1. Carr G: The human genome project. Nat Genet 26:21, 2000. 2. Joshi VR: Impact of the human genome project on medical practice. J Assoc Physicians India 50:856, 2002. 3. Wiggs JL: Molecular genetics and ocular disease. In Jakobiec FA, Lamkin JC (eds): International Ophthalmology Clinics: Advances in ophthalmic genetics and
heritable eye diseases. Chap 1. Boston, Little, Brown and Company, 1993. 4. Wald KJ, Hirose T: Clinical methods for detecting the carrier state of X-chromosome-linked
retinal disorders. In Jakobiec FA, Lamkin JC (eds): International Ophthalmology Clinics: Advances in ophthalmic genetics and
heritable eye diseases. pp 203–217. Boston, Little, Brown and Company, 1993. 5. Heckenlively JR: Discussion of juvenile retinoschisis: model for molecular diagnostic testing
of X-linked ophthalmic disease. Trans Am Ophthalmol Soc 67:464, 1999. 6. Barsh GS, Epstein CF: Gene structure and function in eukaryotic organisms. In Thompson MW, McInnes RR, Willard HF (eds): Principles and Practice of Medical Genetics, pp 7–29, Philadelphia, WB Saunders, 1999. 7. Lyon M: Gene action in the X-chromosome of the mouse. Nature 190:372, 1961. 8. Maumenee IH, Murphree AL: Genetic Disease of the Eye. New York, Oxford University Press, 1998. 9. Thompson MW, McInnes RR, Willard HF: Genetics in Medicine. 5th ed. Philadelphia, WB Saunders, 1991. 10. Wang MX, Carlsen AJR, Liu JC: X-linked opthalmic disorders. In Tasman W Jaeger EA (eds): Duane's Foundations of Clinical Ophthalmology. Vol 3, Chap 57. Philadelphia, Lippincott-Raven, 1999. 11. Fatt HV, Griffin JR, Lyle WM (eds): Genetics for Primary Eye Care Practitioners, pp 1–11, Boston,Butterworth-Heinemann, 1992. 12. Botstein D, White RL, Skolnick M et al: Construction of a genetic linkage map using restriction fragment length
polymorphisms. Am J Hum Genet 32:314, 1980. 13. Wiggs JL: Molecular Genetics of Ocular Disease, pp 1–29, New York, Wiley-Liss, 1995. 14. Gorin MB, Wright AF: Background to molecular genetic principles and techniques. In Wright AF, Barrie J (eds): Molecular Genetics of Inherited Eye Disorders. pp 1–28, Chur, Switzerland, Harwood Academic Publishers, 1994. 15. Seabra MC, Brown MS, Goldstein JL: Retinal degeneration in choroideremia: Deficiency of rab geranylgeranyl
transferase. Science 259:377, 1993. 16. Mauthner H: Ein Fall von Choroideremia. Ber Naturw-med Ver Insbruck 2:191, 1872. 17. Cremers FPM, Ropers H: Choroideremia. In Scriver CR, Beaud et al, Sly WS, Valle D (eds): The etabolic and Molecular Basis of Inherited Disease, pp 5935-59. 8th ed. New York, McGraw-Hill, 2001. 18. Kanski JJ: Clinical Ophthalmology: A Systematic Approach. Oxford, UK: Butterworth-Heinemann, 1999. 19. Hirakawa H, Iijima H, Gohdo T et al: Progression of defects in the central 10-degree visual field of
patients with retinitis pigmentosa and choroideremia. Am J Ophthalmol 127:436, 1999. 20. Szlyk JP, Seiple W, Laderman DJ et al: Use of bioptic amorphic lenses to expand the visual field in patients with
peripheral loss. Optom Vis Sci 75:518, 1998. 21. Lewis AR, Nussbaum RL, Ferrell R: Mapping X-linked ophthalmic diseases: Provisional assessment of
the locus for choroideremia to Xq3-q29. Ophthalmology 92:800, 1985. 22. Yannuzzi LA, Guyer DR, Green R: The Retina Atlas. Mosby–Year Book, Inc 1995. 23. Ghosh M, McCulloch JC: Pathological findings from two cases of choroideremia. Can J Ophthalmol 15:147, 1980. 24. Ghosh M, McCulloch C, Parker JA: Pathological study in a female carrier of choroideremia. Can J Ophthalmol 23:181, 1988. 25. MacDonald IM, Chen MH, Addison DJ et al: Histopathology of the retinal pigment epithelium of a female carrier of
choroideremia. Can J Ophthalmol 32:329, 1997. 26. Flannery JG, Bird AC, Farber DB et al: A histopathologic study of a choroideremia carrier. Invest Ophthalmol Vis Sci 31:229, 1990. 27. Cameron JG, Fine BS, Sharpiro I: Histopathologic observations in choroideremia with emphasis on vascular
changes of the uveal tract. Ophthalmology 94:187, 1987. 28. Rodrigues MM, Ballintine EJ, Wiggert BN et al: Choroideremia: A clinical, electron microscopic, and biochemical report. Ophthalmology 91:873, 1984. 29. van Dorp DB, van Balen ATM: Fluorescein angiography in potential carriers of choroideremia. Ophthalmic Paediatr Genet 5:25, 1985. 30. Syed N, Smith JE, John SK et al: Evaluation of retinal photoreceptors and pigment epithelium in a female
