CLINICAL EVIDENCE
Retinoblastoma affects approximately one infant in 15,000 to 20,000 live births in the United States each year.1–4 The incidence of retinoblastoma among the various geographic populations is relatively constant indicating that environmental influences play little role in the development of this malignant intraocular tumor. Before the 1860s when the role of enucleation in the management of retinoblastoma became known, most cases of retinoblastoma proved fatal. At that time little was suspected about the inheritance patterns of this tumor because few patients, if any, survived to reproductive age. Later, as more patients survived and had children of their own, more evidence arose suggesting the hereditary nature of retinoblastoma.5 It is now known that retinoblastoma can be inherited as a familial tumor in which the affected child has a positive family history of retinoblastoma or as a nonfamilial (sporadic) tumor in which the family history is negative for retinoblastoma. Approximately 6% of newly diagnosed retinoblastoma cases are familial and 94% are sporadic. All patients with familial retinoblastoma are at risk to pass the predisposition for the development of the tumor to their offspring.
Retinoblastoma is generally classified in three different ways: familial or sporadic, bilateral or unilateral, and heritable or nonheritable. Clinically, the first two classification schemes are fused.6 Thus, a case may be classified as unilateral sporadic, bilateral sporadic, unilateral familial, or bilateral familial. About two thirds of all cases are unilateral and one third are bilateral. Genetically it is simpler to discuss retinoblastoma with the latter classification of heritable or nonheritable. The three classification schemes, however, are interrelated. It is recognized that bilateral and familial retinoblastoma is caused by a germline mutation and is, thus, a heritable tumor. Unilateral retinoblastoma is usually not heritable. Sporadic retinoblastoma is heritable in approximately 25% of cases, especially if it is a sporadic bilateral retinoblastoma.
In 1971, Knudson7 proposed the “two hit” hypothesis to explain the events that are necessary for both heritable and nonheritable retinoblastoma. His theory was based on a comparative analysis of unilateral and bilateral retinoblastoma cases. He proposed that the development of any retinoblastoma was caused by two complementary chromosomal mutations. Each of these genetic events can occur randomly with a frequency of 2 × 10-7 per year. In the case of familial retinoblastoma, the initial event or “hit” is a germinal mutation that is inherited and found in all cells of the offspring. The second hit occurs sometime during development, and if it occurs in a somatic cell such as a retinal cell then retinoblastoma develops. Therefore, in familial cases of retinoblastoma, all cells in the body are predisposed to possible tumor development because germline mutation (“first hit”) has been inherited in all cells of the body, including the ovaries and testes. This may help to explain the high incidence of second nonocular tumors, such as osteosarcoma, seen in patients with familial retinoblastoma or bilateral sporadic retinoblastoma.8–11 The offspring in cases of familial retinoblastoma will likewise be predisposed because their germinal mutation will be passed on. By contrast, in most cases of unilateral sporadic retinoblastoma, the two hits occur during development of the retina and both hits are somatic mutations.12 The rest of the body theoretically carries no higher risk to develop other tumors because these patients presumably have normal chromosomal structure elsewhere in the body.
Knudson's theory provides an explanation for the similarities and differences between heritable and nonheritable retinoblastoma. Ophthalmoscopically and histopathologically, heritable and nonheritable retinoblastoma are indistinguishable.13 The major differences between heritable and nonheritable retinoblastoma are that the heritable tumor usually occurs at a younger age,1 is more likely to be bilateral and multicentric,1 and the affected patient is at higher risk for nonocular tumors than is the nonheritable case.8–11 The rationale for this finding is that the probability for the one hit necessary for tumor formation in heritable cases is 2 × 10-7, whereas the probability for two independent hits to occur as in the nonheritable retinoblastoma case is 2 × 10-7 times 2 × 10-7. It undoubtedly takes longer for the two unlikely events to occur in nonheritable cases, and it is highly unlikely that these two events will occur again at another retinal location, thereby explaining the older age at clinical presentation and the lack of multicentricity in nonheritable retinoblastoma. The reason for the higher likelihood of second nonocular tumors in heritable retinoblastoma is that all of the cells of the body already have inherited a single hit or germinal mutation on chromosome 13, which may predispose them to other cancers found in association with chromosome 13 defects.14 These patients are predisposed to nonocular tumors if the second hit occurs. In nonheritable retinoblastoma, both hits need to occur in a solitary cell and this is statistically less likely.
Excess cancer in relatives of patients with heritable retinoblastoma and advanced paternal age support the findings of the genetic influence in retinoblastoma and other solid childhood tumors.15–17 Excess nonocular cancer in relatives provides strong evidence that the gene that controls retinoblastoma is a generalized cancer gene.15 Fathers of children with bilateral sporadic retinoblastoma are generally older than are fathers of patients with unilateral sporadic cases.16
PHYSICAL PROPERTIES
The first clue as to the location of the retinoblastoma gene was recognized by Stallard18 in 1962 when he reported the case of an infant with retinoblastoma that had a deletion in one of the D-group chromosomes. The D-group chromosomes include numbers 13, 14, and 15. It was difficult to distinguish these three chromosomes without the cytogenetic techniques that evolved in the 1960s. Additional cases of D-group deletion retinoblastoma were recognized, and retinoblastoma became known as a part of the D-deletion syndrome. In the 1970s, high-resolution chromosomal banding showed that the affected chromosome in patients with retinoblastoma was chromosome 13.19 Because of this, the syndrome was renamed the 13-deletion syndrome. In 1978, the locus of the deletion was found to be the q14 band, that is the 14 band on the long arm (q) of the 13th chromosome.
