RANGE AND CHARACTER OF EYE MOVEMENTS
It is through familiarity with the normal that we may distinguish the abnormal. Therefore, this discussion begins with a brief description of typical and normal eye movement. As with the determination of visual function, observation of ocular motility begins as the patient enters the examination room and continues during history taking. The physician should note normal or abnormal spontaneous gaze and eye position. It is especially rewarding to observe the spontaneous eye movements of infants and young children, before the “threat” of actual physical examination.
Fixation and Ocular Stabilization
In the human, eye movements occur so that a focused image is projected onto the central fixational area of the retina, the fovea. Here visual function is greatest because of the concentration of receptor cells. A stimulus that falls on the peripheral retina provokes an eye movement that realigns the visual axis of the eye so that the fovea “acquires” the visual direction of that stimulus; that is, the eye “fixates” the stimulus that now becomes the object of regard. The eye movement that acquires the target is the “fixational reflex,” and the steady “lock on” of the fovea is “fixation.”
It is obvious that ocular fixation, in acquiring and holding a target, requires reasonably good visual acuity, although more peripheral parts of retina have some fixational capacity. Therefore, the afferent visual pathways from retina to cortex play a major role in fixation, but there must be a counterpart motor efferent that precisely guides the fixational eye movement to target and then maintains punctilious gaze. The ability of one or both eyes accurately and steadily to fixate a target (muscle light, finger, or pencil) is a prerequisite for evaluating other types of eye movement. For example, if one eye fixates poorly due to dense central scotoma, the cover-uncover or alternate-cover test has limited value.
Fixational movements may be voluntary (the quick eye movements as one looks about a room) or involuntary (when a bright light suddenly appears in the far peripheral field). The involuntary fixational reflex may alternately be called a “visually elicited eye movement”; that such eye movements may be provoked, is used as evidence of peripheral field function in infants (see below). In either case, when the object of regard is acquired by the fovea, fixation is steady, and the eye continues smoothly to track (a pursuit movement) a target that is slowly moved, much as a radar system acquires a target, locks on, and smoothly tracks.
The stability of fixation is quickly determined simply by having a patient (even a baby) gaze at a target (light, pencil, small cartoon character, or hand puppet). Fixation should be steady and unwavering. If the target is slowly moved from side to side, the eyes move smoothly to maintain fixation without “slippage” off the target. These ocular pursuit movements are discussed below. Fixation should be tested binocularly as well as with each eye separately.
Conjugate Gaze: Versions
The ability of the eyes to move symmetrically and synchronously in the same direction, horizontally or vertically, is termed “conjugate” (yoked together) gaze. These versional movements may be rapid (a saccade), as in the command to “look left,” or during the reflex to take up fixation (see above), or may be slow as in the smooth tracking (pursuit) of a slowly moving target. Saccadic and pursuit sub-systems are discussed in some detail in Volume 2, Chapters 9 through 11.
Conjugate gaze is tested by asking the patient to “look right … left.” The patient is then asked to “follow” (“let your eyes follow”) some target such as the examiner's hand-held light (Fig. 1). Command horizontal eye movements should be rapid and symmetric in the two eyes. Binocular excursions (both eyes open) are generally smaller than monocular (one eye covered), and the eyes move through approximately 45° to either side of the straight forward position (i.e., primary gaze). Abduction is difficult to sustain beyond 35° of eccentricity, and a physiologic end-gaze nystagmus is quite common (see Volume 2, Chapter 11). In the abducting eye, the temporal aspect of the corneal margin should almost approximate the outer angle (lateral canthus) of the lids, with little sclera visible. In elderly patients with lax facial tissues, the external lid angle may be displaced so that a minimal rim of sclera is still visible in the abducted position. The adducting eye is positioned such that 1 mm to 2 mm of the nasal aspect of the cornea may be buried in the folds of conjunctiva and caruncle at the inner canthal angle.
|

Vertical conjugate gaze is somewhat more difficult to assess. Downward gaze should be at least 45°, but since the lower lid is indented by the cornea, there is no constant landmark. Upward gaze is even more difficult to assess. Chamberlain1 reported a progressive restriction in upward ocular versions with advancing age: 95 patients aged 5 to 34 years had 36° to 40° of elevation; 125 patients aged 65 to 94 years had 16° to 22° of elevation.
Conjugate versions may also be tested without the patient's active cooperation, by directing attention to a distant target (e.g., a wall-mounted light, or even while an acuity chart is being read), and passively rotating the patient's face to the right, left, up, and down.
Convergence
Convergence is an associated, but disjugate, movement, in that the eyes move toward each other, rather than turning toward the same side (i.e., conjugate). It is usually accomplished by synchronous action of both medial recti muscles as in reading or other near-visual tasks, but convergence may be blended with a conjugate gaze, for example, fixating a near object directly in front of one eye. The neural mechanisms responsible for adduction during lateral conjugate gaze, and during convergence, are separate and may be dissociated in pathologic states, for example, in patients with internuclear ophthalmoplegia.
It is helpful to test convergence capability in the following neuro-ophthalmologic situations: (1) if pupillary light reactions are absent or sluggish, the pupils are observed during convergence (more precisely, during the near-effort reflex); (2) if there is lag of the adducting eye on lateral conjugate gaze suggesting an internuclear ophthalmoplegia; (3) any bilateral external ophthalmoplegia; and (4) in acquired exotropia, in which monocular ductions are full, suggesting convergence paralysis.
Convergence strength varies considerably and depends principally on the cooperation of the patient. A sure sign that the patient is indeed attempting to converge is simultaneous pupillary constriction. Of course, there are pathologic states that involve both convergence and pupillary constriction (e.g., Parinaud's dorsal midbrain syndrome). It is helpful if the patient's own finger is used as a convergence target. Thus, even in a blind patient, convergence may be examined using proprioceptive clues to the nearness of one's finger or hand. Reticence to attempt to gaze at one's finger, or histrionic avoidance, may signal a functional disorder or dissembling.
