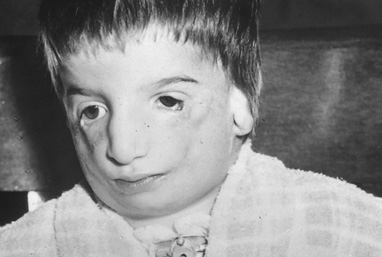
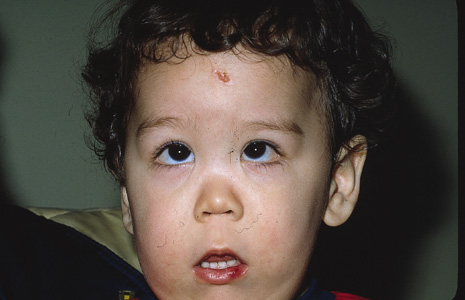
MARFAN SYNDROME
A description of what we now call Marfan syndrome was probably first made by Williams, an ophthalmologist, in 1895, who did not include ectopia lentis as a manifestation. Ironically, it was a pediatrician who first published that association in 1914. Marfan's description of a 5½-year-old child with long, thin extremities, published in 1896, was more widely seen than Williams' the year before, and hence this syndrome now bears his name.
Marfan syndrome is an autosomal dominant condition with complete penetrance and a high degree of expressivity. It has an incidence of 1:66,000 with a gender ratio of unity.1 Advanced paternal age is associated with the sporadic cases.2
The long arm of chromosome 15 has been identified as harboring the genetic defect for this disorder,3 which results from a point mutation in one of the fibrillin genes.4 Fibrillin is an important component of connective tissue microfibrils that form the framework for elastin deposition and are found in lens zonules, the aorta, and other connective tissue structures. The abnormal fibrillin leads to structural weakness resulting in ectopia lentis, aortic dissection, and other disorders and has been shown to cause syndactyly in mice.5 Recent work has suggested the possible role of matrix metalloproteinases (proteolytic enzymes) as well.6
Marfan syndrome's nonocular systemic manifestations are found in the skeletal and cardiovascular systems.
Cardiovascular abnormalities are the more significant systemic manifestations. In more than 95% of the cases in which a cause of death can be established, a cardiovascular problem is at fault.7 Dilatation of the aortic root (with or without aortic regurgitation), mitral valve prolapse, and mitral regurgitation are all seen. When aortic root dilatation is demonstrated, therapy with a β-blocker has been suggested to reduce cardiac contractility in an attempt to halt progression of the aortic root dilatation and prevent aortic dissection.8 More definitive work with β-blockers is still needed.
Abnormalities of the skeletal system include scoliosis, arachnodactyly, anterior chest deformities, increased arm span to total height ratio, joint laxity, and a high arched palate. Scoliosis is the most disabling skeletal abnormality. Attempts have been made in female adolescent patients to alleviate this problem with estrogen therapy. These hormones can suppress the adolescent growth spurt that greatly aggravates the scoliosis.
Ocular Manifestations
Of the many ocular abnormalities, by far the most common is ectopia lentis, occurring in 50% to 80% of affected individuals (Fig. 1). It is usually bilateral, symmetric, and nonprogressive. It may be subtle and detectable only by observing phacodonesis or iridodonesis, sometimes visible by gonioscopy. Lens dislocation is usually superotemporal. There can also be posterior displacement, leaving a gap between lens and iris pigment epithelium. When zonules can be seen, a reduced number is usually not observed.
|

Patients with Marfan syndrome may have poor vision. This often results from delayed and inadequate correction of the extremes of refractive error9 (especially moderate to high myopia). These and other factors, which compromise visual input, may be responsible for the increased incidence of strabismus seen in patients with Marfan syndrome.10
In Marfan syndrome, an increased axial length may also be seen. A slightly higher than normal axial length is common in patients with Marfan syndrome who have no ectopia lentis or retinal detachment. However, the axial length is higher in those patients with ectopia lentis and is even higher in those patients with retinal detachment.11 Other retinal associations include lattice degeneration and atrophic holes.