carrier of choroideremia. Ophthalmology 108:711, 2001. 31. Goedbloed J: Mode of inheritance of choroideremia. Ophthalmology 104:308, 1942. 32. Shastry BS: Recent developments in certain X-linked genetic eye disorders. Biochim Biophys Acta 1182:119, 1993. 33. Nussbaum RL, Lewis RA, Lesko JG et al : Choroideremia is linked to restriction fragment length polymorphism DXYS1 at
Xq13. Am J Hum Genet 37:473, 1985. 34. Lasko JG, Lewis RA, Nussbaum RL: Multipoint linkage analysis of loci in the proximal long arm of the human
X chromosome : Application to mapping the choroideremia locus. Am J Hum Genet 40:303, 1987. 35. Beaufrere L, Rieu S, Hache JC et al: Altered rep-1 expression due to substitution at position +3 of
the IVS13 splice-donor site of the choroideremia (CHM) gene. Curr Eye Res 17:726, 1998. 36. Seabra MC, Goldstein JL, Sudhof TC et al: Rab geranylgeranyl transferase. A multisubunit enzyme that prenylates GTP-binding proteins terminating
in Cys-X-Cys or Cys-Cys. J Bio Chem 267:14497, 1992. 37. Seabra MC, Ho YK, Anant JS: Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Bio Chem 270:24420, 1995. 38. van den Hurk JA, Swartz M, van Bokhoven H et al: Molecular basis of choroideremia (CHM): Mutations involving the
Rab escort protein-1(REP-1) gene. Hum Mutat 9:110, 1997. 39. Sankila EM, Tolvanen R, van den Hurk JA et al: Aberrant splicing of the CHM gene is a significant cause of choroideremia. Nat Genet 1:109, 1992. 40. van Bokhoven H, Schwartz M, Andersson S et al: Mutation spectrum in the CHM gene of Danish and Swedish choroideremia patients. Hum Mol Genet 3:1047, 1994. 41. Schwartz M, Rosenberg T, van den Hurk JA et al: Identification of mutations in Danish choroideremia families. Hum Mutat 2:43, 1993. 42. Fujiki K, Hotta Y, Hayakawa M et al : REP-1 gene mutations in Japanese patients with choroideremia. Graefes Arch Clin Exp Ophthalmol 237:735, 1999. 43. van den Hurk JA, van Zandvoort PM, Brunsmann F: Prenatal exclusion of choroideremia. Am J Med Genet 44:822, 1992. 44. Schwartz M, Rosenberg T: Prenatal diagnosis of choroideremia. Acta Ophthalmol Scand 74:33, 1996. 45. Beaufrere L, Tuffrey S, Hamel C et al : The protein truncation test (PTT) as a method of detection for
choroideremia mutations. Exp Eye Res 65:849, 1997. 46. MacDonald IM, Mah DY, Ho YK et al: A practical diagnostic test for choroideremia. Ophthalmology 195:9, 1998. 47. Duncan JL, Aleman TS, Gardner LM et al: Macular and lutein supplementation in choroideremia. Exp Eye Res 74:371, 2002. 48. Darnall HJA, Bowmaker JK, Mollon JD: Human visual pigments: Microspectrophotometric results from the eyes of
seven persons. Proc R Soc Lond B Biol Sci 220:115, 1983. 49. Kalmus H: Types of defective color vision. In Kalmus H: Diagnosis and Genetics of Defective Color Vision, pp 14–21. New York, Pergamon Press, 1965. 50. Birch J: Efficiency of the Ishihara test for identifying red-green colour
deficiency. Ophthalmic Physiol Opt 17:403, 1997. 51. Nathans J, Thomas D, Hogness DS: Molecular genetics of human color vision: The genes incoding blue, green
and red pigments. Science 232:193, 1986. 52. Nathans J, Piantanida TP, Eddy RL et al: Molecular genetics of inherited variations in human color vision. Science 232:203, 1986. 53. Haynie GD, Mukai S: Genetic Basis of Color Vision. In Jakobiec FA, Lamkin JC (eds): International Ophthalmology Clinics: Advances in Ophthalmic Genetic and
Heritable Eye Diseases, pp 141–152, Boston, Little, Brown and Company, 1993. 54. Neitz M, Neitz J, Jacobs GH: Spectral tuning of pigments underlying red-green color vision. Science. 252:971, 1991. 55. Wolf S, Sharpe LT, Schmidt HJ et al: Direct visual resolution of gene copy number in the human photopigment
gene array. Invest Ophthalmol Vis Sci 40:1585, 1999. 56. Bowmaker JK: Visual pigments and molecular genetics of color blindness. News Physiol Sci 13:63, 1998. 57. Jagla WM, Jagle H, Hayashi T et al. The molecular basis of dichromatic color vision in males with multiple
red and green pigment genes. Hum Mol Genet 11:23, 2002. 58. Winderickx J, Battisti L, Motulsky AG, Deeb SS: Selective expression of human X chromosome–linked green opsin genes. Proc Natl Acad Sci U S A 89:9710, 1992. 59. Sjoberg SA, Neitx M, Balding SD, Neitz J: L-cone pigment genes expressed in normal colour vision. Vision Res 38:3213, 1998. 60. Zhang Q, Kensei M: Detection of congenital color vision defects using heteroduplex-SSCP
analysis. Jpn J Ophthalmol 40:79, 1996. 61. Merbs SL, Nathans J: Absorption spectra of the hybrid pigments responsible for anomalous color
