As a group, the mucopolysaccharidoses are characterized by a rather distinctive spectrum of clinical manifestations. Skeletal deformity, resulting from changes in both the bones and the joints, is a prominent feature. There is also a characteristic facies with coarse, often somewhat grotesque features. Visceromegaly, cardiac disease, respiratory problems, deafness, and mental deficiency occur in certain of the syndromes. The principal ocular manifestations of the various mucopolysaccharidoses are progressive corneal clouding, pigmentary retinal degeneration, optic atrophy, and in some cases glaucoma (Table 1).
TABLE ONE. The Systemic Mucopolysaccharidoses
| Designation | Metabolic Features | General Clinical Manifestations | Ophthalmologic Manifestations |
| MPS IH: Hurler Syndrome | Profound deficiency of α-L-iduronidase | MPS prototype. Coarse facial features. Severe dysostosis multiplex. Moderate dwarfism, contractures; semi-crouched habitus, kyphoscoliosis, gibbus, claw-hand deformity. Visceromegaly, protruberant abdomen, hernias. Pulmonary and cardiovascular disease. Mental deficiency. Hearing impairment. Early death, usually before age 10 years. | Prominent wide-set eye (shallow orbits, hypertelorism), prominent supraorbital ridges, heavy brows, puffy lids. Prominent corneal clouding, of early onset, progressive and severe. Retinal degeneration; arteriolar attenuation, pigmentary changes, abnormal ERG. Nerve head swelling. Optic atrophy. Progressive vision impairment. In some cases, megalocornea and/or glaucoma. |
| MPS accumulation in virtually every system of the body, producing marked somatic and visceral abnormalities | |||
| Urinary excretion of dermatan sulfate and heparan sulfate | |||
| Autosomal recessive | |||
| MPS IS: Scheie Syndrome (formerly MPS V) | α-L-iduronidase deficiency | Minimal to moderate somatic and visceral signs of MPS, less severe than in Hurler prototype. Somewhat coarse facial features. Stature, habitus relatively normal. Prominent joint stiffness, claw-hand deformity, carpal tunnel syndrome, and aortic valve disease. Intellect normal or nearly normal. Hearing impairment common. Life span relatively normal. | Corneal clouding, early and progressive, often more dense peripherally. “Retinitis pigmentosa-like” retinal degeneration, with progressive vision impairment and reduced ERG. In some cases glaucoma. |
| Urinary excretion of dermatan sulfate and heparan sulfate | |||
| Autosomal recessive | |||
| MPS IH/S: Hurler-Scheie Compound | α-L-iduronidase deficiency | Phenotype intermediate between that of Hurler and Scheie syndrome. Dwarfing, progressive joint stiffness, claw-hand deformity, hypertelorism, progressive coarsening of facial features, micrognathia. Hepatosplenomegaly. Cardiovascular disease. Intellectual impairment. Hearing impairment. Survival into teens or twenties. | Progressive corneal haze and vision impairment. In some cases retinal degeneration. Nerve head swelling. Optic atrophy. Possibly glaucoma. |
| Urinary excretion of dermatan sulfate and heparan sulfate | |||
| Autosomal recessive | |||
| MPS II: Hunter Syndrome | Iduronate sulfatase deficiency | Coarse facial features. Dwarfing and skeletal deformities, similar to but less severe than in Hurler prototype. Hepatosplenomegaly, cardiac and respiratory disease. Hydrocephalus. Motor paralysis in some. Hearing impairment. Nodular or pebbly skin lesions. Rapid psychomotor and physical deterioration and early death (often by age 15 years) in severe form. Slower deterioration and longer survival (even into fifth or sixth decade) in milder form. | Progressive retinal degeneration, usually severe, with pigmentary changes, arteriolar attentuation, disc pallor, vision impairment, and abnormal ERG. Often nerve head swelling. Possibly glaucoma. Clinically clear cornea, but microscopic changes reported. |
| Urinary excretion of dermatan sulfate and heparan sulfate. | |||
| X-linked recessive | |||
| MPS III: Sanfilippo Syndrome | Four biochemically different forms: | Clinical manifestations similar in all four forms. Early and severe progressive mental deterioration. Less severe somatic changes. Coarse facial features, megalocephaly, hypertelorism. Moderate skeletal changes. Hepatosplenomegaly. | Corneas clinically clear; some microscopic changes reported. Attentuation of retinal arterioles, some pigmentary changes, vision impairment, and ERG changes documented. Possibly optic atrophy. Rarely nerve head swelling. |
| Type A: Heparan N-sulfatase deficiency | Survival into third decade. | ||
| Type B: α-N-Acetylglucosaminidase deficiency | |||
| Type C: Acetyl-Co: α-Glucosaminide-N-acetyl transferase deficiency | |||
| Type D: N-Acetyl- glucosamine 6-sulfatase deficiency | |||
| Urinary excretion of heparan sulfate in all four forms | |||
| Autosomal recessive | |||
| MPS IV: Morquio Syndrome | Two biochemically different forms | In classic form (IV A), severe dwarfing, skeletal dysplasia, with kyphosis, sternal bulging. Joint laxity rather than stiffness. Odontoid hypoplasia, atlantoaxial instability; spinal cord and medullary compression, long tract signs and respiratory paralysis may occur. Prominent joints, knock-knees. Semicrouching stance, waddling gait. Somewhat coarse facial features. Hypoplastic dental enamel. Occasionally hepatomegaly. Protruberant abdomen. Cardiopulmonary complications. Hearing impairment. Intellect normal or mildly impaired. In milder variant (IV B) findings similar to those of classic form, but often less severe dwarfism, less tendency to atlantoaxial instability, and usually normal dental enamel. | Corneal clouding, usually mild or fine haze, in A and B. Subcortical lens opacities in A. Fundi usually normal; in some cases optic atrophy, disc blurring, arteriolar narrowing, reduced scotopic ERG. |
| N-acetylgalactosamine 6-sulfatase deficiency in classic form (MPS IV A) | |||
| β-galactosidase deficiency in later onset variant (MPS IV B) | |||
| Urinary excretion of keratan sulfate in both forms | |||
| Autosomal recessive | |||
| MPS V: (Vacant, Formerly Scheie) | |||
| MPS VI: Maroteaux-Lamy Syndrome | N-acetylgalactosamine-4-sulfatase (arylsulfatase B) deficiency | Striking dwarfism and skeletal deformities. | Progressive corneal clouding, often dense, usually evident within first few years of life. Graft may accumulate MPS. Papilledema, abducent palsy secondary to hydrocephalus. Optic atrophy. Retinal vascular tortuosity. Typically no signs of retinal degeneration, but pigmentary and ERG-VEP changes noted in milder variant. |
| Urinary excretion of dermatan sulfate | Coarse facial features. Visceromegaly and cardiac disease. Atlantoaxial subluxation, spinal cord compression, hydrocephalus in some cases. Normal intellect. Granular inclusions in circulating leukocytes. Milder variants occur. | ||
| Autosomal recessive | |||
| MPS VII: Sly Syndrome | β-glucuronidase deficiency | Variable, often moderate, clinical manifestations. Coarse facial features. Short stature and skeletal deformity. Hepatosplenomegaly, hernias, cardiovascular and respiratory problems. Intellectual impairment. Inclusions in circulating lymphocytes. | Corneal clouding in some cases. Nerve head swelling also documented. |
| Urinary excretion of dermatan sulfate and heparan sulfate | |||
| Autosomal recessive |
To date, deficiency of ten specific lysosomal enzymes has been demonstrated in the various mucopolysaccharidoses. All are recessively inherited; one mucopolysaccharidosis, MPS type II (Hunter syndrome), is X-linked; the others are autosomal.