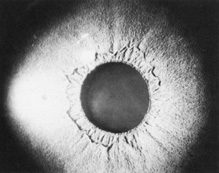
Abnormalities may also be seen in the uveal tract. Choroidal thinning has been described in myopic eyes. The iris morphology is often striking. There can be a complete absence or marked reduction in the number of furrows or crypts, giving the anterior iris surface a smooth, velvety appearance (Fig. 2). Hypopigmentation of the iris pigment epithelium has been described12,13 which probably corresponds to the transillumination of the iris base that is seen in 10% of cases.
|
The cornea can also be affected. Patients with Marfan syndrome can have flatter than average keratometric readings.11,14 Megalocornea15 may also be present. In eyes with ectopia lentis (with or without retinal detachment), keratometric readings may show steepening in the meridian of the lens dislocation.
Other, nonspecific findings have been described in the few ocular histologic studies that have been performed in Marfan syndrome. Separation of individual zonular fibers into their composite filaments9 and angle anomalies (such as pectinate ligaments) have been reported.12,13,16 Common to several pathologic studies is the large size of the globe corresponding to the larger axial lengths measured clinically.
THE PHACOMATOSES
The phacomatoses are a group of heredofamilial conditions that all have as a hallmark various hamartomas. The term comes from the Greek term for birthmark (“mother spot”). Hereditary hamartomatosis has been suggested as a better name for these diseases.17 We only consider the autosomal dominant disorders in this chapter (neurofibromatosis, tuberous sclerosis, and angiomatosis retinae). Despite the lack of hard evidence for autosomal dominant transmission, encephalotrigeminal angiomatosis (Sturge-Weber syndrome) is also discussed for completeness.
Neurofibromatosis
Manifestations of this condition had been observed for ages before being described by Robert William Smith in 1849.18 The more classic description was given by the German pathologist, Friedrich Daniel von Recklinghausen, who accurately described the diverse findings as a single entity in 188219; it is often referred to as von Recklinghausen's disease. Probably because of the complex effects on these patients' personality as a result of the sometimes severe disfigurement, neurofibromatosis maintains a high profile in the lay press20–24 and literature (e.g., The Elephant Man, a 1979 Broadway production written by Bernard Pomerance, and a 1980 British motion picture, although the correct diagnosis for the patient portrayed in those works was probably Proteus syndrome25).
Neurofibromatosis is a proven autosomal dominant condition with variable penetrance and a high degree of expressivity. It is estimated to occur once per 3,300 live births.26 Children of affected mothers are more severely affected than children of affected fathers; no clear mechanism is known for this maternal effect.
Recent work has allowed the division of this disorder into two major autosomal dominant conditions. Neurofibromatosis 1 (a defect on chromosome 17)27 now refers to the much more common, classic syndrome with the wide-ranging systemic and ocular manifestations described below. Neurofibromatosis 2 results from an abnormality on chromosome 22.28,29 This less common variant denotes the syndrome of bilateral acoustic neuromas that may also include neurofibromas and café-au-lait spots.
Although clinical findings are primarily neurocutaneous in nature, any organ system can be involved. The diagnosis requires six or more café-au-lait spots, each larger than 1.5 cm in diameter (Fig. 3). Axillary freckling is also highly suggestive of the diagnosis.30,31(pp508–509) Areas of hypopigmentation or hyperpigmentation can also be seen.
|

The cutaneous tumors, which are common and can be extremely disfiguring, take one of three forms. A fibroma molluscum is usually referred to as the common neurofibroma. This hamartoma is the proliferation of peripheral nerve elements at the distal end of a cutaneous nerve. A plexiform neurofibroma (Fig. 4) has the appearance of a “bag of worms” and represents a diffuse proliferation of tissue within the nerve sheath. The most marked cutaneous change results from diffuse proliferation outside the sheath, elephantiasis neuromatosa. When a hamartoma develops in a restricted space, a compromise in that nerve's function can occur.
|
Neurofibromas may occur anywhere in the central nervous system. They can produce a neurologic deficit corresponding to the affected nerve. Although these neurofibromas are generally benign, malignant change has occurred.32(p1) Meningiomas can also be seen with a higher incidence than the general population, especially involving the orbital portion of the optic nerve.33 When they occur in this location, they are more aggressive than their benign counterparts found in other locations.