SPECIAL TECHNIQUES
Examination of Infants
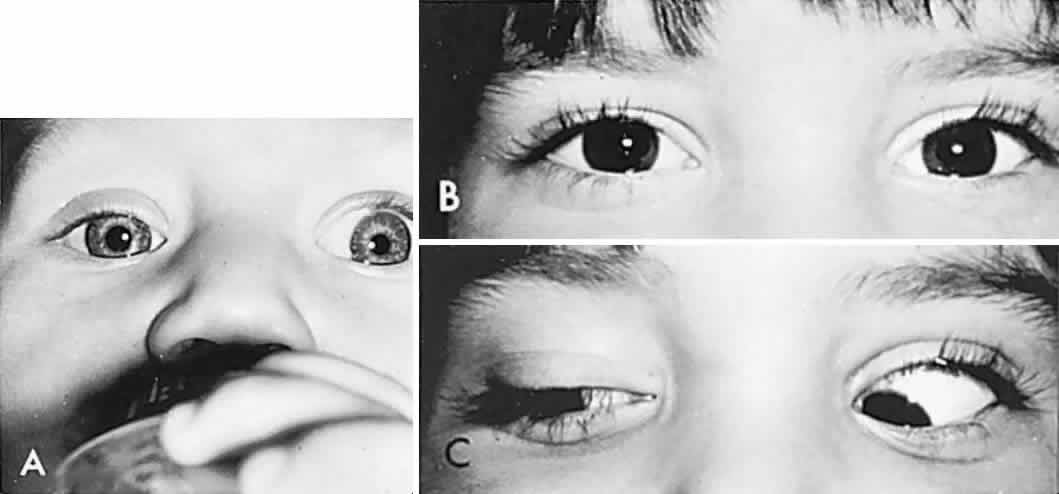
In order to determine the extent of conjugate eye movements in infants, it is often necessary to resort to special clinical techniques and manipulations. However, initial observation of spontaneous versions and fixation may suffice, and such observations should begin before the infant or young child is startled or anxious. Otherwise, visually elicited eye movements are provoked by objects that attract an infant's attention (see Volume 2, Chapter 2, Fig. 9) and therefore also provide a gross estimation of visual function. Similarly, a child turns head and eyes toward a sound (jingling keys). This is an acoustically elicited eye movement that does not require sight, however, and so no conclusions may be drawn regarding vision. If the child accurately fixates and follows any silent object, that is good evidence of reasonably intact vision.
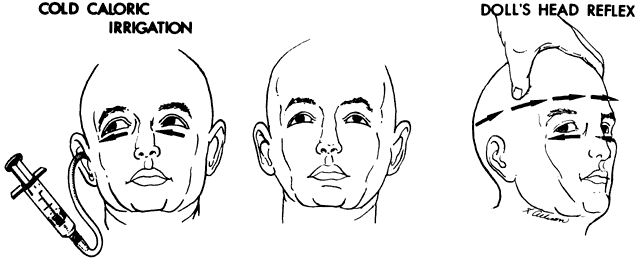
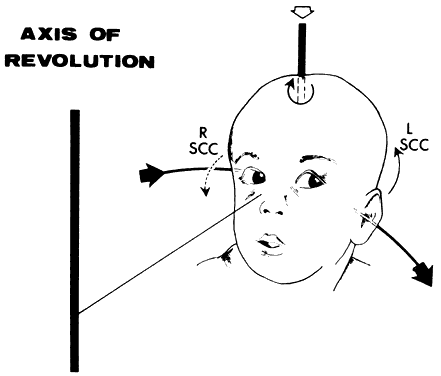
If the examiner holds a neonate upright, face to face, and turns, the neonate's eyes undergo a tonic deviation in the direction of the turn (Fig. 2). This tonic, conjugate eye deviation is a result of labyrinthine stimulation and does not depend on vision. The fast phase (jerk) of such rotation induced nystagmus may be absent in the premature neonate, but it is usually present before 1 week of age. Rotational maneuvers are helpful in determining the state of brain stem systems for conjugate horizontal gaze. Failure to elicit eye movement on rotational testing infers a major pontine or extraocular muscle abnormality. Brisk rotation of the head, vertically and horizontally, on the long axis of the body (“doll's head” phenomenon), produces tonic contralateral horizontal deviation of the eyes in the neonate.
|
Optokinetic nystagmus (OKN; see below) may be used to determine ocular motility, but induced OKN movements are less extensive than with rotational stimulation. Disparity between vertical and horizontal response or between optokinetic and rotational responses may indicate a disorder of supranculear gaze mechanisms, such as in congenital ocular motor apraxia or congenital nystagmus (see Volume 2, Chapter 13).