vision. Science 258:464, 1992. 62. Deeb SS, Lindsey DT, Hibiya Y et al: Genotype-phenotype relationships in human red/green color-vision
defects: Molecular and psychophysical studies. Am J Hum Genet 51:687, 1992. 63. Zhang Q, Mao W, Ma Q et al: Molecular basis of congenital color vision defects in Chinese patients. Jpn J Ophthalmol 36:479, 1992. 64. Sharpe LT, Stockman A, Jagle H et al: Red, green, and red-green hybrid pigments in the human retina: Correlations
between deduced protein sequences and the psychophysical measured
spectral sensitivities. J Neurosci 18:10053, 1998. 65. Winderickx J, Sanocki E, Lindsey DT et al: Defective colour vision associated with a missense mutation in the human
green visual pigment gene. Nat Genet 1:251, 1992. 66. Ueyama H, Kuwayama S, Imai H et al: Novel missense mutations in red/green opsin genes in congenital color-vision
deficiencies. Biochem Biophys Res Commun 294:205, 2002. 67. Reyniers E, Van Thienen NM, Meire F et al: Gene conversion between red and defective green opsin gene in blue cone
monochromacy. Genomics 29:323, 1995. 68. Oda S, Ueyama H, Tanabe S et al: Detection of female carriers of congenital color-vision deficiencies
by visual pigment gene analysis. Curr Eye Res 21:767, 2000. 69. Lang A, Good GW: Color discrimination in heterozygous deutan carriers. Optom Vis Sci 78:584, 2001. 70. Nagy AL, MacLeod DI, Heyneman NE, Eisner A: Four cone pigments in women heterozygous for color deficiency. J Opt Soc Am 71:719, 1981. 71. Zhang Q, Xiao X, Shen H et al: Correlation of gene structure and psychophysical measurement in red-green
color vision deficiency in Chinese. Jpn J Ophthalmol 44:596, 2000. 72. Crognale MA, Teller DY, Motulsky AG, Deeb SS: Severity of color vision defects: Electroretinographic (ERG), molecular
and behavioral studies. Vision Res 38:3377, 1998. 73. Kayser B: Megalokornea oder hydrophthalmus? Klin Monatsbl Augenheilkd 52:226, 1914. 74. Goldberg MF: Clinical manifestations of ectopia lentis et pupillae in 16 patients. Ophthalmology 95:1080, 1988. 75. Meire FM, Delleman JW: Autosomal dominant congenital miosis with megalocornea. Ophthalmic Paediatr Genet 13:123, 1992. 76. De Luise VP, Anderson DR: Primary infantile glaucoma (congenital glaucoma). Surv Ophthalmol 28:1, 1983. 77. Yanoff M, Fine B: Ocular Pathology: A Color Atlas, p 90. Philadelphia: JB Lippincott, 1988. 78. Vail DT: Adult hereditary anterior megalophthalmos sine glaucoma: A definite disease
entity. Arch Ophthalmol 6:39, 1931. 79. Meire FM: Megalocornea: Clinical and genetic aspects. Doc Ophthalmol 87:1, 1994. 80. Meire FM, Bleeker-Wagemakers EM, Oehler M et al: X-linked megalocornea: Ocular findings and linkage analysis. Ophthalmic Paediatr Genet 12:153, 1991. 81. Mackey DA, Buttery RG, Wise GM et al: Description of X-linked megalocornea with identification of gene
locus. Arch Ophthalmol 109:829, 1991. 82. McKusick VA: Mendelian Inheritance in Man, A Catalog of Human Genes and Genetic Disorders. 12th ed. Baltimore, The Johns Hopkins University Press, 1998. 83. Meire FM, Van Egmond J, Hanssens M: Congenital Marfan syndrome with contractures: A clinicopathological report. Bull Belg Ophtalmol 245:91, 1992. 84. Koenekoop RK, Rosenbaum KN, Traboulsi EI: Ocular findings in a family with Sotos syndrome (cerebral gigantism). Am J Ophthalmol 119:657, 1995. 85. Shiono T, Tsunoda M, Chida Y et al: X linked ocular albinism in Japanese patients. Br J Ophthalmol 79:139, 1995. 86. Mansour AM, Traboulsi EI, Frangieh GT et al: Unilateral megalocornea in lamellar ichthyosis. Ann Ophthalmol 17:466, 1985. 87. Hoyt CS, Brown RA: Megalocornea in nonketotic hyperglycinemia. J Pediatr Ophthalmol Strabismus 15:85, 1978. 88. Anton M, Vrba M, Rehurek J et al: Mesodermal dysgenesis. Cesk Oftalmol 46:69, 1990. 89. Meire FM, Delleman JM: Biometry in X linked megalocornea: Pathognomonic findings. Br J Ophthalmol 78:781, 1994. 90. Chen JD, Mackey D, Fuller H et al: X-linked megalocornea: Close linkage to DXS87 and DXS94. Hum Genet 83:292, 1989. 91. Mann I: Developmental Abnormalities of the Eye, pp 352. London, JB Lippincott, 1957. 92. Norrie G: Causes of blindness in children. Acta Ophthalmol 5:357, 1927. 93. Warburg M: Norrie disease. Birth defects 7:117, 1971. 94. Warburg M: Norrie disease: Differential diagnosis and treatment. Acta Ophthalmol 53:217, 1975. 95. Shastry BS, Hiraoka M, Trese DC, Trese MT: Norrie disease and exudative vitreoretinopathy in families with affected