Pheochromocytoma has been noted more frequently in patients with neurofibromatosis than in the general population, although it may be difficult to distinguish the clinical features of neurofibromatosis from multiple endocrine neoplasia (MEN) type IIb, in which pheochromocytomas also occur along with neuromas, café-au-lait spots, and prominent corneal nerves.34(p31–32) Other tumors, such as rhabdomyosarcoma and liposarcoma, have rarely been reported with this disorder which may represent an unrelated association.32(p1)
Skeletal deformities, unrelated to the neural tumors, may also occur. Scapular elevation, asymmetries or absences of long bones, misshapen sphenoid or absence of its greater wing, scoliosis, spina bifida, rib fusion, and many others have all been described (Fig. 5).26,31(pp508–509),32(p1)
|
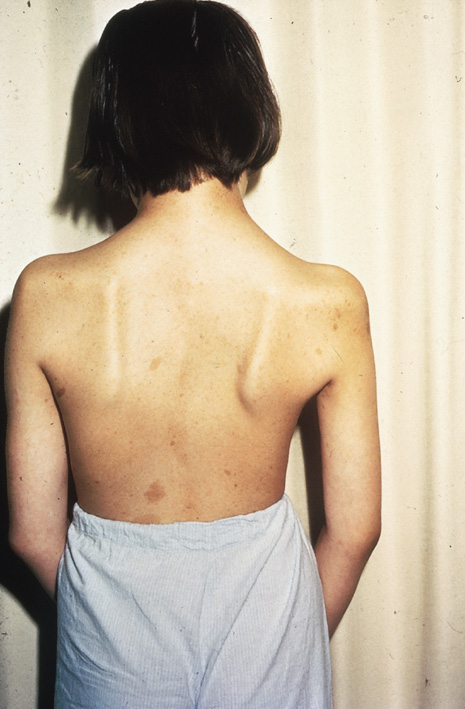
Individuals affected with neurofibromatosis 2 often have bilateral acoustic neuromas, which are Schwann cell tumors arising from the vestibular nerves, producing hearing loss or unsteadiness of gait in the second or third decade of life.28 Café-au-lait spots, cutaneous neurofibromas, and plexiform neurofibromas may also be seen in these patients and may even predate the acoustic nerve symptoms.
OCULAR MANIFESTATIONS.
Ocular involvement in neurofibromatosis involves the cornea, uvea, trabecular meshwork, optic nerve, and retina. Corneal nerves can be thickened; the nerves in the conjunctiva may be thickened as well. Along with an increased incidence of pheochromocytoma, corneal nerve hypertrophy is also seen in multiple endocrine neoplasia. The significance of this commonality is as yet unknown.
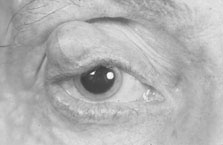
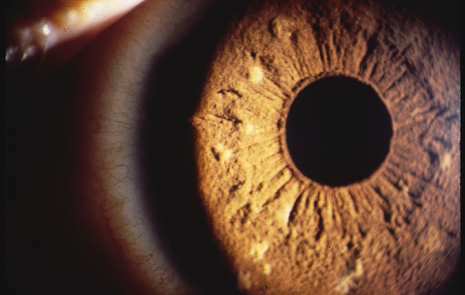
Hamartomas of the iris (melanocytic nevi) can be seen and are called Lisch nodules (Fig. 6). They are variable in size and have a smooth, domeshaped configuration.35 One series35 found these nodules in 92% of their affected population above the age of 6 years; this may mean that their absence prior to that age does not rule out their later occurrence. Hamartomas may also be seen in the trabecular meshwork.34(p30) In a more recent series, the incidence of Lisch nodules was 100% of patients with neurofibromatosis beyond the second decade of life.36
|
Congenital glaucoma may occur in neurofibromatosis. If the upper lid is affected by a neurofibroma, then the eye on that side has a 50% chance of having glaucoma.34(p30)
The retina and optic nerve head may also be involved with hamartomas; in contradistinction to the uveal hamartomas, which are usually melanocytic nevi, the retinal and optic nerve head hamartomas are generally of glial elements.
Posterior to the papilla, the optic nerve may be involved by a glioma, which is different from the acquired gliomas of adulthood. The optic nerve glioma of neurofibromatosis is a true hamartoma, more accurately described34(p30) as a juvenile pilocytic astrocytoma. They are generally benign, but malignant degeneration has been known to occur following radiotherapy.37 Gliomata of the anterior visual pathway (nerve and chiasm) may occur in 15% of patients with neurofibromatosis using computed tomography (CT) scan criteria.38 In all patients with optic nerve gliomata, the incidence of neurofibromatosis is 25%. The glioma may produce proptosis and visual loss.39
The hamartomas may also be found in the nonocular neural elements of the orbit, most commonly as plexiform neurofibroma or neurilemmoma, possibly causing proptosis. Also, absence of the greater wing of the sphenoid may occur, resulting in pulsatile exophthalmos.