Optokinetic Nystagmus
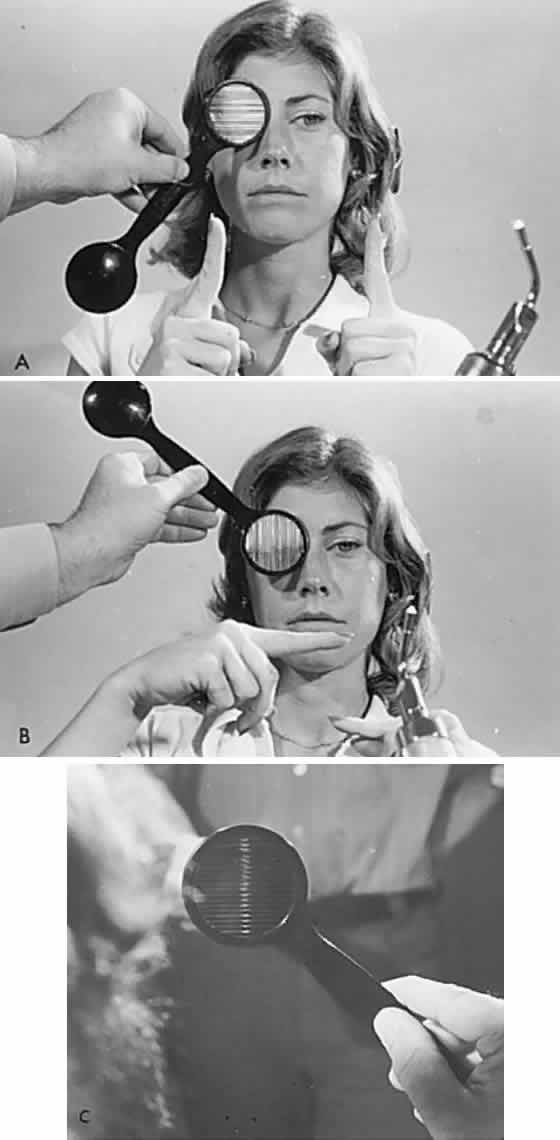
If a series of vertical bars or other such patterned contours is passed before the eyes (Fig. 3), a visually induced nystagmus occurs. This nystagmus was apparently first adequately described by Helmholtz, who observed the eyes of passengers gazing out of train windows at the passing countryside. This phenomenon is easily noted in riders of subway trains, buses, and so forth. There is a slow following phase toward the side of target movement (or at the passing scene) and a rapid jerk return in the opposite direction. The character of OKN is partially dependent on the speed of target movement, the pattern density, and the subject's visual function and state of attentiveness. Because the slow phase is initiated by vision, OKN may be used as a rough parameter of visual function. As noted by Linksz,2 OKN is more closely related to movement perception or contour recognition than to visual acuity. Actually, suppression of OKN response by interposition of a fixation target of variable size is best correlated with “acuity.” It should be recognized that OKN represents an innate and highly complex ocular motor reflex evoked by perception of moving contours, which is only distantly related to visual acuity. In clinical practice, only a rough estimate of visual “acuity” is obtained with the usual optokinetic devices. Examples of the minimum visual requirement with a stimulus at 2 to 4 feet (see Fig. 3) are presented below:
|

- Striped drum: finger count 3 to 5 feet.
- Squares on flag: finger count 3 to 5 feet.
- Tailor's tape: 20/400 to 3/200.
OKN is an especially useful technique in infants as a test of gross visual function, and it should be demonstrable to some degree even in newborns. It is positive evidence of sight in malingerers and hysteric patients, and it is similarly productive in evaluating uncooperative, semiobtunded, or dysphasic patients. Central vision need not be good to have intact OKN response if gross pattern stimuli are used (see Fig. 3). It is evident that visual impulses trigger a cerebral mechanism that more or less regulates the rhythmicity of the nystagmus. OKN is affected by nonvisual factors and may even be observed in hypnotic states, although the character of the OKN is not strictly parallel to that induced by actual visual stimuli. An optokinetic after-nystagmus may be demonstrated on cessation of target rotation, especially if fixation is inhibited by dark environment or lid closure. Further evidence of nonlinearity of visual input and optokinetic response was demonstrated in the rabbit by Collewijn,3 but it is also true in the human patient who meets the criteria for conducting the demonstration; if a gross OKN stimulus is presented monocularly before a seeing but immobile eye, optokinetic response is observed in the occluded or blind, but mobile, fellow eye.
Optokinetic response as a useful clinical tool was first popularized by Kestenbaum,4 and later by Cogan and Smith,5 and others. Cerebral lesions deep in the parietal lobe cause homonymous hemianopias and also interrupt the descending occipitomesencephalic system for slow (pursuit) eye movements.
The occipital optomotor fibers descend in the internal sagittal stratum and are medial to the geniculocalcarine radiations in their posterior course through the parietal lobe. Lesions here consistently produce a homonymous hemianopia (contralateral) and a defective OKN when targets are moved toward the side of the lesion. The defective OKN (a “positive OKN sign”) is not due to targets coming out of the hemianopia, as originally thought by Barany, because strictly occipital or temporal lobe lesions producing profound hemianopias are not associated with a positive (i.e., asymmetric) OKN response. Therefore, in the presence of a homonymous hemianopia, a defective OKN response indicates deep parietal involvement. This sign is considerably more helpful when the nondominant (right) parietal lobe is involved (left hemianopia). When the dominant (usually left) parietal lobe is involved, there are consistently other localizing signs and symptoms, such as motor and sensory dysphasias.
OKN may be helpful in elucidating volitional (saccadic) gaze palsies, with intact pursuit movement. For example, a patient with a right frontal lesion may show an inability to make command eye movements to the left, but slow-moving targets will produce smooth pursuit toward the left. Because pursuit to the left is intact and saccades to the right are normal, with targets rotated toward the left a normal OKN response is seen. However, with targets toward the right, the fast phase leftward cannot be generated, and a tonic deviation toward the right will result, as though the eyes are drawn in that direction by the rotating drum or tape.
OKN is a useful mechanism to amplify muscle pareses by inducing an asymmetric response of yoke muscles, best observed in the fast (saccadic) phase. For example, a subtle weakness of adduction (as seen in internuclear ophthalmoplegia) may be uncovered by observing a grossly dissociated OKN response when the paretic muscle is challenged to make a saccade (i.e., with targets rotated opposite the field of action of the paretic muscle). Thus, the adduction lag of the left eye with left internuclear ophthalmoplegia is enhanced by target rotation toward the left that induces fast phase to the (defective) right. Similarly, a command saccade from left to right also enhances the adduction lag; that is, a grossly slowed rightward saccade of the left eye is observed.6 Despite the complexities of the neurophysiologic basis for OKN, it is a simple and practical clinical maneuver with multiple applications in elucidating defects in the ocular motor system (see Volume 2, Chapter 10).