The diagnosis of the various mucopolysaccharidoses is made on the basis of the distinguishing clinical features, the presence of excessive mucopolysaccharide substances in tissue and urine, and demonstration of the enzyme deficiency using fibroblasts, leukocytes, or serum. Prenatal diagnosis is also possible by analysis of cultured amniotic fluid cells or chorionic villi. Identification of heterozygotes is becoming more available. There has been some success in altering the course of certain of the mucopolysaccharidoses by bone marrow transplantation. Some improvement in the ocular findings after marrow transplantation has been documented.1
In reviewing the mucopolysaccharidoses, reference should be made to comprehensive discussions of the clinical, pathologic, biochemical, and genetic features of these disorders.2–5
MPS TYPE IH: HURLER SYNDROME
Hurler syndrome (MPS IH) is the prototype of the mucopolysaccharidoses. The disorder is severe and progressive. There is accumulation of acid mucopolysaccharide in virtually every system of the body, producing both somatic and visceral abnormalities and leading to early death, usually by age 10 years.
In MPS IH there is profound deficiency of α-L-iduronidase, with excessive urinary excretion of both dermatan sulfate and heparan sulfate in a ratio of approximately 70:30. The disorder is autosomal recessive. It occurs in many races and is probably the most frequent of the mucopolysaccharidoses. The gene encoding α-L-iduronidase, previously assigned to chromosome 22, has been localized to chromosome 4 p 16.3.
Manifestations develop in infancy and early childhood and become more apparent with increasing age. The head tends to be large and misshapen. Scaphocephaly due to premature closure of the sagittal suture is common; there is often a prominent longitudinal ridge along the sagittal suture. The facial features characteristically are coarse and the expression dull (Fig. 1). Hypertelorism is usual and the orbits are shallow; the eyes appear wide-set and prominent. The lids tend to be puffy, the brows prominent. The nose is broad, with wide nostrils and a flat bridge. The ears may be large and low-set. The lips usually are patulous; the tongue is large and protuberant. The teeth generally are small, stubby, and widely spaced; the gums are hyperplastic.
|
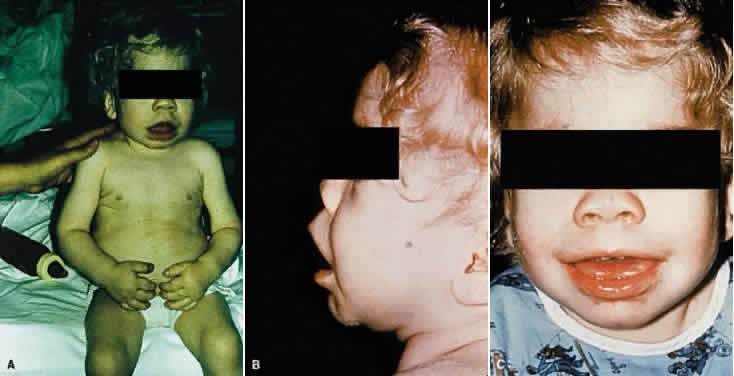
Characteristically there are marked skeletal changes. Moderate dwarfism, short neck, kyphoscoliosis, and gibbus are typical. On plain film examination, the vertebral bodies (particularly those of the lower dorsal and upper lumbar region) are wedge-shaped with an anterior hooklike projection referred to as beaking. The extremities are short, the hands and feet are broad, and the phalanges are short and stubby. Radiologically, the tubular bones show expansion of the medullary cavity and thinning of the cortex. The terminal phalangeal bones commonly appear hypoplastic. The joints are stiff and flexion contractures develop; clawlike deformity of the hands is especially characteristic. The posture is semicrouching and the gait is awkward.
Thoracic deformity is another regular feature of the syndrome; the chest appears large and wide with flaring of the lower ribs over the abdomen. On plain film examination, the ribs appear spatulate or saber-shaped. Typically, the medial end of the clavicle is widened. The many radiologic findings in this condition are commonly described by the term dysostosis multiplex.
The abdomen is protuberant owing to abnormalities in supporting tissues and to visceromegaly. As a rule there is enlargement of both the liver and spleen. Diastasis recti, umbilical hernia, and inguinal hernias are common.
The skin tends to be thick. There is usually generalized hypertrichosis.
Manifestations of cardiac involvement, including murmur, angina, myocardial infarction, and congestive heart failure, are common. Pathologic changes in the heart due to mucopolysaccharide deposition can be extensive. The great vessels and peripheral vessels also are affected.
Respiratory problems develop in virtually every patient. Recurrent upper respiratory tract infection, bronchitis, and chronic nasal congestion are common, and the patients almost always are noisy mouth breathers. A number of factors, including deformity of the facial and nasal bones, narrowing of the passages, abnormalities of the tracheobronchial cartilage, and deposition of mucopolysaccharide in the lungs, contribute. In addition to the abnormalities of the respiratory passages and lungs, cardiac disease and thoracic deformity may contribute to respiratory difficulties.
The principal neurologic manifestation is mental deficiency. There may also be motor signs. Pathologic changes can be found throughout the nervous system. Hydrocephalus may develop. A special feature in some cases is the presence of leptomeningeal cysts. Shoe-shaped deformity of the sella is common. The optic foramina also may be enlarged.