female carriers. Eur J Ophthalmol 9:238, 1999. 96. Schuback DE, Chen Z-Y, Battinelli EM et al: Mutations in the Norrie disease gene. Hum Mutat 5:285, 1995. 97. Donnai D, Mountford RC, Read AP: Norrie's disease resulting from a gene deletion: Clinical features
and DNA studies. J Med Genet 25:73, 1988. 98. Holmes LB: Norrie's disease: An X-linked syndrome of retinal malformation, mental
retardation and deafness. J Pediatr 89:89, 1971. 99. Moreira-Filho CA, Neustein I: A presumptive new variant of Norrie's disease. J Med Genet 16:125, 1979. 100. Goodyear HM, Sonksen PM, McConachie H: Norrie's disease: A prospective study of development. Arch Dis Child 64:1587, 1989. 101. Warburg M: Norrie's disease: A congenital progressive oculo-acoustico-cerebral
degeneration. Acta Ophthal Suppl 89:1, 1966. 102. Enyedi LB, Juan E: Ultrastructural study of Norrie disease. Am J Ophthalmol 111:439, 1991. 103. van Nouhuys CE: Ultrastructural study of Norrie's disease [letter comment]. Am J Ophthalmol 111:439, 1991. 104. Berger W, Meindl A, van de Pol TJR et al: Isolation of a candidate gene for Norrie disease by positional cloning. Nature Genet 1:199, 1992. 105. Chen ZY, Hendirks RW, Jobling MA et al: Isolation and characterization of a candidate gene for Norrie disease. Nat Genet 1:204, 1992. 106. Zhu D, Maumenee IH: Mutations in the ND gene in families with Norrie disease. Am J Hum Genet 53:1260, 1993. 107. Berger W, Ropers H: Norrie Disease. In Scriver CR, Beauet AL, Sly WS et al (eds): The metabolic and molecular basis of inherited disease, pp 5977–5985. 8th ed. New York, McGraw-Hill, 2001. 108. Meindl A, Berger W, Meitinger T et al: Norrie disease is caused by mutations in an extracellular protein resembling
C-terminal globular domain of mucins. Nature Genet 2:139, 1992. 109. Meitinger T, Meindl A, Bork P et al: Molecular modeling of the Norrie disease protein predicts a cysteine knot
growth factor tertiary structure. Nature Genet 5:376, 1993. 110. Fuchs S, van de Pol D, Beudt U et al: Three novel and two recurrent mutations of the Norrie disease gene in patients
with Norrie syndrome. Hum Mut 8:85, 1996. 111. Shastry BS, Hejtmancik JF, Trese MT: Identification of novel missense mutations in the Norrie disease gene associated
with one X-linked and four sporadic cases of familial
exudative vitreoretinopathy. Hum Mut 9:396, 1997. 112. Fuchs S, Kellner U, Wedemann H, Gal A: Missense mutation (Arg 121 Trp) in the Norrie disease gene associated
with X-linked exudative vitreoretinopathy. Hum Mutat 6:257, 1995. 113. Shastry BS, Hejtmanick JF, Plager DA et al: Linkage and candidate gene analysis of X-linked familial exudative
vitreoretinopathy. Genomics 27:341, 1995. 114. King RA, Hearing VJ, Creel DJ et al: Albinism. In Scriver CR, Beaudet AL, Sly WS et al (eds): The metabolic and molecular basis of inherited disease, pp 5587–5627. 8th ed. New York, McGraw-Hill, 2001. 115. Charles SJ, Green JS, Grant JW et al: Clinical features of affected males with X-linked ocular albinism. Br J Ophthalmol 77:222, 1993. 116. Apkarian P: A practical approach to albino diagnosis: VEP misrouting across the age
span. Ophthalmic Paediatr Genet 13:77, 1992. 117. Worobec-Victor SM, Bain MAB: Oculocutaneous genetic diseases. In Renie WA (ed): Goldberg's Genetic and Metabolic Eye Disease, pp 489–541. Boston, Little, Brown and Company, 1986. 118. Schnur RE, Wick PA, Bailey C et al: Phenotypic variability in X-linked ocular albinism: Relationship
to linkage genotypes. Am J Hum Genet 55:484, 1994. 119. Wong L, O'Donnell Jr FE, Green WR: Giant pigment granules in the retinal pigment epithelium of a fetus with
X-linked ocular albinism. Ophthalmic Paediatr Genet 2:47, 1983. 120. Nakagawa H, Hori Y, Sato S et al: The nature and origin of the melanin macroglobule. J Invest Dermatol 83:134, 1984. 121. Shen B, Samaraweera P, Rosenberg B et al: Ocular albinism type 1: More than meets the eye. Pigment Cell Res 14:243, 2001. 122. Garner A, Jay BS: Macromelanosomes in X-linked ocular albinism. Histopathology 4:243, 1980. 123. Schnur RE, Nussbaum RL, Anson-Cartwright L et al: Linkage analysis in X-linked ocular albinism. Genomics 9:605, 1991. 124. Ballabio A, Andria G: Deletions and translocations involving the distal short arm of the human
X chromosome: Review and hypotheses. Hum Mol Genet 1:221, 1992. 125. Schaefer L, Ferrero GB, Grillo A et al: A high resolution deletion map of the human chromosome Xp22. Nat Genet 4:272, 1993. 126. Bassi MT, Schiaffino MV, Renieri A et al: Cloning of the gene for ocular albinism type 1 from the distal short arm