Diplopia
Double vision is a disturbing symptom that most patients cannot ignore, but many are unable to describe adequately. Diplopia may be intermittent at first, as the patient's fusional mechanism is not overtaxed, or it may first be noticed only for distant vision (suggesting lateral rectus paresis) or principally during near-vision tasks (suggesting medial rectus or superior oblique paresis). The issue may be clarified by the following questions:
- When and how was the double vision first noticed?
- Were there other symptoms when the double vision began: light-headedness, dizziness, or
spinning; weakness or tingling in face, arm, or leg; difficulty
with speech or swallowing (or other signs and symptoms of vertebrobasilar
ischemic episode); facial or ocular pain?
- Did one or both lids droop? Before or since?
- Are the images side by side, one above the other, or a combination?
- Is one image tilted (suggests superior oblique palsy)?
- Is it worse in the distance or while reading?
- Is the double vision the same throughout the day?
- Since first noticed, has it gotten worse, better, or stayed about the same?
- Can the head and face be turned or tilted to avoid the double vision?
- Is there a medical history of trauma, diabetes, hypertension, or dysthyroidism?
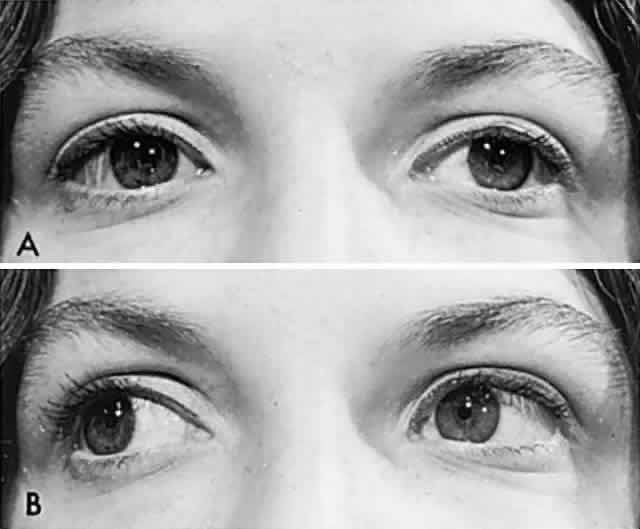
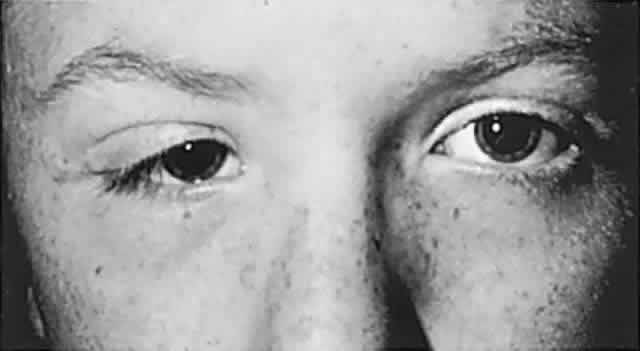
General inspection of the patient should include observation for spontaneous head tilt or face turn. While taking the history, the physician notes the position of the lids and looks for partial or total ptosis (myasthenia, third nerve palsy, Horner's syndrome) or unilateral or bilateral lid retraction (dysthyroidism). Signs of local orbital disease should be sought: proptosis, lid edema, and inflamed or hypervascular conjunctiva (see Volume 2, Chapters 12 and 14).
Almost all diplopia occurs as the result of an acquired paresis or palsy of one or more extraocular muscles. However, a child who insidiously develops a nonparalytic comitant squint may rarely complain of diplopia. In addition, in instances of fusional disturbances, such as convergence insufficiency, double vision may be a major symptom (see Volume 2, Chapter 13). These conditions are defined by appropriate examination techniques that demonstrate their nonparalytic characteristics; that is, monocular ductions are full, and angle of deviation remains the same regardless of which eye is made to fixate a target. In other words, in nonparalytic strabismus, the angle of muscle imbalance (squint) is essentially constant, and each eye has a full range of motion if tested separately.
Monocular diplopia (or perhaps more accurately, a “ghost image”) is an uncommon complaint and is usually the result of opacities of the cornea or lens, a high degree of astigmatism, or a partially dislocated lens. The use of a pinhole should solve this diagnostic dilemma (see Volume 2, Chapter 1). Kommerell7 described monocular diplopia caused by pressure of the upper lid that deforms the regular curvature of the cornea. The diagnosis is confirmed by an abnormal retinoscopic reflex, the “Venetian blind phenomenon.”
Bender8 reported instances of polyopia and monocular diplopia of cerebral origin (cerebral polyopia) accompanied by major field defects and other signs of parietal dysfunction, including: spatial disorientation, fluctuating extinction of images, extinction of cutaneous sensation, and disturbances of ocular fixation, which he attributed to occipital lobe disease (see Volume 2, Chapter 7).
In general, the complaint of diplopia indicates an acquired cranial nerve or extraocular muscle disorder that must be confirmed and investigated in an orderly, step-by-step analysis of ocular motility. It is beyond the scope of this work to present a detailed discussion of the anatomy and physiology of the extraocular muscles. However, a brief outline of basic ocular motility follows, primarily for the convenience of readers not formally trained in ophthalmology.
Each eye is maneuvered by six extraocular muscles, each of which has a partner in the contralateral orbit for coordinated simultaneous movement of the fellow eye. Muscles working in pairs for conjugate gaze are called “yoke” muscles. If we consider six cardinal positions of gaze, there are six pairs of yoke muscles that act as the prime movers in those six positions of gaze. Thus, on gaze rightward, the right lateral rectus and left medial rectus are yoke muscles. Each extraocular muscle has an ipsilateral antagonist that must relax if the globe is to move. Therefore, as the lateral rectus contracts to abduct the globe, its antagonist, the medial rectus, is simultaneously inhibited.