Ocular involvement in neurofibromatosis 2 is much less frequent and extensive than in type 1. Approximately half of affected patients have cortical or posterior subcapsular opacities.40 Lisch nodules, which can be diagnostic of neurofibromatosis 1 when multiple, are rarely seen in neurofibromatosis 2.36
TUBEROUS SCLEROSIS
This disorder is occasionally referred to as Bourneville's disease, after Desiré Maglorie Bourneville, the French physician who, in 1880, described the triad of seizures, mental retardation, and cutaneous changes.41 The multiple “potatolike” tumors found in the brain gave rise to the term tuberous sclerosis, as named by Bourneville.
The reported incidence varies from 1 to 5 to 7 per 100,000.42,43 Similar to other autosomal dominant conditions, there is no gender predilection. Most cases appear to be new mutations; this occurrence is thought to be as high as 86% by some, although McKusick2(p1834) casts doubt upon such a high figure. A high paternal age is not associated with these new mutations. Penetrance is low and expressivity is variable; tumors have been reported in many organs and locations44–46 and can be caused by different mutations.47
Systemic Manifestations
The major systemic manifestations occur in the central nervous system, although other organ systems are involved as well. Seizures (motor, focal, petit mal, and psychomotor) occur in 93% of affected individuals. Mental retardation occurs in 62% of all patients; virtually all patients with mental retardation also have seizures.
Multiple intracranial hamartomas usually occur only in patients with seizures. They form in the cerebrum, basal ganglia, and periventricular gray matter and can protrude into the ventricle. They are detectable by plain skull roentgenography.
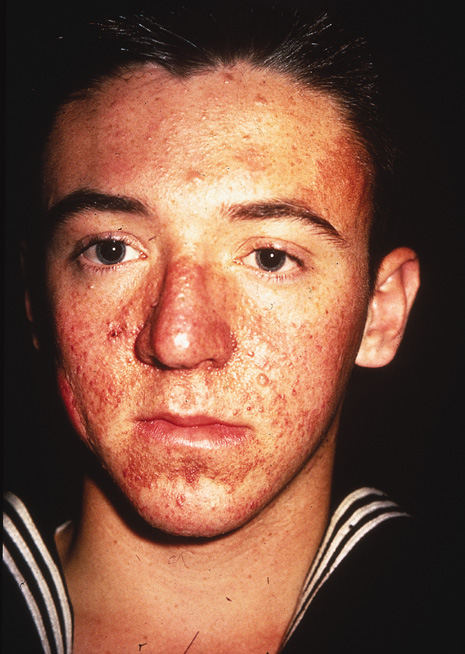
The most widely recognized cutaneous manifestation is adenoma sebaceum, found in a butterfly distribution of the face. It occurs in 83% of affected individuals (Fig. 7). The term adenoma sebaceum is a misnomer because the lesion is an angiofibroma, not an adenoma, and has nothing to do with sebaceous glands. These small dermal tumors appear at approximately 2 years of age and resemble multiple papules or nodules.
|
The ash-leaf lesion is another characteristic cutaneous sign on the trunk and limbs. This lesion can be seen in 86% of patients and may be present at birth; some feel these to be pathognomonic of Bourneville's disease. Appreciating these small, ovoid patches is much easier under a Wood's lamp. Histologically, these lesions contain melanocytes without melanin; hence their depigmented appearance.
Shagreen patches are another highly characteristic cutaneous lesion. They are usually seen on the back as a slightly raised, corrugated area, resembling a pigskin or orange peel. A less frequently seen but characteristic skin lesion is the subungual or periungual fibroma. There are other nonspecific signs involving the skin and hair that include café-au-lait spots, poliosis, and subcutaneous nodules.