Deafness is frequent; it may be of mixed or sensorineural type. Middle ear infections are common.
The course is one of progressive mental and physical deterioration. Death most frequently results from cardiac or respiratory disease.
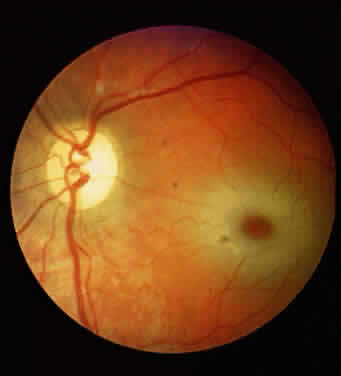
The principal ophthalmologic manifestations of MPS IH are progressive corneal clouding, retinal degeneration, optic atrophy, and vision loss. It appears that the corneal and retinal changes relate somewhat to the pattern of mucopolysacchariduria—that is, corneal changes are greater in those conditions characterized by higher levels of dermatan sulfate in the urine, as in Hurler syndrome. The retinal degeneration appears to correlate with the degree of heparan sulfaturia; the retinal changes are more severe in Hunter and Sanfilippo syndromes, less in Hurler syndrome.6,7
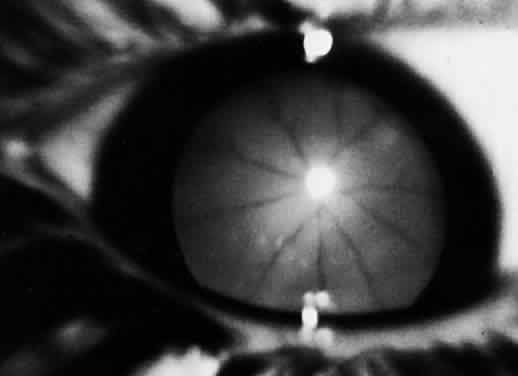
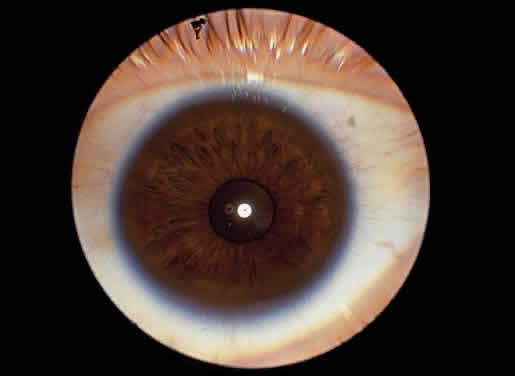
Corneal clouding was recognized early to be an important feature of this disorder, and classic clinical descriptions of the corneal changes are to be found in the older literature.8–12 Clouding of the cornea is usually evident by age 2 to 3 years, often by age 1 year; in some cases it is seen at birth.12,13 Photophobia is a common early symptom. With time there is progression from a generalized haziness or steamy appearance to a dense, milky ground-glass opacification (Fig. 2). On slit-lamp examination, one sees fine granular opacities in the corneal stroma, often increasing in density from the anterior stroma and subepithelial region to the posterior stromal layers.10–12
|

In 1939, Berliner provided what is probably the first significant histopathologic study of the eye in Hurler syndrome, describing the corneal changes in detail. He found large vacuolated cells under Bowman's layer, fragmentation of Bowman's layer, separation of the corneal lamellae, and deposits of granular material in the stromal spaces.10 Subsequent reports confirmed these findings, and in 1944 Hogan and Cordes noted in addition fine granules in the cytoplasm of the corneal corpuscles.14–16 Later studies documented these histopathologic changes and provided further evidence for mucopolysaccharide accumulation in the cornea.13,17–19 The epithelium may be intact or may show edema and cytoplasmic vacuolization, with accumulation of metachromatic material in and around cells. Bowman's layer usually shows thinning and lamellar splitting or fragmentation, with infiltration of vacuolated cells containing metachromatic material. In the stroma there is swelling and vacuolization of keratocytes, intracellular and extracellular deposition of metachromatic material, and lamellar separation. Descemet's membrane and endothelium usually are described as normal, although cytoplasmic vacuolization and metachromatic staining of the endothelium have been noted. Histochemical techniques provide evidence for mucopolysaccharide accumulation in the vacuolated cells. In their ultrastructural study of the eyes in two cases of MPS IH, Chan and coworkers documented the presence of numerous fibrillogranular inclusions in corneal epithelium, keratocytes, and endothelium, the presence of multimembranous inclusions in keratocytes, and the presence of extracellular fibrillogranular material in corneal stroma.20
Progressive corneal clouding may prevent visualization of the fundus, but signs of retinal involvement and optic atrophy have been documented in Hurler syndrome. Gills and coworkers reported the absence of the foveal reflex and optic atrophy in several patients.21 In addition, the fundus appeared “albinoid” in one, and the retinal arterioles were narrowed in another. The electroretinogram (ERG) is abnormal, usually markedly reduced, in Hurler syndrome.21–23
Mailer also emphasized the association of optic atrophy in Hurler syndrome and reviewed the possible causes.19 It would seem that the optic atrophy can be secondary to any of the following, singly or in combination: mucopolysaccharide infiltration, hydrocephalus, retinal degeneration, and even glaucoma. Papilledema also has been observed, in some cases in association with hydrocephalus.16,24
With regard to retinal degeneration and optic atrophy, related histopathologic findings include enlargement and vacuolization of cells of the nuclear layer of the retina, vacuolization of the ganglion cells, atrophy of the optic nerve, and thickening and infiltration of the arachnoid with foam cells.13,17,19 By electron microscopy, Chan and coworkers documented the presence of fibrillogranular inclusions in retinal pigment epithelium and ganglion cells and multimembranous inclusions in retinal ganglion cells and optic nerve astrocytes.20
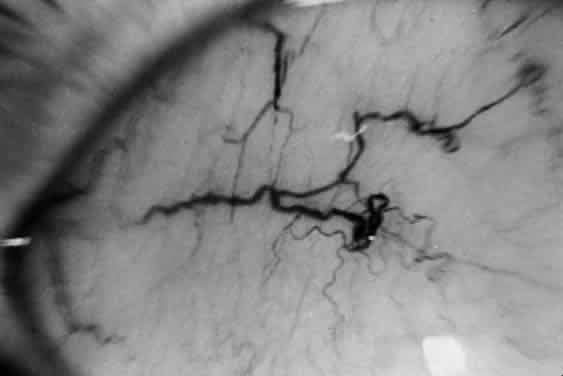
In addition to corneal, retinal, and optic nerve changes, there may be histopathologic evidence of mucopolysaccharide accumulation in the epithelium of the ciliary body, in the walls of the iris capillaries, in the sclera, and in the conjunctiva.13,17,19 Ultrastructural changes have been found in uveal melanocytes and fibrocytes, ciliary epithelium, smooth muscle cells of ciliary body, pericytes, trabecular endothelium, lens epithelium, and sclerocytes.20 Conjunctival biopsy had been used as a diagnostic procedure. The characteristic ultrastructural changes and histochemical reactions of mucopolysaccharidosis have been well documented in conjunctival specimens.20,25 The findings include the presence of single membrane-limited cytoplasmic vacuoles containing fibrogranular material in epithelium, vacuoles containing predominantly fibrogranular material and occasionally membranous lamellar inclusions in fibroblasts and histiocytes in subepithelial connective tissue, vacuolization of the lymphatic endothelium and perithelial cells, and membranous lamellar vacuolization of the Schwann cells of the conjunctival nerves. There is evidence that the fibrogranular vacuoles and the membranous lamellar vacuoles are lysosomes that contain, respectively, accumulated acid mucopolysaccharide and glycolipid.
