of a single ancestral
CYP1B1 mutation in Slovak Gypsies (Roms) affected with primary
congenital glaucoma. J Med Genet 1999;36:290 128. Bejjani BA, Stockton DW, Lewis RA et al. Multiple CYP1B1 mutations and
incomplete penetrance in an inbred population segregating primary congenital
glaucoma suggest frequent de novo events and a dominant modifier
locus Hum Mol Genet 2000;9:367 129. Kakiuchi T, Isashiki Y, Nakao K et al. A novel truncating mutation of cytochrome
P4501B1 (CYP1B1) gene in primary infantile glaucoma. Am J Ophthalmol 1999;128:370 130. Graham-Lorence S, Peterson JA. P450s: Structural similarities and functional
differences. FASEB J 1996;10:206 131. Chen CD, Kemper B. Different structural requirements at specific proline
residue positions in the conserved proline-rich region of cytochrome
P450 2C2. J Biol Chem 1996;271:28607 132. Yamazaki S, Sato K, Suhara K et al. Importance of the proline-rich region
following signal-anchor sequence in the formation of correct conformation
of microsomal cytochrome P-450s. J Biochem 1993;114:652 133. Nelson DR, Koymans L, Kamataki T et al. P450 superfamily: Update on new
sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics 1996;6:1 134. Nebert DW. Proposed role of drug-metabolizing enzymes: Regulation of steady
state levels of the ligands that effect growth, homeostasis, differentiation, and
neuroendocrine functions. Mol Endocrinol 1991;5:1203 135. Stoilov I, Jannson I, Sarfarazi M et al. Role of cytochrome P450 in development. Drug
Metab Drug Interact 2001;18:33 136. Lichter PR, Farley F, Stringham HM et al. Co-segregation of open-angle
glaucoma and the nail-patella syndrome. Am J Ophthalmol 1997;124:506 137. Vollrath D, Jaramillo-Babb VL, Clough MV et al. Loss-of-function mutations
in the LIM-homeodomain gene, LMX1B, in nail-patella. Hum Mol Genet 1998;7:1091 138. Chen H, Lun Y, Ovchinnikov D et al. Limb and kidney defects in Lmx1b mutant
mice suggest an involvement of LMX1B in human nail patella syndrome. Nat
Genet 1998;19:51 139. Dreyer SD, Morello R, German MS et al. LMX1B transactivation and expression
in nail-patella syndrome. Hum Mol Genet 2000;9:1067 140. Dreyer SD Zhou G, Baldini A et al. Mutations in LMX1B cause abnormal skeletal
patterning and renal dysplasia in nail patella syndrome. Nat Genet 1998;19:47 141. Clough MV, Hamlington JD, McIntosh I. Restricted distribution of loss-of-function
mutations within the LMX1B genes of nail-patella syndrome patients. Hum
Mutat 1999;14:459 142. Farley FA, Lichter PR, Downs CA et al. An orthopaedic scoring system for
nail-patella syndrome and application to a kindred with variable expressivity
and glaucoma.J Pediatr Orthop 1999;19:624 143. Zimmerman CC, Lingappa VR, Richards JE et al. A trabecular meshwork glucocorticoid
response (TIGR) gene mutation affects translocational processing. Mol
Vis 1999;5:19 144. Stoilov I., Akarsu AN, Sarfarazi M. Identification of three different truncating
mutations in cytochrome P4501B1 (CYP1B1) as the principal cause
of primary congenital glaucoma (buphthalmos) in families linked to
the GLC3A locus on chromosome 2p21. Hum Mol Genet 1997;6:641 145. Akarsu AN, Turacli ME, Aktan SG, et al. A second locus (GLC3B) for primary
congenital glaucoma (buphthalmos) maps to the 1p36 region. Hum Mol
Genet 1996;5:1199 |