of the X chromosome. Nat Genet 10:13, 1995. 127. Schiaffino MV, Baschirotto C, Pellegrini G et al: The ocular albinism type 1 (OA1) gene product is a membrane glycoprotein
localized to melanosomes. Proc Natl Acad Sci U S A 93:9055, 1996. 128. Schiaffino MV, d'Addio M, Alloni A et al: Ocular albinism: Evidence for a defect in an intracellular signal transduction
system. Nat Genet 23:108, 1999. 129. Samaraweera P, Shen B, Newton JM et al: The mouse ocular albinism 1 gene product is an endolysosomal protein. Exp Eye Res 72:319, 2001. 130. Incertl B, Cortese K, Pizzigoni A et al: Oa1 knock-out: New insights on the pathogenesis of ocular albinism
type 1. Hum Mol Genet 9:2781, 2000. 131. Honing S, Sandoval LV, von Figura K: A di-leucine-based motif in the cytoplasmic tail of LIMP-II
and tyrosinase mediates selective binding of AP-3. EMBO J 17:1304, 1998. 132. Orlow SJ: Melanosomes are specialized members of the lysosomal lineage or organelles. J Invest Dermatol 105:3, 1995. 133. Jimbow K, Park JS, Kato F et al: Assembly, target-signaling and intracellular transport of tyrosinase
gene family proteins in the initial stage of melanosome biogenesis. Pigment Cell Res 13:222, 2000. 134. Setaluri V: Sorting and targeting of melanosomal membrane proteins: Signals, pathways, and
mechanisms. Pigment Cell Res 13:128, 2000. 135. Akeo K, Tanaka Y, Okisaka S: A comparison between melanotic and amelanotic retinal pigment epithelial
cells in vitro concerning the effects of L-dopa and oxygen on
the cell cycle. Pigment Cell Res 7:145, 1994. 136. Wick MM, Byers L, Frei E: L-Dopa: Selective toxicity for melanoma cells in vitro. Science 197:468, 1977. 137. O'Donnell Jr FE, Green WR, Fleischman JA et al: X-linked ocular albinism in blacks, ocular albinism cum pigmento. Arch Ophthalmol 96:1189, 1978. 138. Shiono T, Tsunoda M, Chida Y et al: X-linked ocular albinism in Japanese patients. Br J Ophthalmol 79:139, 1995. 139. Szymanski KA, Boughman JA, Nance WE et al: Genetic studies of ocular albinism in a large Virginia kindred. Ann Ophthalmol 16:183, 1984. 140. Schiaffino MV, Bassi MT, Galli L et al: Analysis of the OA1 gene reveals mutations in only one-third of
patients with X-linked ocular albinism. Hum Mol Genet 4:2319, 1995. 141. Rosenberg T, Schwart M: X-linked ocular albinism: Prevalence and mutations—a national
study. Eur J Hum Genet 6:570, 1998. 142. Oetting WS, King RA: Molecular basis of albinism: Mutations and polymorphisms of pigmentation
genes associated with albinism. Hum Mutat 13:99, 1999. 143. Schnur RE, Gao M, Wick PA et al: OA1 mutations and deletions in X-linked ocular albinism. Am J Hum Genet 62:800, 1998. 144. Winship I, Gericke G, Beighton P: X-linked inheritance of ocular albinism with late-onset sensorineural
deafness. Am J Med Genet 19:797, 1984. 145. Winship IM, Babaya M, Ramesar RS: X-linked ocular albinism and sensorineural deafness: Linkage to
Xp22.3. Genomics 18:444, 1993. 146. Rosenberg T, Schwartz M, Simonsen SE: Aland eye disease (Forsius-Eriksson-Miyake syndrome) with
probability established in a Danish family. Acta Ophthalmologica 68:281, 1990. 147. Weleber RG, Pillars DM, Powell BR et al: Aland Island eye disease (Forsius-Eriksson syndrome) associated
with contiguous deletion syndrome at Xp21: Similarity to incomplete
congenital stationary night blindness. Arch Ophthalmol 107:1170, 1989. 148. Glass IA, Good P, Coleman MP et al: Genetic mapping of a cone and rod dysfunction (Aland Island eye disease) to
the proximal short arm of the human X chromosome. J Med Genet 30:1044, 1993. 149. Bunker CH, Berson EL, Bromley WC, et al: Prevalence of retinitis pigmentosa in Main. Am J Ophthalmol 97:357, 1984. 150. Newsome DA: Retinitis pigmentosa, Usher's syndrome, and other pigmentary retinopathies. In Newsome DA (ed). Retinal Dystrophies and Degenerations, pp 161–194. New York, Raven Press, 1988. 151. Berson EL: Retinitis pigmentosa and allied diseases: Application of electroretinographic
testing. Int Ophthalmol 4:7, 1981. 152. Fishman GA, Farber MD, Derlacki DJ: X-linked retinitis pigmentosa. Profile of clinical findings. Arch Ophthalmol 106:369, 1988. 153. Stone J, Maslim J, Valter-Kocsi K, et al: Mechanism of photoreceptor death and survival in mammalian retina. Prog Retin Eye Res 18:689, 1999. 154. Fishman GA: Retinitis pigmentosa. Genetic percentages. Arch Ophthalmol 96:822, 1978. 155. Berson EL, Sandberg M, Rosner B et al: Natural course of retinitis pigmentosa over a three-year interval. Am J Ophthalmol 99:240, 1985. 156. Bhattacharya SS, Wright AF, Clayton JF et al: Close genetic linkage between X-linked retinitis pigmentosa and