Megalocornea has been described in many cases; in most cases intraocular pressure has been normal, but glaucoma has been documented in some patients with MPS IH.9–13,26,27 Electron microscopy of the trabeculectomy specimen from a child with Hurler syndrome and open-angle glaucoma showed membrane-bound cytoplasmic inclusions consistent with mucopolysaccharide accumulation in cells of the corneoscleral junction and iris stroma.28
Progressive impairment of vision is usual, secondary to corneal clouding, retinal degeneration, and optic atrophy, singly or in combination; glaucoma, the effects of cerebral mucopolysaccharide accumulation, and the development of hydrocephalus may also contribute. In view of the extensive systemic and neurologic abnormalities in MPS IH, the poor prognosis for life, and the probability of concurrent retinal degeneration and optic atrophy, corneal transplant in an effort to improve vision had not often been recommended in the past. However, the possibility of altering the course of Hurler syndrome with treatment may alter the prospects for successful keratoplasty. Corneal transplantation several years after successful bone marrow transplantation in a child with MPS IH has been reported; Huang and colleagues in 1996 documented the ultrastructural changes in the corneal specimen.29
Whereas Rosen and coworkers described keratoplasty and ultrastructural changes of the cornea in a patient with “Hurler's disease,” it is doubtful that the patient had MPS type IH.30 In the case of a successful keratoplasty in a patient with “atypical mucopolysaccharidosis” reported by Gollance and D'Amico, it is difficult to determine the type of mucopolysaccharidosis involved.31
As mentioned in the description of the facies, the eyes tend to be wide-set and prominent owing to hypertelorism and shallow orbits, the lids tend to be puffy, and the brows are heavy and the lashes coarse. These features are seen to some degree in other mucopolysaccharidoses also.
MPS TYPE IS: SCHEIE SYNDROME
MPS type IS was first described by Scheie, Hambrick, and Barness in their classic study of ten patients reported in 1962.18
In the Scheie syndrome, as in the Hurler syndrome, there is deficiency of the lysosomal enzyme α-L-iduronidase and urinary excretion of both dermatan sulfate and heparan sulfate. The condition is autosomal recessive. The predominant manifestations are corneal clouding, joint stiffness, claw-hand deformity, carpal tunnel syndrome, and aortic valve disease, principally aortic stenosis and regurgitation. The facial features are coarse; the mouth is broad. Other somatic and visceral changes characteristic of mucopolysaccharidosis tend to be minimal. Stature is normal, and the patients do not develop the distorted habitus characteristically seen in Hurler syndrome. Intellect is normal or nearly normal, although psychiatric disturbances have been reported. There may be hearing impairment. The life span is relatively normal.
Histopathologic changes are similar to those of the prototype MPS IH, but in MPS IS the cortical neurons appear normal.
Corneal clouding is a prominent manifestation of the Scheie syndrome.18 Developing early in life, sometimes present at birth, the corneal clouding tends to worsen with age and may ultimately interfere with vision. The corneal involvement is diffuse but tends to be most dense peripherally. Clinically the hazy cornea may appear enlarged, edematous, and thickened, initially raising suspicion of glaucoma, particularly when telltale somatic signs of mucopolysaccharidosis are minimal.
Scheie and coworkers showed that the pathologic corneal and conjunctival changes were similar to those in Hurler syndrome and presented evidence for the presence of acid mucopolysaccharide in the abnormal vacuolated cells of these tissues.18 Subsequently, Quigley and Goldberg and others described the ultrastructural changes of the conjunctiva in Scheie syndrome, documenting the presence of single membrane-limited vacuoles containing granulofibrillar material and occasional membranous inclusions in the conjunctival fibroblasts and similar vacuolization of epithelial cells.32 Kenyon and associates showed similar changes and demonstrated histochemical reactions for acid mucopolysaccharide in several of the mucopolysaccharidoses, including Scheie syndrome.25
A study by Quantock and colleagues suggests that variation in collagen fibril diameter in corneal stroma, in addition to light scattering from glycosaminoglycan deposits, may contribute to corneal clouding in Scheie syndrome.33
Corneal transplants have been tried with little success.18
In most reported cases, the ocular pressure has been normal or in the upper range of normal, but in some cases glaucoma has been documented.34,35
Although retinal changes have not been documented in all reported cases,18,36 retinal degeneration is a recognized feature of the Scheie syndrome.2,21–23,32,35 Manifestations include vision impairment, particularly progressive night blindness, field changes such as ring scotoma, retinal pigmentary changes (“RP-like”), and subnormal or extinguished ERG responses. Reduced corneal sensitivity also has been noted.34
MPS TYPE I H/S: HURLER-SCHEIE SYNDROME
A number of patients having features intermediate between those of the Hurler and the Scheie syndromes have been reported.2,37–39 As in MPS IH and MPS IS, in MPS I H/S there is deficiency of α-L-iduronidase and urinary excretion of both dermatan sulfate and heparan sulfate. The histopathologic changes are those of mucopolysaccharide accumulation in connective tissue throughout the body, as well as in parenchymal cells of the liver and brain.