The six muscles of each eye may functionally be grouped in three pairs: the two horizontal recti (lateral and medial); the two vertical recti (superior and inferior); and the two obliques (superior and inferior). Each of these pairs represents an agonist-antagonist combination.
Numerous procedures have been devised to assess the function of the extraocular muscles, but if weakness is recent and moderate to marked, a simple test of versions (conjugate gaze) will usually suffice. It is also helpful to note the relative positions of the binocular corneal light reflections, as well as the gross positions of the globes. Mild limitations of movement often can more readily be detected during observation of binocular movements, as the eyes are turned into the “field of action” of the paretic muscle.
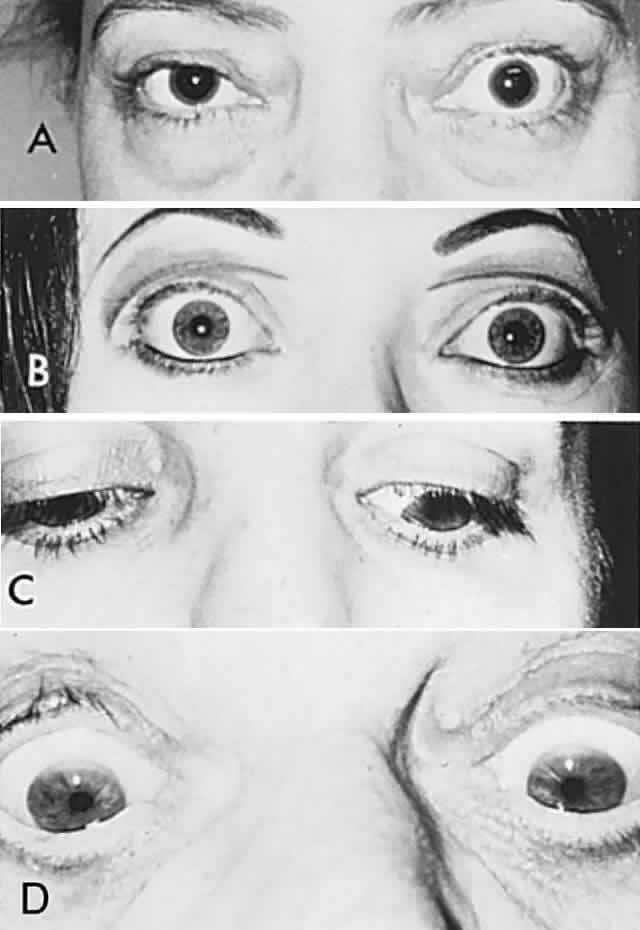
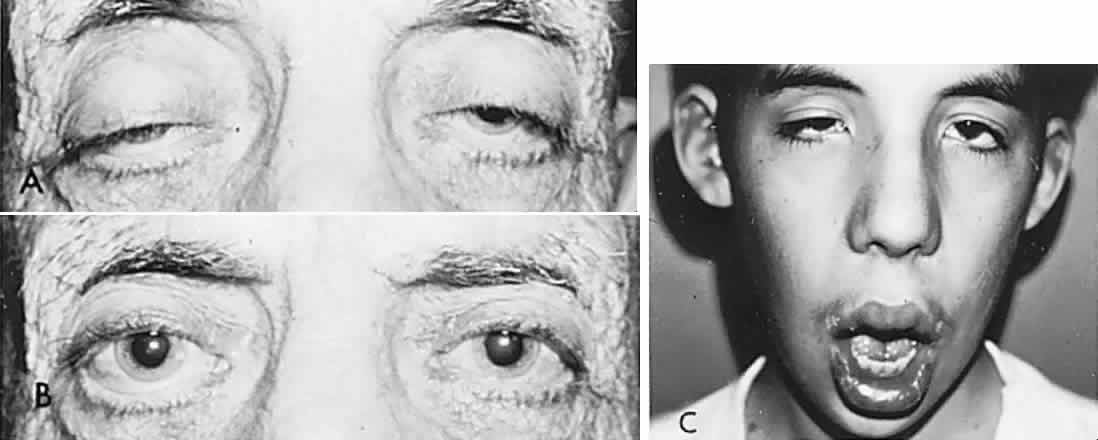
According to Hering's law, yoke muscles receive equal and simultaneous innervation. This phenomenon provides a sensitive mechanism for the detection of subtle muscle pareses. For example, the right lateral rectus and left medial rectus are yoke muscles for right horizontal gaze. If the right lateral rectus is paretic and fusion is consequently broken, the patient must elect to fixate an object with one eye or the other. If the nonparetic left eye fixates, there is an inward deviation (esotropia) of the right eye resulting from paresis of right lateral rectus, as well as relatively unopposed contraction of the medial rectus. This is called the primary deviation (Fig. 4). If, however, the paretic eye fixates, or is forced to fixate (by occluding the intact eye), then additional innervation is required in an attempt to activate the paretic lateral rectus. This excessive innervation determined by the effort of the paretic eye to fixate, according to Hering's law, will also be equally transmitted to the intact left medial rectus, which now “overacts” and excessively adducts the sound eye. This overaction of the nonparetic eye, when the paretic eye is forced to fixate, is termed secondary deviation and is greater than primary deviation.