a restriction fragment length polymorphism identified by recombinant DNA
probe L1.28. Nature 309:253, 1984. 157. Musarella MA, Burghes A, Anson-Cartwright L et al: Localization of the gene for X-linked recessive type of retinitis
pigmentosa (XLRP to Xp21) by linkage analysis. Am J Hum Genet 43:484, 1988. 158. Musarella MA, Anson-Cartwright L, Leal SM et al: Multipoint linkage analysis and heterogeneity testing in 20 X-linked
retinitis pigmentosa families. Genomics 8:286 1990. 159. Ott J, Bhattacharya S, Chen JD: Localizing multiple X chromosome-linked retinitis pigmentosa loci
using multilocus homogeneity tests. Proc Natl Acad Sci U S A 87:701, 1990. 160. Dahl N, Sundvall M, Pettersson U et al: Genetic mapping of the loci for X-linked retinitis pigmentosa. Clin Genet 40:435, 1991. 161. Breuer DK, Affer M, Andreasson S et al: X-linked retinitis pigmentosa:current status. In Anderson RE, LaVail MM, Hollyfield JG (eds): New Insights Into Retinal Degenerative Diseases, pp 11–12. New York, Kluwer Academic/Plenum Publishing, 2001. 162. Hardcastle AJ, Thiselton DL, Zito I et al: Evidence for a new locus for X-linked retinitis pigmentosa (RP23). Invest Ophthalmol Vis Sci 4:2080, 2000. 163. Gieser L, Fujita R, Goring HH et al : A novel locus (RP24) for X-linked retinitis pigmentosa
maps to Xq26-27. Am J Hum Genet 63:1439, 1998. 164. Schwahn U, Lenzner S, Dong J et al: Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet 19:372, 1998. 165. Mears AJ, Gieser L, Yan D et al: Protein-truncation mutations in the RP2 gene in a North American
cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet 64:897, 1999. 166. Miano MG, Testa F, Filippini F et al: Identification of novel RP2 mutations in a subset of X-linked retinitis
pigmentosa families and prediction of new domains. Hum Mutat 18:109, 2001. 167. Chappel JP, Hardcastle AJ, Grayson C et al: Delineation of the plasma membrane targeting domain of the X-linked
retinitis pigmentosa protein RP2. Invest Ophthalmol Vis Sci 42:2015, 2002. 168. Meindl A, Dry K, Herrmann K et al: A gene (RPGR) with homology to the RCC1 guaning nucleotide exchange
factor is mutated in X-linked retinitis pigmentosa (RP3). Nat Genet 13:35, 1996. 169. Roepman R, Bauer D, Rosenberg T et al: Identification of a gene disrupted by a microdeletion in a patient with
X-linked retinitis pigmentosa (XLRP). Hum Mol Genet 5:827, 1996 170. Roepman R, van Duijnhoven G, Rosenbert T et al: Positional cloning of the gene for X-linked retinitis pigmentosa 3: homology
with the guanine-nucleotide-exchange factor
RCC1. Hum Mol Genet 5:1035, 1996. 171. Kirschner R, Rosenberg T, Schultz-Heienbrok R et al: RPGR transcription studies in mouse and human tissues reveal a retina-specific
isoform that is disrupted in a patient with X-linked
retinitis pigmentosa. Hum Mol Genet 8:1571, 1999. 172. Vervoort R, Lennon A, Bird AC et al: Mutational hot spot within a novel RPGR exon in X-linked retinitis
pigmentosa. Nat Genet 25:462, 2000. 173. Bischoff FR, Ponstingl H: Catalysis of guanine nucleotide exchange on Ran by the mitotic regulator
RCC1. Nature 354:80, 1991. 174. Rosa JL, Casaroli-Marano RP, Buckler AJ et al: p619, a giant protein related to the chromosome condensation regulator
RCC1, stimulates guanine nucleotide exchange on ARF1 and Rab proteins. EMBO J 15:4262, 1996. 175. Vervoort R, Wright AF: Mutations of RPGR in X-linked retinitis pigmentosa (RP3). Hum Mutat 19:486, 2002. 176. Linari M, Ueffing M, Manson F et al: The retinitis pigmentosa GTPase regulator RPGR interacts with the delta
subunit of rod cyclic GMP phosphodiesterase. Proc Natl Acad Sci U S A 96:1315, 1999. 177. Hong DH, Yue G, Adamian M et al: Retinitis pigmentosa GTPase regulator (RPGR)-interacting
protein is stably associated with the photoreceptor ciliary axoneme
and anchors RPGR to the connecting cilium. J Biol Chem 276:12091, 2001. 178. Boylan JP, Wright AF: Identification of a novel protein interacting with RPGR. Hum Mol Genet 9:2085, 2000. 179. Roepman R, Bernoud-Hubac N, Schick DE et al: The retinitis pigmentosa GTPase regulator (RPGR) interacts with