The retinoblastoma locus on chromosome 13 encodes for a large gene of 4.73 kilobase message.20 An intact gene protects against expression of retinoblastoma. It is believed that the gene is a recessive suppressor gene and may play a role in cell growth and development.21 In order for retinoblastoma to develop, both copies of the gene at the 13q14 locus must be lost, deleted, mutated, or inactivated.21 If either the maternal or paternal copy of the gene that is inherited by an individual is defective, then that individual is heterozygous for the mutant allele. Tumor formation requires both alleles of the gene to be mutant or inactive. These two mutations correlate to the two hits theorized by Knudson.7,12
The retinoblastoma gene comprises at least 27 exons over a genomic locus of 200 kilobases. The coding portion of the gene consists of 4.73 kilobases. The retinoblastoma gene has been demonstrated to be missing or rearranged on approximately 40% to 100% of retinoblastoma tumor tissue.14,22,23 Even when no deletion is found, the message from the gene has been found to be altered or absent. In these cases, it is likely that some minute structural changes on the gene are present and are too submicroscopic to be identified with routine karyotyping methods.
Only 5% to 6% of patients with retinoblastoma are found to have a visible deletion in chromosome 13 when studied by peripheral blood sampling.24,25 Approximately 6% of our patients with retinoblastoma have been found to have a chromosomal abnormality involving 13q, and one half were unilateral cases and one half were bilateral cases.24 It is hypothesized that many more patients have chromosomal deletions that are not detected by normal banding chromosomal studies because the deletions are too small for identification. High-resolution banding studies may detect chromosomal mosaicism, that is, the deletion on some but not all cells. More recently, newer techniques of analysis of the deoxyribonucleic acid (DNA) of chromosome 13 has enabled investigators to identify very small deletions that would have been otherwise too small to detect on chromosomal banding.26,27
The increasing cytogenetic technology and the development of DNA probes for loci in the vicinity of the retinoblastoma gene locus (RB1) has allowed detection of subtle genomic rearrangements. The RB1 gene encodes a nuclear phosphoprotein p110RB1 with a molecular weight of 105 to 110 kilodaltons. Evidence suggests that this protein regulates cell proliferation and differentiation. In a review of 192 patients with retinoblastoma studied with DNA analysis, various germline mutations in the RB1 gene were found including nonsense mutation in 43%, frameshift in 35%, intron mutation in 12%, missense mutation in 6%, in-frame deletion in 3%, and promoter mutation in 2%.28 Mutations were distributed throughout 24 of the 27 exons of the RB1 gene with no singe hotspot of mutation. Based on this knowledge, the spectrum of germline RB1 mutations in patients with