|
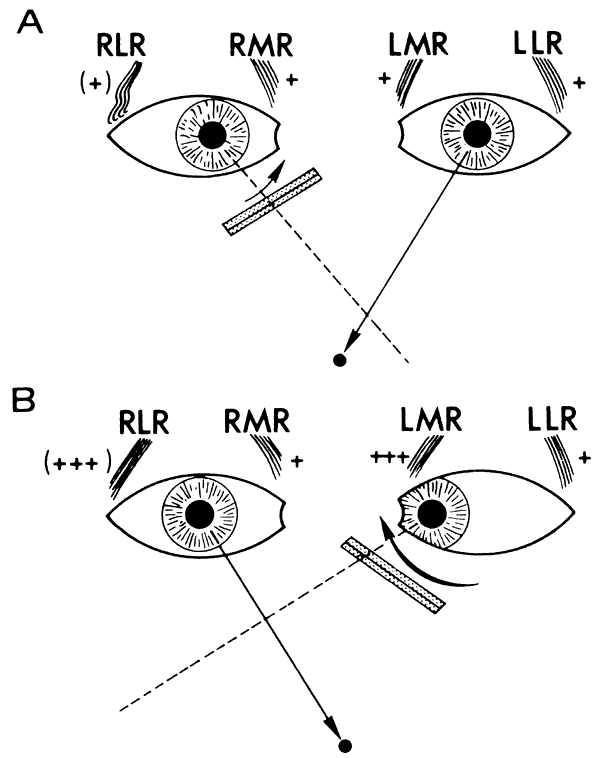
The difference in size of primary and secondary deviation is evident when the alternate cover test is used. This is an excellent objective and reproducible method to analyze paretic ocular muscles, but it requires sufficient visual function for adequate central fixation with each eye. The previous discussion (and see Fig. 4) showed that the occluded eye deviates; for example, the right eye with a lateral rectus paresis deviates inward behind the occluder. If the occluder is now quickly switched to the other eye, the right eye will make an outward movement to “take up fixation,” and this movement is easily observed. Rapid alternation of occluder from one eye to the other (approximately 1 to 2 seconds) provokes an easily discernible and reproducible pattern of fixational eye movements. The alternate cover test may be performed in the six cardinal positions of gaze to discover (1) the field of gaze in which deviation is greatest and (2) in that field of gaze, which eye fixation produces the greater deviation. The difference in the angle of deviation from one field of gaze to the other, and with each eye fixating, may be roughly estimated or accurately measured by prism neutralization.
Diplopia “fields” may be performed by placing a red glass over one eye (by convention, the right) and having the patient indicate the field of gaze of greatest subjective separation of images. This test demonstrates the relative position of the two eyes with respect to one another. With an esotropic deviation (inward turn) the images are uncrossed, that is, the red (right eye) image is toward the right; with an exotropic deviation, the images are crossed, the red image toward the left. The more peripheral-appearing image belongs to the paretic eye. It is helpful if the patient holds up two fingers to demonstrate the relative position of the two images, which should change as the eyes are moved into different gaze directions (Fig. 5). The Maddox rod is a series of parallel cylindrical bars that transform a point source of light into a line perpendicular to the cylinder axes (see Fig. 5). If a red Maddox rod is used, the right eye sees a straight vertical or horizontal red line, which may be compared with the position of a white point source of light seen with the left eye. Torsional deviations, such as those that occur with superior oblique palsies, may be uncovered by subjective appearance of tilting of the Maddox rod image. Special techniques for testing function of the superior oblique are discussed in the section related to fourth nerve palsy (see Volume 2, Chapter 12).
|
Every patient with diplopia, the cause of which is not obvious, and that is not accompanied by pain or involvement of the pupil, should raise the possibility of myasthenia, for which an intravenous edrophonium (Tensilon) test should be considered. Myasthenia gravis can affect the extraocular muscles singly or in any combination, with or without ptosis, and it is most assuredly a commonly missed diagnosis unless the physician keeps the disease in mind and a syringe at hand (see Volume 2, Chapter 12).
The local orbital inflammatory reaction that consistently accompanies dysthyroid states is a common cause of extraocular muscle restriction. The situation of a middle-aged woman with isolated vertical diplopia represents a monotonous clinical picture with which neurologists and ophthalmologists alike should be thoroughly familiar. If, for good measure, there is unilateral or bilateral lid retraction, the diagnosis is made regardless of the absence of other clinical or chemical signs of dysthyroidism (see Volume 2, Chapters 12 and 14).
In order to establish the presence of a restrictive local orbital myopathy, the forced duction test is used. This test determines the presence of mechanical resistance by actually taking hold of the globe and attempting to move it in the direction that the patient cannot (Fig. 6; see Volume 2, Chapter 14). It should be pointed out that in any long-standing ocular deviation, whether due to sixth nerve palsy, ocular myasthenia, or congenital esotropia, there may be secondary changes in muscles with contractures and therefore a “positive forced duction test.” The conclusions of Kommerell and Oliver9 are apparently valid: any paresis of an extraocular muscle ultimately results in a contracture of the ipsilateral antagonist; and secondary changes in the nonparetic eye (contracture of yoke muscle) occur only if the paretic eye fixates continuously.
|
The characteristics of paralytic strabismus, that is, differences in primary and secondary deviation and variation of squint angle with different fields of gaze, are most marked when the paresis is recent. These differences become less marked with time, and the strabismus becomes more symmetric, that is, less incomitant. This phenomenon is the so-called spread of comitance. A sequence of events takes place such that comitance emerges; for example, if the right lateral rectus muscle is paretic, the following occur in some degree: (1) overaction (secondary deviation) of the contralateral yoke, the left medial rectus; (2) contracture of the ipsilateral antagonist, the right medial rectus, such that the angle of the inward turn increases; (3) in the presence of hypoactivity of the antagonist, the right medial rectus, its contralateral yoke muscle, the left lateral rectus, will become apparently paretic, especially if the paretic eye is used for fixation (inhibitional palsy of Chavasse); and (4) therefore, the esotropic deviation increases in the field of gaze of the left lateral rectus, as well as in the field of gaze of the originally paretic right lateral rectus.