The prominent clinical manifestations are skeletal changes (dysostosis multiplex) with dwarfing and progressive joint stiffness, scaphocephaly, hypertelorism, and progressive coarsening of facial features. In addition, a receding chin (micrognathia) appears to be a distinctive feature. Other manifestations include hepatosplenomegaly, pulmonary and cardiovascular involvement, mental retardation, and hearing impairment. Significant manifestations (destruction of the sella, cerebrospinal fluid rhinorrhea, and loss of vision) related to the presence of arachnoid cysts have also been reported in MPS I H/S. Patients with the Hurler-Scheie syndrome may survive into the teens or twenties.
As in both MPS IH and MPS IS, corneal clouding occurs in MPS I H/S.2,37–39 The corneal haze is diffuse (sometimes more dense peripherally) and progressive; it may be evident in childhood and ultimately may interfere with vision. Keratoplasty has been tried; a lamellar graft in one patient with MPS I H/S had remained clear for 4 years at the time of the report.38
Retinal degeneration also occurs in MPS I H/S, although the true incidence is unknown because corneal clouding may obscure the fundus findings. Chijiiwa and associates reported two patients with night blindness, decreased visual acuity, constricted visual fields, and reduced ERG; both had retinal pigmentary changes with scattered spicules and arteriolar attenuation.40 Jensen and associates also had documented fundus and ERG abnormalities.41 In their ERG study, Caruso and colleagues found variable abnormalities in MPS I H/S.23
Blurring of the disc margins has been noted, and in one case this finding was associated with increased intracranial pressure, with dilatation of the ventricles, enlargement of the sella, and pathologic documentation of mucopolysaccharide accumulation in the central nervous system.42
Mullaney and coworkers reported chronic angle-closure glaucoma in an 11-year-old boy with MPS I H/S.43 Electron microscopy of tissues obtained at trabeculectomy showed the presence of vacuoles containing fibrillogranular material consistent with mucopolysaccharide deposition; there was marked intracellular and extracellular vacuolar formation in the iris, scattered vacuolar formation in sclera and Tenon's capsule, and little extracellular or intracellular vacuolar formation in trabecular meshwork.
MPS TYPE II: HUNTER SYNDROME
In Hunter syndrome (MPS II), the metabolic defect is deficiency of the lysosomal acid hydrolase iduronate sulfatase. There is urinary excretion of both dermatan sulfate and heparan sulfate in a ratio of approximately 1:1. In contrast to the other mucopolysaccharidoses, MPS II is X-linked recessive. Its locus has been mapped to Xq27-28.
Phenotypically Hunter syndrome closely resembles the Hurler prototype. The manifestations of MPS II, however, are generally less severe than those of MPS IH, and Hunter syndrome is distinguished clinically by longer survival and the absence of gross corneal clouding.
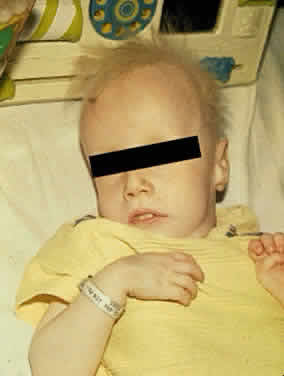
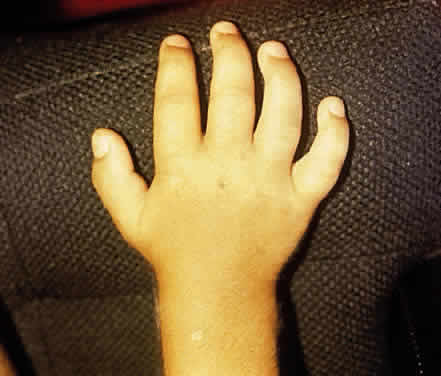
In Hunter syndrome, the facial features are coarse, the supraorbital ridges tend to be prominent, the tongue is large, and the teeth are widely spaced. Dwarfing and stiffness of the joints are prominent features. Claw-hand deformity is common (Fig. 3). Lumbar gibbus may develop but is usually not severe. As a rule there is hepatosplenomegaly. The abdomen is protuberant. Hernias are common. Cardiac involvement is a regular feature of the syndrome; congestive heart failure and coronary artery disease are major causes of death. Respiratory disability also is evident in most patients. Neurologic manifestations vary. Spastic quadriplegia may develop from impingement on the cervical spinal cord. Hydrocephalus may develop. Mental deterioration occurs, but the severity and the rate of regression vary. Progressive deafness occurs in most patients.
|
A distinctive feature of Hunter syndrome is the occurrence of nodular or pebbly ivory-colored skin lesions, most frequently on the back extending from the inferior angle of the scapula toward the axillary line, less often in the pectoral area, nape of the neck, and lateral aspect of the upper arms and thighs. Adults with Hunter syndrome also tend to have a rosy or ruddy complexion.
Within Hunter syndrome there is a broad spectrum of severity.44,45 At least two major clinical forms, differentiated primarily on the basis of central nervous system involvement, are recognized. Patients with the more severe form, type A, show more rapid neurologic deterioration and usually die before age 15 years. The milder form, type B, is characterized by slower mental deterioration and is compatible with survival into the fifth or sixth decade of life.
