1913: Pagenstecher noted the inheritance of “solutio retinae” in
hypermetropic children.25
1932: Anderson localized the “anterior dialysis” to the inferotemporal
quadrant in nonmyopic young males.26
Thomson emphasized macular involvement.27
1938: Mann and MacRae theorized that the “congenital vascular veils” in
the vitreous are the result of retinal splitting.28
1948: Juler described retinal detachment in children with thinning and
ruptures of the inner linings of the cysts.29 It was then assumed that the pathology is retinal but involves secondary
changes in the vitreous.
1950: Sorsby and colleagues recognized the sex-linked inheritance of retinoschisis.1 Combining this finding with the previous views, they postulated that the
veils represent ruptured retinal cysts, which, at a later stage, probably
progress to congenital falciform detachment and pseudoglioma.
1953: Jager coined the term “retinal schisis.”30
1953/4: Kleinert31 and MacRae32 agreed with the concept of central and peripheral splitting of the retina.
1960/1: Balian and Falls33 and Gieser and Falls34 emphasized that the congenital vascular veils in the vitreous are combined
with macular changes.
1964/6: Sarin and associates35 and Sabates36 identified members of the same families who had “hereditary retinal
schisis.”
1973: Forsius and colleagues confirmed the X-linkage of retinal schisis
and found that the maculopathy is present in virtually all cases.37
1976: Harris and Yeung classified retinoschisis into stages based on its
degree of severity with respect to the occurrence of striation and atrophic
and pigmentary changes.38
1983: Wieacker and coworkers assigned the gene to band Xp 22.2 of the human
X chromosome by linkage analysis.13
X-linked retinoschisis1 can be called a retinovitreal dystrophy because retinal schisis, not vitreous syneresis, is the predominant sign. The common denominator of the retinal changes is thought to be Mueller cell degeneration,39 the sequelae of which are most visible in the fovea and in the periphery of the inferotemporal fundus.40 Peripheral changes only may be present in the rare autosomal dominant form.41 The wide range of biomicroscopic findings changes with the duration of the condition and age of the patient.1 The posterior vitreous adhesion (that is, premacular and prepapillary, as well as perivascular) is well developed in the young.
Vitreous attachments to the inner layer of the schisis can cause progression of schisis through traction, particularly if vitreous contraction is accelerated by vitreous hemorrhage. Vitreous traction may lead to nasal dragging of the retina.42 The inner schisis layer consists of glia, nerve fiber layer, and vessels.40 Ultrastructural studies confirm the split along the ganglion cell and nerve fiber layer separating cell processes from their bodies in the inner nuclear layer and resulting in a picture of “decapitated and degenerated Mueller cells.”43 Whereas the inner layer of the schisis consists mostly of glial cells, the outer layer also provides a glial lining of the schisis cavity.43 Because vascularization of the inner retinal layers extends to the middle limiting membrane,44 schisis, however, takes place internal to the inner nuclear layer; the deep retinal capillaries may be disrupted, appearing angiographically as areas of nonperfusion.45 Inner layer atrophy may also ensue as a result of chronic inner layer ischemia and glial cell decapitation. Atrophic holes in the inner layer and perivascular gliosis may be the biomicroscopic corollaries. The outer layer may undergo atrophy or may be invaded by proliferations of the pigment epithelium.39,40