Ocular Manifestations
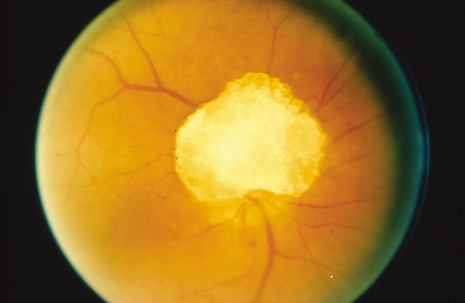
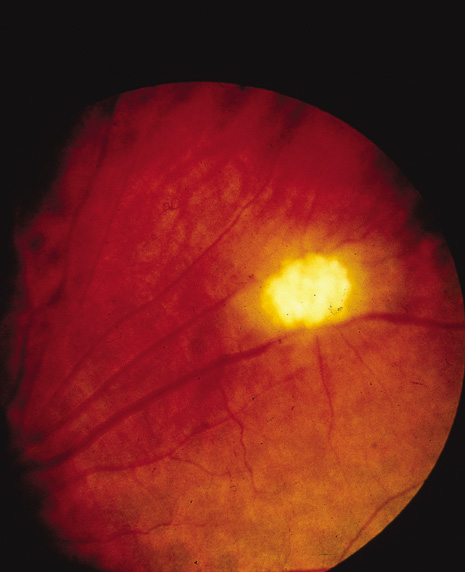
The eyes are involved in approximately half of affected individuals.48 Giant drusen (glial or astrocytic hamartoma) of the optic nerve are common and have a multilobulated appearance resembling a mulberry, tapioca grains, or fish eggs (Fig. 8). When visual symptoms are produced, it is usually in the form of a field defect. The lesion may also bleed resulting in a vitreous hemorrhage. Giant drusen are also in the differential diagnosis of pseudopapilledema.
|
Glial hamartomas of the retinal nerve fiber layer may also occur (Fig. 9). Similar to their counterparts on the optic nerve and in the central nervous system, they can calcify and hence be identified by plain radiography.
|
VON HIPPEL-LINDAU DISEASE
Retinal angiomas as a distinct entity was defined by the German ophthalmologist Eugen von Hippel in 1904.49 The association of angiomatous retinas and cerebellar capillary hemangiomas was made in 1926 by the Swedish neurologist, Arvid Lindau.50 Hamartomas in these two areas remain the hallmarks of von Hippel-Lindau disease.
Because von Hippel-Lindau disease is a rare disorder, prevalence and occurrence data are unavailable. Approximately one-fifth of cases show an autosomal dominant pattern with incomplete penetrance.43(p1) The balance of the cases are sporadic. The major systemic manifestation is cerebellar hemangioblastoma (capillary hemangioma), which usually becomes manifest in the fourth decade of life. It occurs in 20% of patients with retinal tumors.50 Multiple hemangioblastomas are seen in 10% of cases. Cerebellar hemangioblastoma may cause increased intracranial pressure or neurologic symptoms specific for the cerebellum.
The hamartomas may be found in the spinal cord, usually involving the lower cervical and thoracic areas. They may cause a spastic paraparesis, which can progress to a full-fledged spinal cord compression syndrome. Tumors have also been reported in the medulla.
Tumors and cysts of other internal organs also occur. An increased incidence of pheochromocytoma in these patients is seen; the associated hypertension in the presence of a vascular brain tumor may lead to a subarachnoid hemorrhage. Hypertension may also be caused by the cerebellar tumors; in these cases, urinary catecholamines are normal.2(p1889)
Kidney tumors or cysts may also occur in tuberous sclerosis. The renal tumors can produce a hematopoietic substance resulting in polycythemia. However, 10% to 20% of patients with cerebellar hemangioblastoma may have polycythemia without a renal tumor.32(p3) The pathogenesis of the polycythemia without a renal tumor is unknown. Metastatic renal cancer has also been reported.2(p1889)
Papillary cystadenoma of the epididymis can occur bilaterally in von Hippel-Lindau disease; the bilateral occurrence of this rare tumor is very suggestive of von Hippel-Lindau disease. This tumor can occur in the broad ligament of the uterus in affected women.
Cystic and other benign lesions have also been reported in the pancreas, liver, spleen, adrenals, bones, and omentum.
