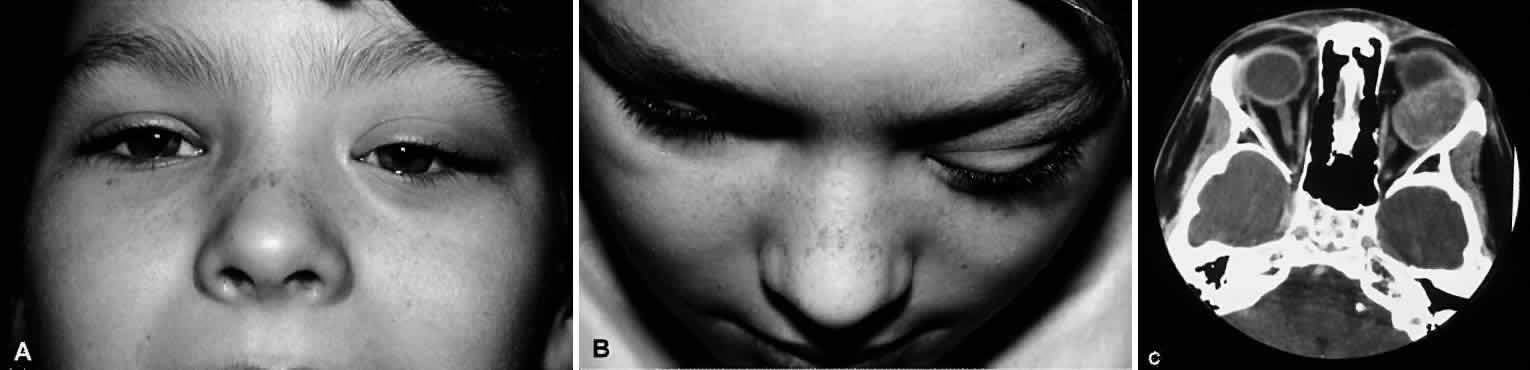
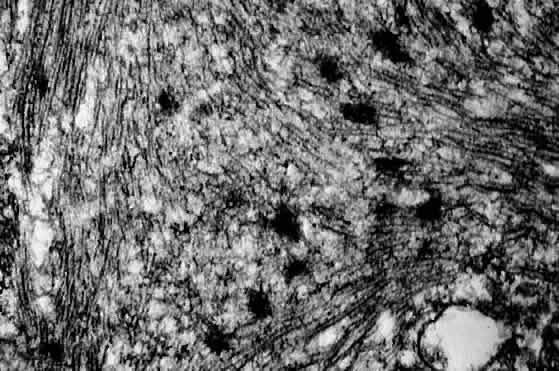
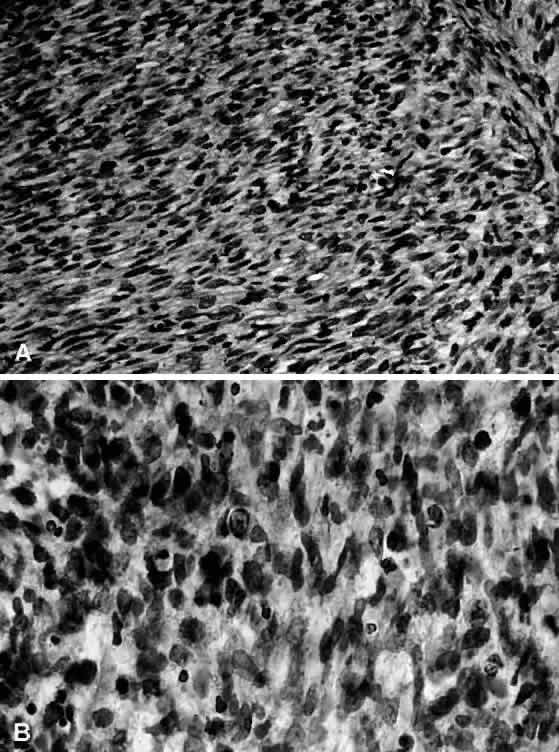
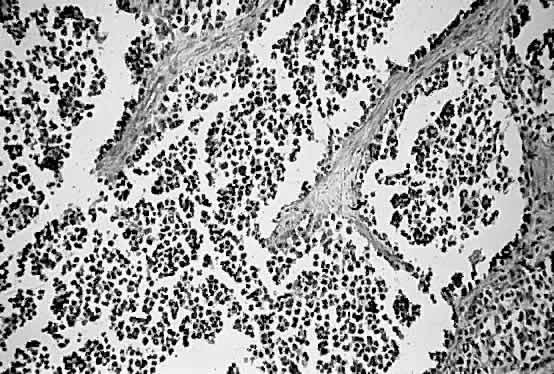
1. Miller RW: Frequency and environmental epidemiology of childhood cancer. In
Pizzo PA, Poplack DG (eds): Principles and Practice of Pediatric
Oncology, pp 3–18. Philadelphia, JB Lippincott, 1988 2. Porterfield JF, Zimmerman LE: Rhabdomyosarcoma of the orbit: a clinicopathologic study of 55 cases. Virchows Arch Pathol Anat 335:329, 1962 3. Lederman M: Radiotherapy in the treatment of orbital tumors. Br J Ophthalmol 40:592, 1956 4. Cassady JR, Sagerman RH, Tretter P, Ellsworth RM: Radiation therapy for rhabdomyosarcoma. Radiology 91:116, 1968 5. Heyn RM, Holland R, Newton WA et al: The role of combined chemotherapy in the treatment of rhabdomyosarcoma
in children. Cancer 34:2128, 1974 6. Abramson DH, Ellsworth RM, Tretter P et al: The treatment of orbital rhabdomyosarcoma with irradiation and chemotherapy. Ophthalmology 86:1330, 1979 7. Maurer HM, Beltangady M, Gehan EA et al: The Intergroup Rhabdomyosarcoma Study-I: a final report. Cancer 61:209, 1988 8. Maurer HM, Gehan EA, Beltangady M et al: The Intergroup Rhabdomyosarcoma Study-II. Cancer 71:1904, 1993 9. Crist W, Gehan EA, Ragab AH et al: The Third Intergroup Rhabdomyosarcoma Study. J Clin Oncol 13:610, 1995 10. Stiller CA, Parkin DM: International variations in the incidence of childhood soft-tissue sarcomas. Paediatr Perinat Epidemiol 8:107, 1994 11. Stiller CA, McKinney PA, Bunch KJ et al: Childhood cancer and ethnic group in Britain: a United Kingdom Children's
Cancer Study Group (UKCCSG) study. Br J Cancer 64:543, 1991 12. Bernard JL, Bernard-Couteret E, Coste D et al: Childhood cancer incidence in the southeast of France (abstr). Eur J Cancer 29(A):2284, 1993 13. Knowles DM, Jakobiec FA, Potter GD, Jones IS: The diagnosis and treatment
of rhabdomyosarcoma of the orbit. In Jakobiec FA (ed): Ocular and Adnexal