In contrast to MPS IH, obvious corneal clouding is not a regular feature of MPS II.46,47 However, slight corneal changes may be detected by slit-lamp examination in older patients with Hunter syndrome, and histologic evidence of corneal mucopolysaccharide accumulation has been reported.48,49 Spranger and colleagues documented clinically visible corneal opacities in a child with severe MPS II, in addition to fine corneal opacities in a young adult with mild MPS II.50
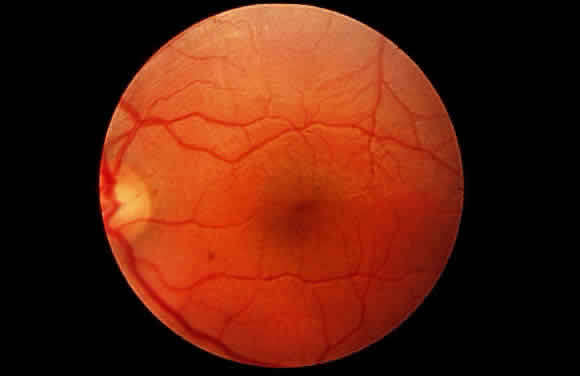
The principal ophthalmologic manifestation of MPS II is progressive retinal degeneration with attendant impairment of vision.21,51 Night vision problems and visual field defects are common. The disorder may lead to blindness. The fundus signs include retinal pigmentary changes, sometimes spicule formation, retinal arteriolar attenuation, and optic disc pallor. The ERG is usually reduced or extinguished21,22,52; in some cases it is normal.22 In addition, bilateral epiretinal membranes with tortuosity of the retinal vessels has been reported as an unusual finding in two brothers with Hunter syndrome type B.53
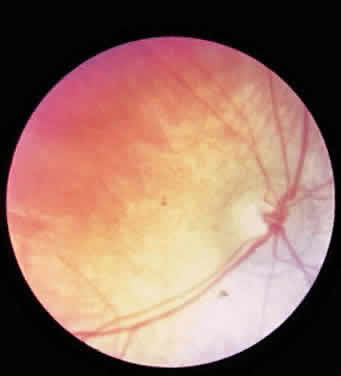
Swelling of the nerve head is a frequent finding in Hunter syndrome; it may be due to increased intracranial pressure or mucopolysaccharide deposition in and around the optic nerve.44,54–56
In their light and electron microscopic study of the eye in type A Hunter syndrome, McDonnell and coworkers found single membrane-bound structures containing fibrillogranular and multimembranous material in conjunctival epithelium, pericytes, and fibrocytes; corneal epithelium, keratocytes, and endothelium; trabecular endothelium; iris pigment epithelium, smooth muscle, and fibrocytes; ciliary pigmented and nonpigmented epithelium and fibrocytes; retinal pigment epithelium and ganglion cells; optic nerve astrocytes and pericytes; and sclerocytes.57
In their histopathologic study of the eye by light microscopy in type B Hunter syndrome, Goldberg and Duke found few corneal abnormalities.48 The corneal epithelium and Bowman's layer were intact, except peripherally where Bowman's layer was split and where eosinophilic material was present beneath the epithelium. Descemet's membrane and endothelium were intact, although eosinophilic granules were present in the endothelial cytoplasm. Fine granular deposits were present in the corneal stroma, chiefly in interlamellar spaces. The nonpigmented epithelium of the ciliary processes appeared foamy. There were significant retinal changes, including pigment migration, paucity of pigment epithelial cells, diminution of rods and cones, reduction in number of ganglion cells, and gliosis of the nerve fiber layer. The sclera was thickened.
In their electron microscopy study of the same patient, Topping and coworkers found fibrillogranular vacuoles and membranous lamellar vacuoles in various tissue of the eye, although the ultrastructure of the cornea was relatively unaltered.49 The nonpigmented epithelium of the ciliary processes was engorged with membranous fibrillogranular vacuoles, and similar vacuoles were present in choroid and scleral fibroblasts. Similar but fewer inclusions of this type were found within the corneal keratocytes and the pigmented epithelium of the ciliary processes. There were membranous lamellar vacuoles in the retinal ganglion cells and in migrated pigment epithelial cells. Some membranous lamellar vacuoles were also present in keratocytes, choroidal and scleral fibroblasts, and nonpigmented epithelium of the ciliary processes. The content of the fibrillogranular vacuoles is probably mucopolysaccharide; that of the membranous vacuoles is probably glycolipid.
Kenyon and coworkers found similar ultrastructural changes in their study of conjunctiva and by histochemical techniques confirmed acid mucopolysaccharide in several of the mucopolysaccharidoses, including Hunter syndrome.25 Kaiden and associates reported chronic angle-closure glaucoma in an adult patient with Hunter syndrome.58 Spranger and colleagues also documented glaucoma in MPS II type B.50
MPS TYPE III: SANFILIPPO SYNDROME
The Sanfilippo syndrome (MPS III), sometimes referred to as polydystrophic oligophrenia, is a mucopolysaccharidosis in which there is severe mental retardation and relatively less severe somatic abnormalities. Four biochemically different but clinically indistinguishable forms of the syndrome occur: in type A there is deficiency of heparan N-sulfatase; in type B there is deficiency of α-N-acetylglucosaminidase; in type C there is deficiency of acetyl-CoA: α-glucosaminide-N-acetyltransferase; and in type D there is deficiency of N-acetyl glucosamine 6-sulfatase, the gene for which has been localized to chromosome 12q14. In all forms there is urinary excretion of heparan sulfate. All forms are autosomal recessive. Mental retardation, the predominant clinical manifestation of MPS III, usually becomes evident in the first few years of life. With increasing age there is progressive deterioration of intellect and behavior. Because the patients usually are strong, management often becomes a problem as they regress; many require institutionalization.
Somatic abnormalities typical of mucopolysaccharidosis tend to be mild or inconspicuous. There is some coarseness of facial features. Synophrys is usual. Generalized hirsutism may be marked. Dwarfing, joint stiffness, and claw-hand deformity are usually evident but are not as severe as in the Hurler prototype. Radiologically, the skeletal changes of dysostosis multiplex are relatively mild. Slight to moderate hepatosplenomegaly develops, and the abdomen tends to be protuberant. Respiratory difficulties are common. Heart involvement may occur but tends to be less severe than in other mucopolysaccharidoses. Hearing loss is common in moderate to severely affected patients. Hydrocephalus also may develop.59
Corneal clouding does not occur in MPS III, although microscopic changes were noted in one of Sanfilippo's patients, and histologic corneal and scleral changes were reported in another patient subsequently.60,61 Jensen found vacuoles and accumulation of granular material in sclera and, to a lesser degree, in cornea and ciliary body; the histochemical findings were believed to be consistent with accumulation of acid mucopolysaccharide.61 In their histopathologic study of the conjunctiva in the various mucopolysaccharidoses, Kenyon and coworkers also found the characteristic light and electron microscopic changes in Sanfilippo syndrome and by histochemical techniques demonstrated accumulation of acid mucopolysaccharide.25
Retinal involvement and progressive vision loss may occur in MPS III. Narrowing of the retinal vessels and pigmentary changes have been noted.21,22,25 Subnormal ERG responses have been recorded in both types A and B.21–23 Optic atrophy may develop.22,62 Significant histopathologic changes of the retina and perineural connective tissue, in addition to signs of mucopolysaccharide accumulation in many other parts of the eye, in Sanfilippo syndrome type A were first documented by Del Monte and colleagues.63 Phase contrast and electron microscopy showed intracellular accumulation of fibrillogranular and membranous lamellar vacuoles in cornea, trabecular meshwork, iris, lens, ciliary body, sclera, retinal ganglion cells, retinal pigment epithelium, and optic nerve glia. There was retinal pigment epithelial hyperplasia and hypopigmentation, vascular attenuation, and marked photoreceptor loss, closely resembling that found in inherited retinitis pigmentosa. Clinically the patient had signs of pigmentary retinal degeneration and optic atrophy, with vision loss and no recordable ERG; the corneas were clear.