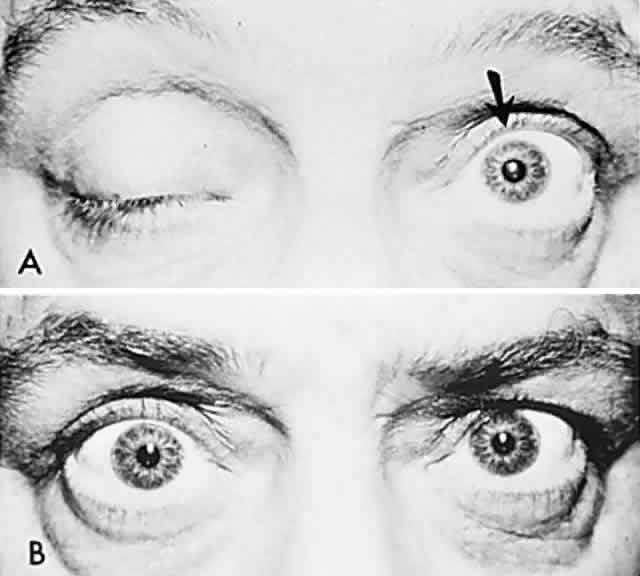
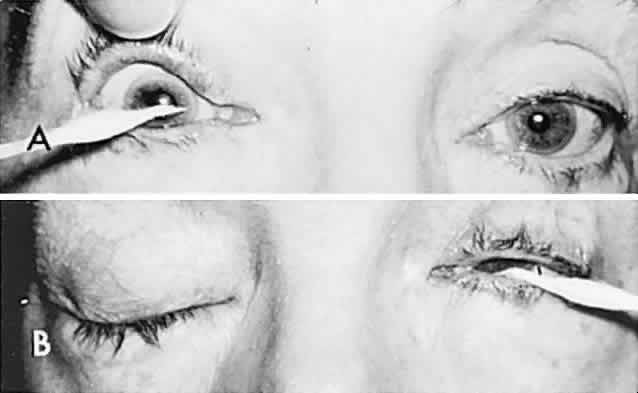
Bell's Phenomenon and Spasticity of Conjugate Gaze
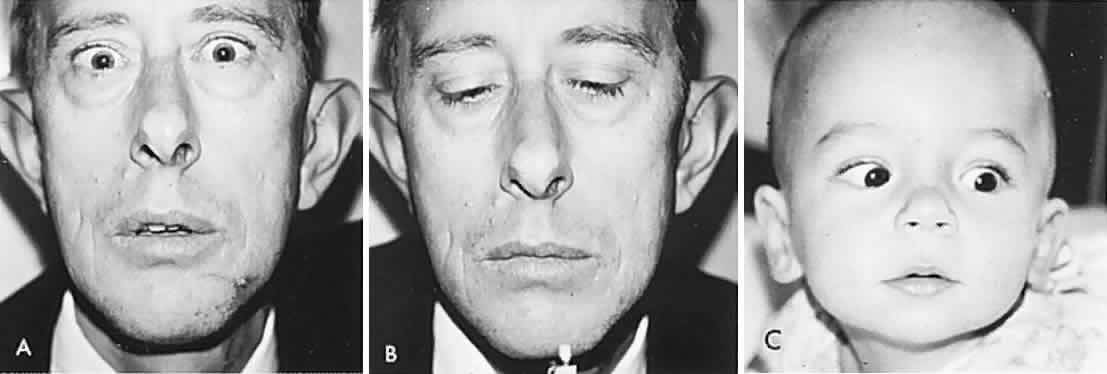
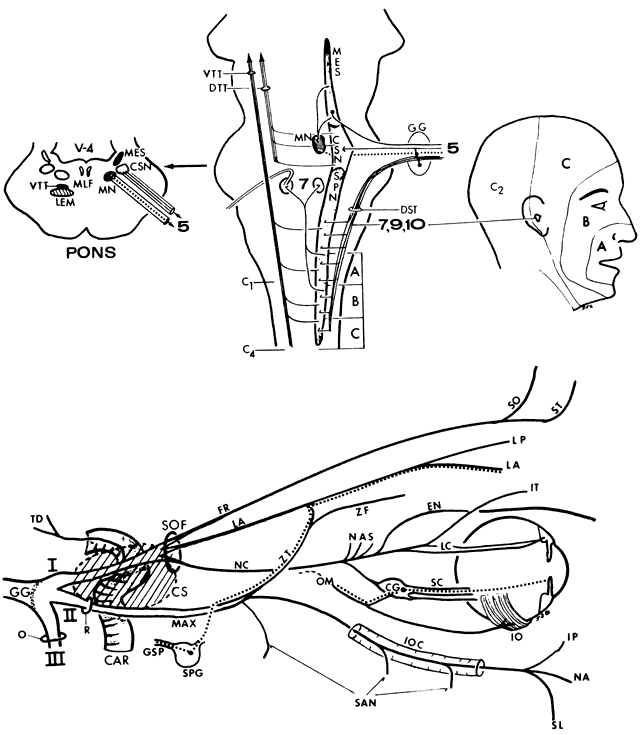
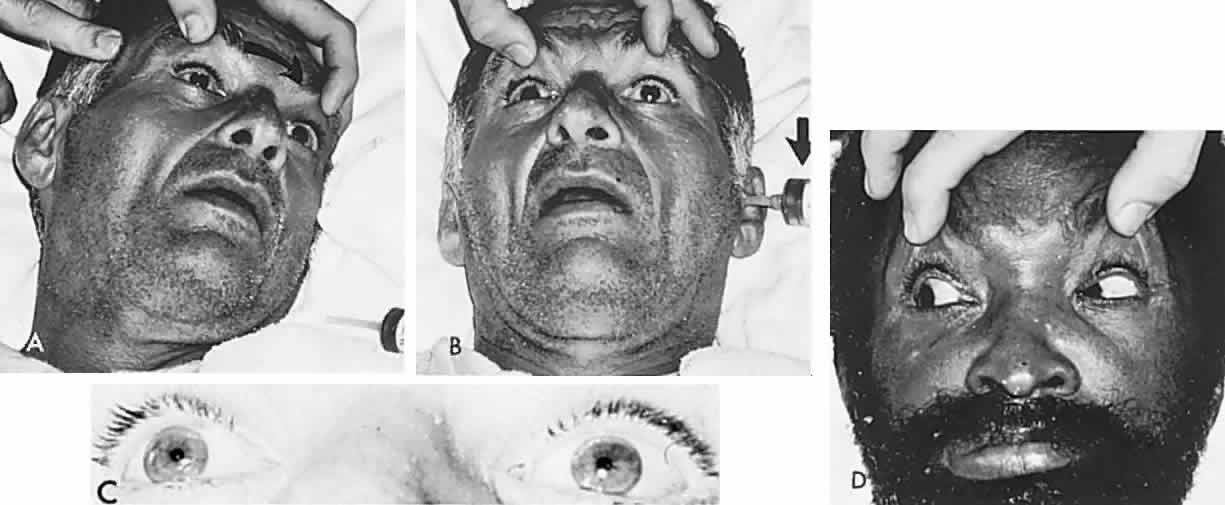
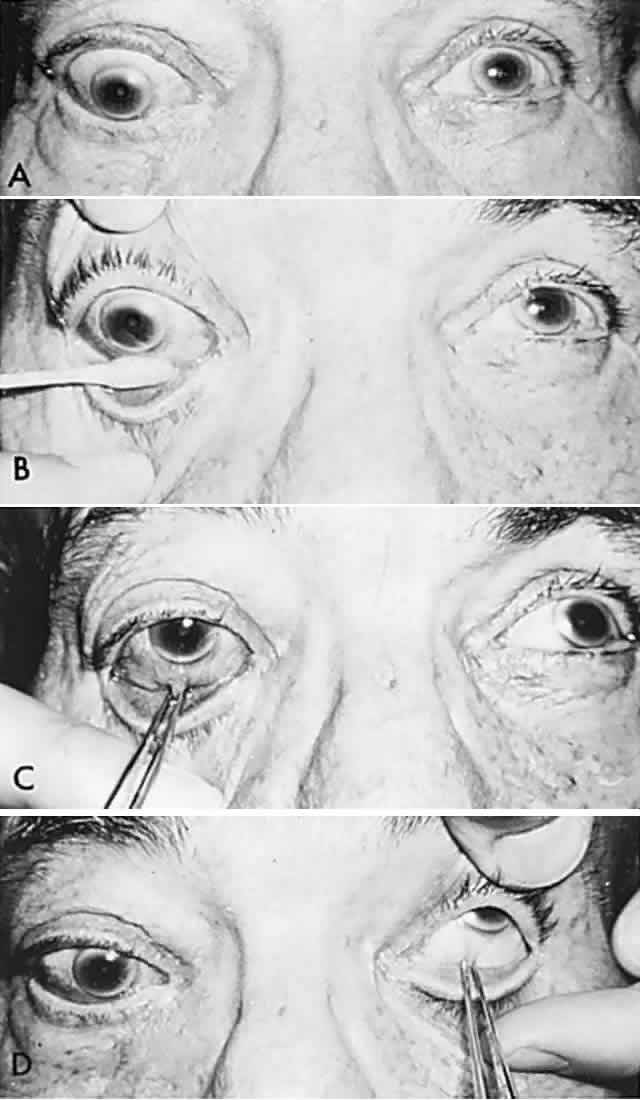
In 1823, Sir Charles Bell reported on the oculogyric phenomenon that accompanies forceful closure of the eyelids, which Bell first noted as an upward deviation of the globe during attempted eyelid closure in the presence of the peripheral facial palsy that now bears his name.10 Bell's phenomenon is an associated movement of eyes and orbicularis oculi, such that the eyes typically roll upward and outward when efforts are made to close the eyelids against resistance (Fig. 7). The exact neural mechanism for this integrated movement is unknown, but it involves brain stem pathways between the seventh nerve nucleus in the pons and the third nerve nuclear complex in the rostral midbrain. In patients who cannot volitionally elevate the eyes, intact Bell's phenomenon (like doll's head deviation upward) indicates that brain stem pathways, the nuclear cell complex for upward gaze, the associated motor neurons, and the extraocular muscles related to eye elevation are functioning, and that the upward-gaze palsy is due to a supranuclear defect.
|

Cogan11 studied the occurrence of abnormal Bell's phenomenon and found that in a series of 156 persons with no known neurologic disease, 132 showed upward or upward and outward deviation of the eyes, that is, the anticipated ocular deviation with forced lid closure. In that presumed normal control group, no deviation from the primary position was