Retinal schisis involves a delicate balance between vitreous traction on the inner layer and the opposing strength of residual intraretinal bridges of tissue and adhesions of the outer layer of schisis to the pigment epithelium. Intraretinal strands of tissue that bridge the schisis cavity have been shown in vivo using the retinal thickness analyzer.46 If the vitreous gel is collapsed, the inner wall of the schisis is found in the retrolental space.33,40 Not surprisingly, such bullous schisis is more common in the young (i.e., in children younger than 10 years),47–49 often accompanied by vitreous and/or intraschisis hemorrhage.47 Visual loss in the young patient is often associated with contraction of adherent vitreous, bullous schisis, and its complications. It is a dynamic process; therefore, with increasing age bullous schisis tends to flatten spontaneously in the vast majority of cases.47,49 Application of a laser beam to the area of schisis has been shown to disrupt the equilibrium of vitreous traction and retinal adhesions and to produce rhegmatogenous detachments resulting from outer layer breaks as well as from full-thickness retinal breaks.50
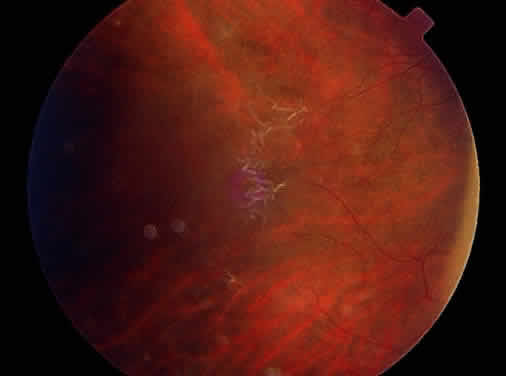
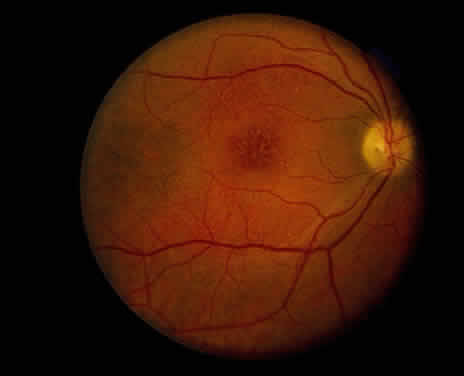
Clinically, retinal schisis presents with decreased central visual acuity, absolute scotomas, or both.1,33,38 Children may have strabismus and nystagmus.25,47 As in FEVR, but in contrast to Wagner and Stickler syndrome, the refraction tends to be hypermetropic (“hypermetropic amblyopia”).1,25,26,31–34,40,48 On biomicroscopic examination, edema and cystic central macular schisis of the retina are apparent in virtually all cases.37,38 Loss of macular reflex, which may be an early sign,34,35 is followed by wheel-like formation,37 a multicystic radiate appearance (Fig. 1),38 pigmentary degeneration, and cystic macular atrophy (Fig. 2).38 These macular changes, not bullous schisis per se, determine visual loss in patients often in the fourth or fifth decade of life.51
|
|
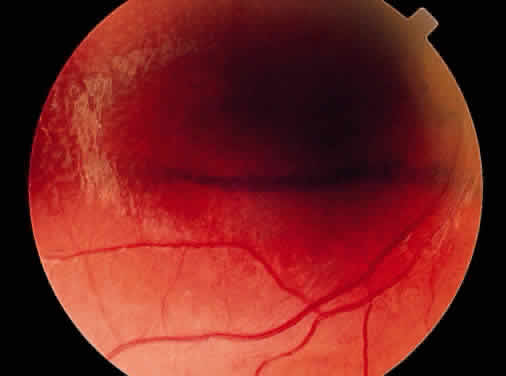
Peripheral retinal schisis is present in half of the patients and in 96% is located in the inferotemporal quadrant.48 At least initially, the inner layer of the schisis is attached to the vitreous and features vessels, internal limiting membrane, and nerve fiber layer. These tissues undergo progressive atrophy as well as reactive gliosis, which in a perivascular location may appear as vascular sheathing (Fig. 3). Total atrophy may result in dehiscences and peripheral arborization of the retinal vasculature.33,34,45 Optic atrophy may follow.40 The outer layer of the schisis cavity also undergoes atrophy, reactive gliosis, intralayer pigment migration, and hole formation. A rhegmatogenous detachment may be the sequela in 4% to 22% of all patients.30,42,49,51–53 With time, the vitreous undergoes syneresis and detachment.31,33,40 Vitreous detachment actually may improve the schisis by releasing traction. Adhesions to retinal vessels are responsible for frequently found vitreous hemorrhages.29,31,40
|
Because progression of the disease cannot be predicted and regression sometimes occurs, patients should be observed regularly, particularly those younger than 10 years of age.47 Indications for surgical intervention are rhegmatogenous retinal detachment and repeated vitreous hemorrhages.
Photocoagulation has not been shown to be beneficial in that it creates external wall holes.50 Such prophylactic treatment has led to retinal detachment in 14% to 43% of cases.48,49 This failure may be related to the fact that most of the mechanical cohesiveness of the retina lies in the inner layer near its basement membrane. The usual energy levels for coagulation may be too intense for the thin outer layer.
Similarly, scleral buckling has been shown to be marginally effective in treating the detachments. Redetachments after buckling were reported in up to 40%.48,53 In cases of vitreous hemorrhage and vitreous attachment to the inner layer of schisis, scleral buckling combined with pars plana vitrectomy techniques may be beneficial. Recent approaches to surgery have centered on removal of the inner layer of the schisis cavity,52,53 if possible by lens-sparing technique.54 The position of the inner layer is determined by vitreous forces in its degree of separation and topography (dragging). In many cases it may be impossible to separate the cortical vitreous from the inner layer of schisis. Inner layer retinectomy most definitely eliminates traction that might lead to redetachment and may account for the superior long-term results reported with this method.54