retinoblastoma is broad. Large chromosomal alterations are detected by cytogenetic analysis in 7% to 8%, and smaller DNA rearrangements are identified by DNA fragment analysis in 16% of patients.28 Despite the sophistication of current techniques, approximately 20% of patients with bilateral retinoblastoma have an existing mutation that is not found.28 It is suggested that a series of complementary tests to rapidly detect mutations is most beneficial for patients with retinoblastoma.
13Q14 SYNDROME
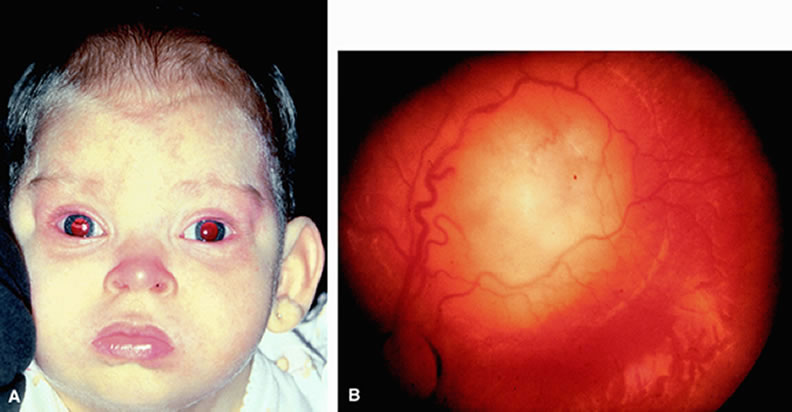
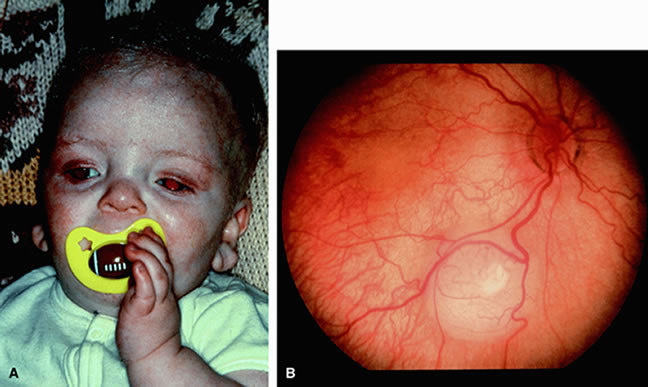
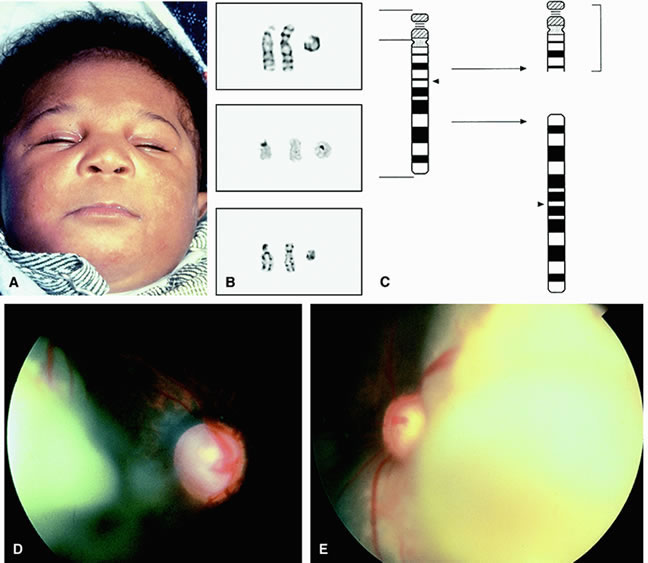
The 13q deletion syndrome may manifested by several phenotypic abnormalities. Many patients have minimal or no visible abnormality.29 The characteristic findings include some degree of the following dysmorphic features: microcephaly, broad prominent nasal bridge, hypertelorism, microphthalmos, epicanthus, ptosis, protruding upper incisors, micrognathia, short neck with lateral folds, large prominent low set ears, facial asymmetry, imperforate anus, genital malformations, perineal fistula, hypoplastic or absent thumbs, toe abnormalities, and psychomotor and mental retardation.30–32 The midface of patients with 13q deletion are notable for prominent eyebrows, broad nasal bridge, bulbous tipped nose, large mouth, and thin upper lip32–34 (Figs. 1, 2, 3, and 4). We recently reported a case of severe midline facial and central nervous system abnormalities in a child with 13q abnormalities that manifested retinoblastoma and holoprosencephaly.35
|
|
|
|
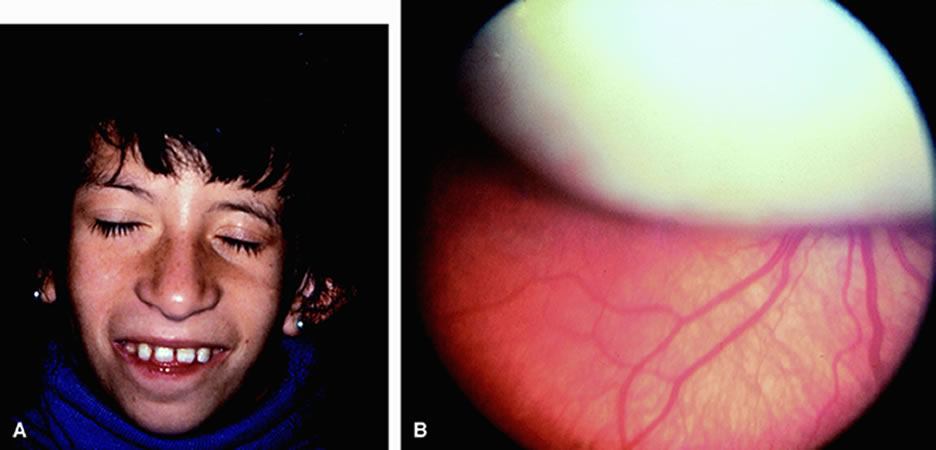
Karyotype analysis of children with these or other dysmorphic features may allow earlier detection of retinoblastoma. We have seen several cases of retinoblastoma that were initially suspected based on the recognition of the previously described dysmorphic features that prompted a karyotype analysis that revealed a deletion in chromosome 13. This finding subsequently prompted a retinal examination, which revealed unilateral multifocal tumors in both cases.32
LINKAGE MARKERS
Before the identification of the retinoblastoma gene, linked markers such as esterase D were used to identify individuals who had the heritable form of retinoblastoma. Esterase D is an enzyme expressed in all cells that is coded on chromosome 13. If both of its alleles are fully active, then the enzyme is measured as 100% activity. If one allele is missing, then the activity drops to 50%. Esterase D has been found to be closely linked to the retinoblastoma gene on the 13th chromosome. In fact it has been found to lay in the proximal portion of the 13q14 locus.36 Because of this tight association with retinoblastoma, screening for the retinoblastoma gene by testing for esterase D levels has been used in the past but is rarely used today. If esterase D is 50% or less than its normal activity, then it suggests that a chromosome 13 deletion and possible retinoblastoma gene deletion is present.