Ceuterick and associates also found membrane-bound electron-lucent inclusions in the retinal ganglion cells and photoreceptors of a 22-week-old fetus with type A Sanfilippo disease; there were also vacuoles containing granular material in other cells and tissues of the eye.64
In their study of the histopathologic and ultrastructural changes in the eye in Sanfilippo type B, Lavery and coworkers found cytoplasmic single membrane-bound vacuoles containing the major storage product, acid mucopolysaccharide, in virtually every ocular tissue.65 There were also lamellar cytoplasmic membranous bodies of complex lipid, the minor storage product, mainly in retinal ganglion cells and lens epithelium. In addition, many tissues contained inclusions of an intermediate type, composed of combined fibrillogranular and lamellar membranous material. There was photoreceptor cell degeneration similar to that seen in some forms of retinitis pigmentosa.
MPS TYPE IV: MORQUIO SYNDROME
The syndrome that bears Morquio's name is characterized by severe dwarfism and skeletal deformity, often with neurologic complications, and a number of extraskeletal abnormalities such as corneal clouding and aortic valve disease. In this mucopolysaccharidosis (MPS IV), there is defective degradation of keratan sulfate. There is excessive urinary excretion of keratan sulfate, although the amount of keratan sulfate in the urine tends to diminish as affected patients grow older.
As with Sanfilippo syndrome, clinically similar but enzymatically different forms of Morquio syndrome occur. The designation MPS IV-A is used to denote classic Morquio syndrome, in which the enzyme defect is a deficiency of N-acetylgalactosamine 6-sulfatase; the gene encoding this enzyme has been localized to chromosome 16q24. The designation MPS IV-B is used to denote a later-onset variant of Morquio syndrome in which the enzyme defect is a deficiency of β-galactosidase. Both mild and severe forms of MPS IV-A and MPS IV-B occur. All forms are autosomal recessive.
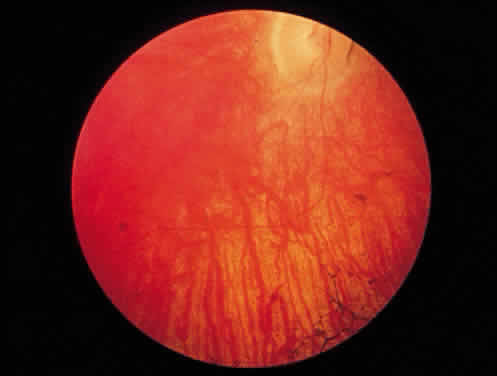
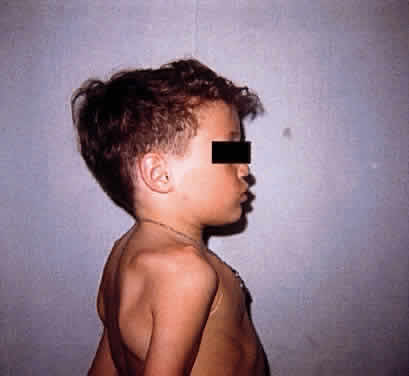
Patients with Morquio syndrome appear normal in the first months of life, although radiographic signs may be present early. With growth during the first years of life, abnormalities such as retarded growth, knock knees, flat feet, prominent joints, dorsal kyphosis, sternal bulging, flaring of the rib cage, and awkward gait become evident. The deformities worsen with age. Affected persons characteristically are markedly dwarfed and develop a semicrouching stance. Joint stiffness is not a feature, however; rather, joints may be excessively loose, leading to instability. Barrel chest and pigeon breast deformity are common (Fig. 4). The neck typically is short. The face is abnormal, with somewhat coarse features, a broad-mouthed appearance, prominent jaw, and widely spaced teeth. The dental enamel is often thin, giving the teeth a grayish appearance and leading to flaking and fracturing of the enamel and multiple cavities. In some cases aortic regurgitation develops. Progressive hearing loss occurs in almost all patients. Invariably there is absence or severe hypoplasia of the odontoid process, and there is usually ligamentous laxity of the spinal column. Atlantoaxial subluxation and spinal cord and medullary compression are frequent complications; manifestations may be acute, subacute, or chronic, and subtle or severe, ranging from minimal long tract signs to spastic paraplegia, respiratory paralysis, and death. In general the course is one of progressive incapacitation. Intelligence usually is normal or mildly impaired.
|
Corneal clouding is a feature of Morquio syndrome,66 although some of the earlier literature would suggest otherwise.26 The corneal clouding in Morquio syndrome is relatively mild, having the appearance of a fine haze rather than the dense ground-glass opacification common to Hurler syndrome. The changes may not become clinically evident to the unaided eye for several years, often not before age 10 years. In the early stages the corneal involvement may be overlooked unless careful slit-lamp examination is performed. The biomicroscopic appearance is that of diffuse involvement of the stroma with punctate or granular opacities but usually sparing of the epithelium, Bowman's layer, and endothelium. Depending on the density of the corneal haze, there may be some impairment of vision but usually not of a severe degree. Corneal clouding is seen in MPS IV-B as well as in MPS IV-A.67,68
In their histologic examination of the cornea in Morquio syndrome, Ghosh and McCulloch found intracytoplasmic vacuolization of keratocytes and the presence of fibrillar, granular, and lamellar substance having histochemical properties consistent with those of acid mucopolysaccharide.69 Subsequently, Iwamoto and associates documented the presence of fibrillogranular and multimembranous membrane-bound inclusions distributed primarily in the cornea and trabecular meshwork, to a milder degree in the conjunctiva and sclera, and sparsely in the retinal pigment epithelium.70 Intracytoplasmic vacuoles limited by single-unit membranes containing fine fibrillogranular material characteristic of the mucopolysaccharidoses have also been documented in conjunctival biopsy.68
Olsen and coworkers found cataracts in addition to corneal stromal clouding in three siblings with Morquio syndrome (MPS IV-A).71 On slit-lamp examination there were innumerable small spherical grayish opacities of about identical size subcortically, in a zonular arrangement; the nuclei and lens capsules were clear. The lenses and corneas of the parents and healthy siblings were clear.