Ocular Manifestations
The only primary ocular manifestation of this condition is the retinal hemangioma, originally described by von Hippel. The hemangioma is composed of endothelial cells and pericytes. The tumors can be found anywhere in the retina but are usually located in the midperiphery. These vascular masses often leak, producing hemorrhages or exudates; it is from these secondary effects that patients complain of visual symptoms, usually by their mid-twenties or early thirties. The exudation can be quite severe, producing an exudative retinal detachment. Organization of the retinal detachment can lead to anterior chamber shallowing and secondary glaucoma.
STURGE-WEBER SYNDROME (ENCEPHALOTRIGEMINAL ANGIOMATOSIS)
As with many eponymic syndromes, those whose names are primarily associated with the disorder were not the first to describe it. Schirmer first described the association of buphthalmos and facial angioma in 1860.51 In 1879, Sturge added seizures to this association.52 A distinct clinical entity was further defined and established by Weber in 1929, when he added cerebral hemangiomas and contralateral hemiplegia to the syndrome.53 Reliable prevalence and occurrence data are unavailable. There does not appear to be a clear sex or ethnic predilection.
Firm evidence for autosomal dominant transmission, as in repeated observation of affected kindreds, is lacking. One report of an affected father and son appeared in 1979.2(p1719)
Systemic Manifestations
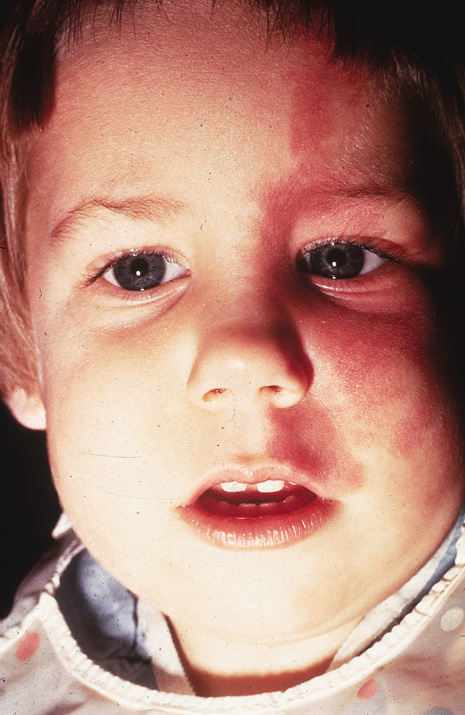
Similar to other phacomatoses, this disorder is primarily neurocutaneous in its systemic manifestations. The hallmark of Sturge-Weber syndrome is the nevus flammeus or port-wine stain (or birthmark) which is found in the first, first and second, or first, second and third divisions of the trigeminal nerve (Fig. 10). It is most often unilateral and respects the midline. It is present at birth and sometimes darkens with age. Histologically, the nevus flammeus is a cavernous hemangioma. Vascular lesions may also occur in the oral mucosa and lips.
|
Nonspecific central nervous system manifestations occur in a high proportion of affected individuals. Grand mal or focal motor seizures can occur in 80% and mental retardation in 60% of patients.32(p5)
It is unclear whether these findings are directly related to the meningeal cavernous hemangiomas which affect many of these patients. The meningeal hemangiomas are usually ipsilateral to the facial nevus flammeus and may cause hemiparesis in 56% of affected individuals.31(pp495–497)
Intracranial calcifications of an odd configuration (convoluted, gyriform, railroad track sign) can be seen on plain radiographs beginning in the second year of life. They are usually underlying the vascular hamartomas and may result from either cortical hypoxia produced by the tumors or calcium binding of transudated proteins.
Occurring less frequently than the neurocutaneous manifestations, the vascular hamartomas have been found in lung, pituitary, pancreas, ovaries, and gastrointestinal tract.
Ocular Manifestations
A choroidal cavernous hemangioma is found in the eye ipsilateral to the facial lesions in 40% of affected individuals. One-half of all choroidal cavernous hemangiomas are found in patients with Sturge-Weber syndrome. In contradistinction to choroidal cavernous hemangiomata in patients without Sturge-Weber syndrome, Sturge-Weber hemangiomata are diffuse and have poorly defined, infiltrative margins. There may also be a cystic degeneration of overlying retina and serous exudation into the subretinal space. Heterochromia irides may be seen in patients who have a choroidal hemangioma; the affected eye can have a darker iris.43(p3)
Approximately 30% of patients with Sturge-Weber syndrome develop glaucoma. Most of the cases of glaucoma are manifest early in infancy, but cases do occur later in childhood and young adulthood. Glaucoma usually occurs when the upper or lower lid is affected by the cutaneous hemangioma, although bilateral and contralateral cases have been documented.43(p3) The glaucoma is felt not to be related to the choroidal hemangioma that may be present.34(p29) The mechanism of the glaucoma is unknown. Other cases have the anterior chamber drainage angle configuration typically found in primary infantile glaucoma.