novel transport-like proteins in the outer segments of rod photoreceptors. Hum Mol Genet 9:2095, 2000. 180. Zito I, Downes SM, Patel RJ et al: Evidence for a new X-linked syndrome involving retinitis pigmentosa. Invest Ophthalmol Vis Sci 42:A3447, 2001. 181. Dryja TP, Adams SM, Grimsby JL et al: Null RPGRIP1 alleles in patients with Leber congenital amauosis. Am J Hum Genet 68:1295, 2001. 182. Yang Z, Peachey NS, Moshfeghi DM et al: Mutations in the RPGR gene cause X-linked cone dystrophy. Hum Mol Genet 11:605, 2002. 183. Liu L, Chen H, Liu M et al: Two novel mutations of the retinitis pigmentosa GTPase regulator gene in
two Chinese families with X-linked retinitis pigmentosa. Chin Med J 115:833, 2002. 184. Fujita R, Buraczynska M, Gieser L et al: Analysis of the RPGR gene in 11 pedigrees with the retinitis pigmentosa
type 3 genotype: Paucity of mutations in the coding region but splice
defects in two families. Am J Hum Genet 61:571, 1997. 185. Rabe VW, Sharon D, Berson EL et al: The muation spectrum of RPGR-ORF15 in North-American patients
with X-linked retinitis pigmentosa (XLRP). Invest Ophthalmol Vis Sci 42:A3450, 2001. 186. Miano MG, Vervoort R, Conte I et al: Mutational analysis of the RPGR exon ORF15 in South European patients with
X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 42:A3446, 2001. 187. Sharon D, Bruns GA, McGee TL et al: X-linked retinitis pigmentosa: Mutation spectrum of the RPGR and
RP2 genes and correlation with visual function. Invest Ophthalmol Vis Sci 41:2712, 2000. 188. Flaxel CJ, Jay M, Thiselton DL et al : Difference between RP2 and RP3 phenotypes in X linked retinitis pigmentosa. Br J Ophthalmol 83:1144, 1999. 189. Bailey CC, Qureshi SH, Hardcastle AJ et al: Phenotype/genotype correlations in X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 42:A417, 2001. 190. Hong DH, Pawlyk BS, Shang J et al: A retinitis pigmentosa GTPase regulator (RPGR)–deficient
mouse model for X-linked retinitis pigmentosa (RP3). Proc Natl Acad Sci U S A 97:3649-3654, 2000 191. Buckley EG: Pediatric cataracts and lens anomalies. In Nelson LB (ed): Harley's Pediatric Ophthalmology, pp 258-282.4th ed. Philadelphia, W.B. Saunders, 1998. 192. He W, Li S: Congenital cataracts: Gene mapping. Hum Genet 106:1, 2000. 193. Goldberg MF, Hardy J, Hussels I: X-linked cataract: Two pedigrees. Birth Defects 7:164, 1971. 194. Francois J: Genetics of cataract. Ophthalmolgica 184:61, 1982. 195. Wirth MG, Russell-Eggitt IM, Craig JE et al : Aetiology of congential and paediatric cataract in an Australian population. Br J Ophthalmol 86:782, 2002. 196. Drill AE, Woodbury G, Bowman JE: X-chromosomal-linked sutural cataracts. Am J Ophthalmol 68:867, 1969. 197. Capella JA, Kaufman HE, Lill FJ: Hereditary cataracts and microphthalmia. Am J Ophthalmol 56:454, 1963. 198. Francis PJ, Berry V, Hardcastle AJ et al: A locus for isolated cataract on human Xp. J Med Genet 39:105, 2002. 199. Abbassi V, Lowe CU, Calcagno PL: Oculo-cerebro-renal syndrome: A review. Am J Dis Child 115:145, 1968. 200. Bateman JB, Harley RD: Genetics of Eye Disease. In Nelson LB (ed): Harley's Pediatric Ophthalmology, pp 15. 4th ed.Philadelphia, W.B. Saunders, 1998. 201. Attree O, Olivos I, Okabe I et al: The Lowe oculocerebrorenal syndrome gene encodes a novel protein highly
homologous to inositol polyphosphate-5-phosphatase. Nature 358:239, 1992. 202. Jefferson AB, Majerus PW: Properties of type II inositol polyphosphate 5-phosphatase. J Biol Chem 2:9370, 1995. 203. Monnier N, Satre V, Lerouge E et al: OCRL1 mutation analysis in French Lowe syndrome patients: Implications
for molecular diagnosis strategy and genetic counseling. Hum Mutat 16:157, 2000. 204. Satre V, Monnier N, Berthoin F et al: Characterization of a germline mosaicism in familes with Lowe syndrome
and identification of seven novel mutations in the OCRL1 gene. Am J Hum Genet 65:68, 1999. 205. Nance W, Warburg M, Bixler D et al: Congenital X-linked cataract, dental anomalies and brachymetacarpalia. Birth Defects 10:285, 1974. 206. Horan M, Billson F: X-linked cataract and Hutchinsonian teeth. Aust Paediatr J 10:98, 1974. 207. Bixler D, Higgins M, Hartsfield J: The Nance-Horan syndrome: A rare X-linked ocular dental trait
with expression in heterozygous females. Clin Genet 26:30, 1984. 208. Stambolian D, Lewis RA, Buetow K et al: Nance-Horan syndrome: Localization within the region Xp21.1-Xp22.3 by linkage analysis. Am J Hum Genet 47:13, 1990. 209. Zhu D, Alcorn DM, Antonarakis SE et al: Assignment of the Nance-Horan syndrome to the distal short arm of