observed in 11, convergence occurred in 5, downward deviation was noted in 3, wandering movements occurred in 2, and conjugate lateral deviation was seen in 3. A second group of patients in Cogan's study was composed of 78 patients with unilateral cerebral or infratentorial lesions, with reasonably definite localizing signs. Of the 54 patients with presumed unilateral cerebral disease, 34 showed a lateral conjugate deviation of the eyes to the side opposite the lesion, 5 to the same side, and 15 showed no deviation, with forced lid closure. Twenty-four patients with unilateral lesions of the cerebellum, brain stem, or labyrinth demonstrated no consistent deviation. Eye movements related to simple blinking are rapid, and the eyes actually deviate and return to their original position even before the blink is completed.12
The pathologic reflex of conjugate deviation of the eyes to the side opposite a cerebral lesion, was called “spasticity of the conjugate oculomotor mechanism” by Cogan. The spastic deviation is not dependent on conjugate-gaze paralysis and, in fact, occurs toward the side opposite that which would be anticipated with a cerebral gaze palsy. Smith and colleagues13 emphasized that spasticity of conjugate gaze has lateralizing rather than localizing significance; this phenomenon has its highest correlation with temporo-parietal lesions, is less frequent with occipital disorders, and is not seen with frontal lobe disease. There is apparently no consistent association of spasticity of conjugate gaze with homonymous hemianopia, long-tract signs, or abnormal optokinetic response. As an isolated finding, spasticity of conjugate gaze should bear no diagnostic weight.