Tumors, pp 708–734. Birmingham, AL, Aesculapius, 1978 14. Olurin O: Orbital rhabdomyosarcoma in pregnancy. Cancer 24:1013, 1969 15. Ellenbogen E, Lasky MA: Rhabdomyosarcoma of the orbit in the newborn. Am J Ophthalmol 80:1024, 1975 16. Sood GC, Sen DK, Diwan R, Aurora AL: Orbital rhabdomyosarcoma in an infant. Br J Ophthalmol 54:203, 1970 17. Jones IS, Reese AB, Krout J: Orbital rhabdomyosarcoma: an analysis of 62 cases. Am J Ophthalmol 61:721, 1966 18. Ashton N, Morgan G: Embryonal sarcoma and embryonal rhabdomyosarcoma of the orbit. J Clin Pathol 18:699, 1965 19. Kassel SH, Copenhaver R, Arean VM: Orbital rhabdomyosarcoma. Am J Ophthalmol 60:811, 1965 20. Li FP, Fraumeni JF, Mulvihill JJ et al: A cancer family syndrome in twenty-four kindreds. Cancer Res 48:5358, 1988 21. Malkin D, Li FP, Strong LC et al: Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and
other neoplasms. Science 250:1233, 1990 22. McKeen EA, Bordurtha J, Meadows AT et al: Rhabdomyosarcoma complicating multiple neurofibromatosis. J Pediatr 93:992, 1978 23. Hartley AL, Birch JM, Marsden HB et al: Neurofibromatosis in children with soft tissue sarcoma. Pediatric Hematol Oncol 5:7, 1988 24. Whang-Peng J, Knutsen T, Theil K et al: Cytogenetic studies in subgroups of rhabdomyosarcoma. Genes Chromosom Cancer 5:299, 1992 25. Pappo AS, Shapiro DN, Crist WM, Maurer HM: Biology and therapy of pediatric rhabdomyosarcoma. J Clin Oncol 13:2123, 1995 26. Mitchell CD, Ventris JA, Carr TJ, Cowell JK: Molecular definition in a somatic cell hybrid of a specific 2;13 translocation
breakpoint in childhood rhabdomyosarcoma. Oncogene 6:89, 1991 27. Stratton MR, Williams S, Fisher C et al: Structural alterations of the RB1 gene in human soft tissue tumours. Br J Cancer 60:202, 1989 28. Trent J, Casper J, Meltzer P et al: Nonrandom chromosome alterations in rhabdomyosarcoma. Cancer Genet Cytogenet 16:189, 1985 29. Granberg I, Mark J: The chromosomal aberration of double-minutes in a human embryonic rhabdomyosarcoma. Acta Cytol 15:42, 1971 30. Grufferman S, Schwartz AG, Ruymann FB, Maurer HM: Parents' use of cocaine and marijuana and increased risk of rhabdomyosarcoma
in their children. Cancer Causes Control 4:217, 1993 31. Ghali MH, Yoo KY, Flannery JT, Dubrow R: Association between childhood rhabdomyosarcoma and maternal history of