the X chromosome. Hum Genet 86:54, 1990. 210. Bergen AAB, Ten Brink J, Schuurman EJM et al: Nance-Horan syndrome: Linkage analysis in a family from the Netherlands. Genomics 21:238, 1994. 211. Toutain A, Ronce N, Dessay B et al: Nance-Horan syndrome: Linkage analysis in 4 families refines localization
in Xp22.21-p22.13 region. Hum Genet 99:256, 1997. 212. Favor J, Pretsch W: Genetic localization and phenotypic expression of X-linked cataract (Xcat) in
Mus musculus. Genet Res (Camb) 56:157, 1990. 213. Stambolian D, Favor J, Silvers W et al: Mapping of the X-linked cataract (Xcat) mutation, the
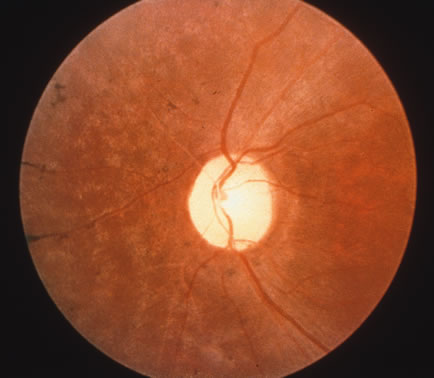
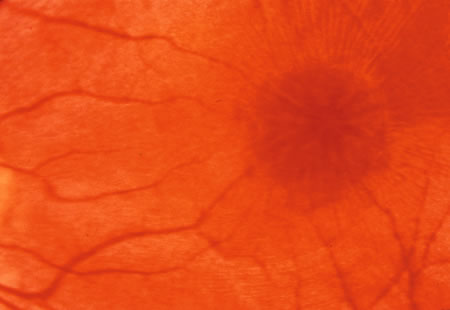
gene implicated in the Nance Horan syndrome, on the mouse X chromosome. Genomics 22:377, 1994. 214. George ND, Yates JR, Moore AT: X-linked retinoschisis. Br J Ophthalmol 79:697, 1995. 215. De la Gaoelle A, Aitalo T, Forsisus H: X-linked juvenile retinoschisis. In Wright A, Jav B. (eds): Molecular Genetics of Inherited Eye Disorders, pp 339–357. Chur, Switzerland: Harwood Academic Publishers, 1994. 216. Forsius H, Krause U, Helve J et al: Visual acuity in 183 cases of X-chromosome retinoschisis. Can J Ophthalmol 8:385, 1973. 217. Sieving PA, Yashar BM, Ayyagari R: Juvenile retinoschisis: A model for molecular diagnostic testing of X-linked
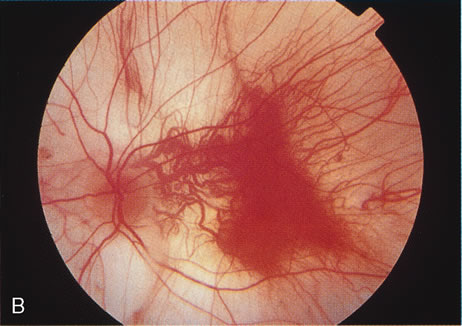
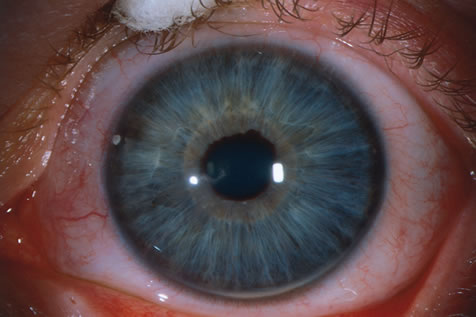
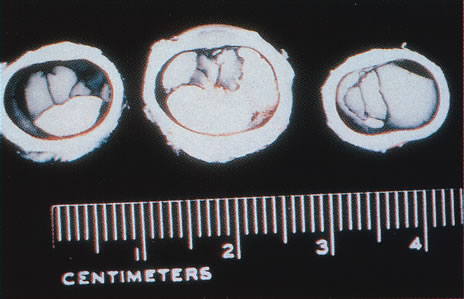
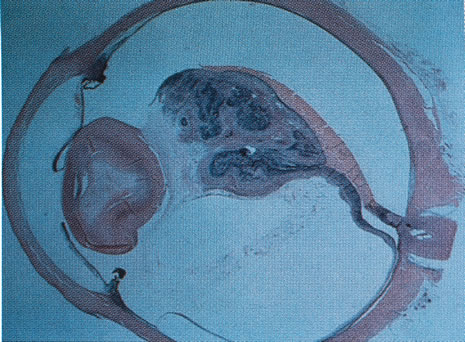
ophthalmic disease. Trans Am Ophthalmol Soc 157:451, 1999. 218. Deutman AF: Sex-linked juvenile retinoschisis. In The Hereditary Dystrophies of the Posterior Pole of the Eye, pp 48–99. The Netherlands, Van Gottum, 1971. 219. Primo SA, Tomasino SF: Progressive retinal changes observed in juvenile X-linked retinoschisis. J Am Optom Assoc 61:548, 1990. 220. Dalmer B, Brummer S, Foerster MH et al: X-linked congenital retinoschisis. Arch Clin Exp Ophthalmol 228:432, 1990. 221. Hardy RA, Crawford JB: Retina. In Vaughan D, Asbury T, Riordan-Eva P (eds): General Ophthalmology, pp 178–199 Stamford, CT, Appleton & Lange, 1999. 222. Ewing CC, Cullen AP: Fluorescein angiography in X-chromosomal maculopathy with retinoschisis (juvenile
hereditary retinoschisis). Can J Ophthalmol 7:19, 1972. 223. Sauer CG, Oehrig A, Warneke-Wittstock K et al: Positional cloning of the gene associated with X-linked juvenile
retinoschisis. Nat Genet 17:164, 1997. 224. Retinoschisis Consorium: Functional implications of the spectrum of mutations found in 234 cases
with X-linked juvenile retinoschisis (XRLS). Hum Mol Genet 7:1185, 1998. 225. Molday LL, Hicks D, Sauer CG et al: Expression of X-linked reinoschisis protein, RS1 in photoreceptors
and bipolar cells. Invest Ophthalmol Vis Sci 42:816, 2001. 226. Manchot WA: Pathology of hereditary juvenile retinoschisis. Arch Ophthalmol 88:131, 1972. 227. Peachey NS, Fishman GA, Derlaci DJ et al: Psychophysical and electroretinographic findings in X-linked juvenile
retinoschisis. Arch Ophthalmol 105:513, 1987. 228. Condon G, Arnwostern S, Wang NS et al: Congenital hereditary (juvenile X-linked) retinoschisis: Histopathological
and ultrastructural findings in three cases. Arch Ophthalmol 104:576, 1986. 229. Gass JD: Müller cell cone, an overlooked part of the anatomy of the fovea centralis: Hypothesis
concerning its role in the pathogenesis of macular
hole and foveomacular schisis. Arch Ophthalmol 117:821, 1999. 230. Mooy CM, van den Born LI, Baarsma S et al: Hereditary X-linked juvenile retinoshcisis: A review of the role
of Müller cells. Arch Ophthalmol 120:979, 2002. 231. Grayson G, Rend SN, Ellis JA et al: Retinoschisin, the X-linked retinoschisis protein, is a secreted
photoreceptor protein and is expressed and released by Wert-Rb 1 cells. Hum Mol Genet 9:1873, 2000. 232. Reid SN, Farber DB: Transport of a photoreceptor-secreted protein, retinoschisin, by
Müller cells. Invest Ophthalmol Vis Sci 42:S647, 2001. 233. Weber BHF, Schrewe H, Molday L et al: Inactivation of the murine X-linked juvenile retinoschisis gene, Rs1h, suggests
a role of retinoschisin in retinal cell layer organization
and synaptic structure. Proc Natl Acad Sci 99:6222, 2002. 234. Eksandh LC, Ponjavie V, Ayyagari R et al: Phenotypic expression of juvenile X-linked retinoschisis in Swedish
families with different mutations in the XLRS1 gene. Arch Ophthalmol 119:1098, 2000. |