WAARDENBERG'S SYNDROME
First described, in part, in 1926 by Mende, Waardenberg's syndrome was fully outlined by the Dutch physician in 1951.54 It is an autosomal dominant disorder with variable expressivity the occurrence rate of which is approximately 1 in 4,000 live births. Advanced paternal age has been associated with spontaneous mutation.31(pp248–249)
Systemic Manifestations
The cutaneous findings are prominent. A white forelock is commonly seen. This may be present at birth but can darken with age. A black forelock has also been described.2(p1901) The hair may become prematurely gray. There may be poliosis. The eyebrows can develop a medial flare and become confluent.
The most disfiguring cutaneous manifestation is vitiligo, which can be widespread. Because of the hypopigmented hair and eye (described below), Waardenberg's syndrome is considered by some to be a form of partial albinism.31(pp248–249)
The most serious manifestation of this disorder is deafness, usually bilateral. The defect seems to develop in the organ of Corti. Animal experimentation has suggested that this defect may be related to poor regulation of endolymphatic fluid.2(p1903) Another neurologic manifestation, occurring infrequently, is aganglionic megacolon (Hirschsprung's disease).
Skeletal malformations including cleft lip/palate, cervical ribs, and Sprengel's deformity occur infrequently.
Ocular Manifestations
Telecanthus, an increased distance between the medial canthi, is a characteristic ophthalmologic finding. Hageman and Delleman have divided Waardenburg's syndrome into two types.55 Type I Waardenburg's syndrome is characterized by telecanthus with deafness occurring in 25% of affected individuals. In type II, telecanthus does not occur, although up to 50% of patients are deaf.
Other findings include iris heterochromia, hypopigmentation and stromal hypoplasia and patchy fundus depigmentation, further adding to the characterizaton of Waardenburg's syndrome as a partial form of albinism.
MYOTONIC DYSTROPHY
The cardinal features of this disorder were first described by Steinert in Germany in 1909; myotonic dystrophy is sometimes referred to as Steinert's disease.56(pp160–161) Approximately one-fourth of the cases are thought to be new mutations. As with neurofibromatosis, children of affected mothers are more severely affected than children of affected fathers.2(p1223) This observation, along with the much more frequent mother-to-child transmission than father-to-child transmission, suggests some maternal factor in this disorder.
Systemic Manifestations
The hallmark of this disorder is myotonia, which is the inability to relax a contracted muscle. This is accompanied by muscle wasting and fatigue, affecting virtually all muscle groups. Symptoms begin usually in the third to fourth decade, although it can range from infancy to the sixties. Myotonia, especially of the forearm or jaw muscles, often precedes the onset of fatigue by several years. Typical facies include a dull, expressionless, myopathic look, with drooping of the lids and mouth and dysarthria. The chest muscles and diaphragm are also affected producing hypoventilation, adding to the generalized fatigue. The electromyogram can show the myotonia as well as myopathic discharges, even in clinically uninvolved muscles.
Also typical is male frontal baldness and testicular atrophy. Ovarian cysts, dysmenorrhea, generalized, mild pituitary hypofunction, and cardiac conduction abnormalities can also be seen. Mental deterioration can occur late. Patients usually succumb to cardiac failure or pneumonia.
Ocular Manifestations
Unique to myotonic dystrophy is the iridescent, polychromatic, Christmas tree–like opacities scattered throughout the anterior and posterior cortical layers of the lens. These may be seen in advance of any systemic symptoms or signs. A diffuse opacification of a thin layer near the posterior capsule can also occur. This can progress to a stellate opacity.
Other ocular manifestations include bilateral ptosis, which is associated with the generalized deficient muscular function, and external ophthalmoplegia. Blepharitis is commonly seen. Decreased corneal sensation and an epithelial dystrophy have also been described. A macular dystrophy which does not appear to cause any visual dysfunction may also occur in this condition.34(p500),57,58