stillbirths. Int J Cancer 50:365, 1992 32. Grufferman S, Wang HH, DeLong ER et al: Environmental factors in the etiology of rhabdomyosarcoma in childhood. J Natl Cancer Inst 68:107, 1982 33. Frayer WC, Enterline HT: Embryonal rhabdomyosarcoma of the orbit in children and young adults. Arch Ophthalmol 62:203, 1959 34. Henderson JW: Neoplasms of muscle. In Henderson JW (ed): Orbital Tumors, pp 497–511. New
York, Thieme-Stratton, 1980 35. Joffe L, Shields JA, Pearah JD: Epibulbar rhabdomyosarcoma without proptosis. J Pediatr Ophthalmol 14:364, 1977 36. Elsas FJ, Mroczek EC, Kelly DR, Specht CS: Primary rhabdomyosarcoma of the iris. Arch Ophthalmol 109:982, 1991 37. Naumann G, Font RL, Zimmerman LE: Electron microscopic verification of primary rhabdomyosarcoma of the iris. Am J Ophthalmol 54:110, 1972 38. Woyke S, Chwirot R: Rhabdomyosarcoma of the iris. Br J Ophthalmol 56:60, 1972 39. Wilson ME, McClatchey SK, Zimmerman LE: Rhabdomyosarcoma of the ciliary body. Ophthalmology 97:1484, 1990 40. Baron EM, Kersten RK, Kulwin DR: Rhabdomyosarcoma manifesting as acquired nasolacrimal duct obstruction. Am J Ophthalmol 115:239, 1993 41. Bozzao L, Fantozzi LM, Rosa M: Childhood orbital pathology: investigation by computed tomography. J Neurosurgical Sci 26:25, 1982 42. Smith RR: The orbit. In Cohen MD, Edwards MK (eds): Magnetic Resonance
Imaging of Children, pp 325–386. Philadelphia, BC Decker, 1990 43. Lueder GT, Burrows PE, Hurwitz JJ et al: Vascular patterns in orbital rhabdomyosarcoma. J Pediatr Ophthalmol Strabismus 31:46, 1994 44. Rothman SLG, Kier EL, Allen WE, Pratt AG: Cerebral angiography in the diagnosis of orbital rhabdomyosarcoma. Neuroradiology 112:353, 1974 45. Sevel D: Intracranial extension of a non-striated embryonal rhabdomyosarcoma of
the orbit. Br J Ophthalmol 53:180, 1969 46. Harris GJ, Beatty RL: Acute proptosis in childhood. In Linberg JV (ed): Oculoplastic
and Orbital Emergencies, p 88. East Norwalk, CT, Appleton & Lange, 1989 47. Tsokos M: The diagnosis and classification of childhood rhabdomyosarcoma. Semin Diagn Pathol 11:26, 1994 48. Wharam MD, Maurer HM: Management of orbital rhabdomyosarcoma. In Jacobs
C (ed): Cancers of the Head and Neck, pp 223–243. Boston, Martinus
Nijhoff, 1987 49. Jakobiec FA, Font RL: Mesenchymal tumors. In Spencer WH (ed): Ophthalmic
Pathology: An Atlas and Textbook, pp 2554–2603. Philadelphia, WB
Saunders, 1986 50. Weber CO: Anatomische untersuchung einer hypertrophische zunge nebst bemerkungen
ueber die neubildung quergestreifter muskelfasern. Virchow Arch Pathol Anat 7:115, 1854 51. Horn RC, Enterline HT: Rhabdomyosarcoma: A clinicopathologic study and classification of 39 cases. Cancer 11:181, 1958 52. Morales AR, Fine G, Horn RC: Rhabdomyosarcoma: an ultrastructural appraisal. Pathol Ann 7:81, 1972 53. Mierau GW, Favara BE: Rhabdomyosarcoma in children. Cancer 46:2035, 1980 54. Newton WA: Classification of rhabdomyosarcoma. Curr Topics Pathol 89:241, 1995 55. Crist WM, Garnsey L, Beltangady MS et al: Prognosis in children with rhabdomyosarcoma: a report of the Intergroup
Rhabdomyosarcoma Studies I and II. J Clin Oncol 8:443, 1990 56. Wijnaendts LCD, van der Linden JC, van Unnik AJM et al: Histopathological features and grading in rhabdomyosarcomas of childhood. Histopathology 24:303, 1994 57. Newton WA Jr, Soule EH, Hamoudi AB et al: Histopathology of childhood sarcomas, Intergroup Rhabdomyosarcoma Studies
I and II: clinicopathologic correlation. J Clin Oncol 6:67, 1988 58. Gehan EA, Glover FN, Maurer HM et al: Prognostic factors in children with rhabdomyosarcoma. Natl Cancer Inst Monogr 56:83, 1981 59. Caillaud JM, Gerard-Marchant R, Marsden HB et al: Histopathological classification of childhood rhabdomyosarcoma: a report
from the International Society of Pediatric Oncology Pathology Panel. Med Pediatr Oncol 17:391, 1989 60. Asmar L, Gehan EA, Newton WA et al: Agreement among and within groups of pathologists in the classification
of rhabdomyosarcoma and related sarcomas: report of an international
study of four pathology classifications. Cancer 74:2579, 1994 61. Heyn R, Haeberlen V, Newton WA et al: Second malignant neoplasms in children treated for rhabdomyosarcoma. J Clin Oncol 11:262, 1993 62. Abramson DH, Notis CM: Visual acuity after radiation for orbital rhabdomyosarcoma. Am J Ophthalmol 118:808, 1994 63. Sagerman RH: Orbital rhabdomyosarcoma: a paradigm for irradiation. Radiology 187:605, 1993. |