Fundus abnormalities have been reported infrequently in Morquio syndrome. Optic atrophy has been noted.72 In a review of reported fundus changes in Morquio syndrome, Dangel and Tsou made reference to blurring of the discs.73 They described narrowing of the retinal arterioles in an adult with Morquio syndrome and documented electrophysiologic changes: the light-adapted ERG was normal, but the dark-adapted ERG was reduced and the electro-oculogram was slightly abnormal in one eye.73 Abraham and coworkers also found slight scotopic abnormalities.74 In most cases the fundi and light-adapted ERG are normal in patients with MPS IV.21,22 Von Noorden and coworkers documented mesodermal anomalies in one patient with MPS IV.66 Mydriasis attributed to sympathetic involvement in Morquio syndrome has also been mentioned.73
MPS TYPE VI: MAROTEAUX-LAMY SYNDROME
The Maroteaux-Lamy syndrome (MPS VI) is characterized by severe dwarfism, visceromegaly, cardiac lesions, and progressive corneal clouding. In some cases hydrocephalus and spinal cord compression develop. Resembling the prototype mucopolysaccharidosis in many ways, the Maroteaux-Lamy syndrome is distinguished by retention of normal intellect, the pattern of mucopolysacchariduria, and the enzyme defect. In MPS VI there is deficiency of N-acetylgalactosamine-4-sulfatase (arylsulfatase B), with urinary excretion of predominantly dermatan sulfate. The gene encoding N-acetylgalactosamine 4-sulfatase has been localized to chromosome 5q 13-q14. Metachromatic granulation of circulating leukocytes is a characteristic finding in MPS VI. The disorder is autosomal recessive. In addition to the classic form of Maroteaux-Lamy, milder variants associated with the same enzyme deficiency are described.
In the severe or classic form of MPS VI, growth retardation affecting both the trunk and limbs is usually evident by age 2 or 3 years. Genu valgum, lumbar kyphosis, and anterior sternal protrusion develop. The lower ribs are flared. Joint movement is restricted. Claw-hand deformity develops; carpal tunnel syndrome is common. The head appears relatively large. The facial features tend to be coarse. There is often mild hypertrichosis. As a rule, hepatomegaly develops in patients older than 6 years of age, splenomegaly develops in about half the patients, and the abdomen usually is protuberant. Cardiac involvement, particularly valve lesions, similar to that of Hurler syndrome may develop. Deafness occurs in some patients.
The principal neurologic complications are hydrocephalus and spinal cord compression secondary to atlantoaxial subluxation consequent to hypoplasia of the odontoid process. Survival is variable; most patients with the severe form of MPS VI die by the second or third decade.
The principal ophthalmologic manifestation of MPS VI is progressive corneal clouding, usually evident within the first few years of life. The appearance is that of ground-glass haze distributed diffusely throughout the stroma, sometimes denser peripherally, and usually of sufficient degree to be seen grossly.75,76 In addition to the stromal opacities, some epithelial and endothelial changes may be seen on slit-lamp examination.76 Corneal opacities have been documented in the mild variant of MPS VI as well in the severe form.77 A decrease in corneal clouding after bone marrow transplantation has been noted.78 In what appears to be the first reported histopathologic study of the eye in Maroteaux-Lamy syndrome, Kenyon and coworkers in 1972 described changes typical of mucopolysaccharidosis.76 On light microscopy they found cytoplasmic vacuolization of corneal epithelium, interruption of Bowman's layer with accumulation of foamy histiocytes, swelling of keratocytes with foamy cytoplasm and separation of stromal lamellae, some cytoplasmic vacuolization of corneal endothelium, but essentially no alteration of Descemet's membrane. Other findings included thickening of sclera with vacuolated cells between the fibers, vacuolated cells in the trabecular meshwork, ballooned histiocytes, vacuolated fibrocytes in connective tissue stroma of the ciliary body, involvement of the basal portion of nonpigmented ciliary epithelium, and some changes in the choroid. By histochemical techniques, they documented accumulation of acid mucopolysaccharide in the affected cells and tissues. The retina appeared normal except for the macular area, where reduction of the ganglion cell population and thinning of the nerve fiber layer were noted. The optic nerve showed atrophy and secondary gliosis. Electron microscopy confirmed the presence of single membrane-limited vacuoles containing predominantly fibrillogranular material, some containing polymorphous material and membranous lamellae, in the cornea, sclera, trabecular meshwork, and uveal tract, but not in the retina.
Similar corneal changes have been found in the mild phenotype of MPS VI.79 Schwartz and associates documented the recurrence of mucopolysaccharide accumulation in the repeat corneal graft specimens from two patients with the mild form of MPS VI.80
Goldberg and coworkers reported the occurrence of papilledema and abducent palsy secondary to the increased intracranial pressure of hydrocephalus in a child with Maroteaux-Lamy syndrome.75 In addition, they documented tortuosity of the retinal vessels not only in the child with papilledema but also in her siblings, whose optic discs were normal. Sheridan and Johnston also documented the occurrence of papilledema secondary to hydrocephalus in Maroteaux-Lamy.56
Except for the above-mentioned disc changes and retinal vascular tortuosity, the fundi in MPS VI generally are normal. As a rule, patients with Maroteaux-Lamy syndrome do not develop ophthalmoscopic signs of pigmentary retinal degeneration, and the ERG usually is normal.76,79 However, in an adult patient with a mild variant of Maroteaux-Lamy with typical diffuse corneal clouding, DiFerrante and colleagues documented alternating areas of hypopigmentation and hyperpigmentation in the parapapillary region, with reduced A-wave response on ERG and increased latency on visual-evoked responses.81
MPS TYPE VII: SLY SYNDROME
In the Sly syndrome (MPS VII), there is deficiency of β-glucuronidase, leading to a block in the degradation of dermatan sulfate and heparan sulfate, with urinary excretion of both dermatan and heparan sulfate. The disorder is autosomal recessive. The human gene encoding β-glucuronidase is localized on chromosome 7q 21.1-q22.
Clinical manifestations within the syndrome vary. The spectrum includes many of the characteristic features of mucopolysaccharidosis, including short stature, progressive skeletal deformity and radiologic signs of dysostosis multiplex, coarse facial features, hypertelorism, hepatosplenomegaly, diastasis recti, protuberant abdomen, hernias, intellectual impairment, cardiovascular involvement, and respiratory problems. Reported patients have shown coarse inclusions in circulating leukocytes. A severe neonatal form associated with fetal hydrops has been reported.
In some patients with Sly syndrome the corneas are clear.82–86 Within the phenotypic variation of this disorder, however, corneal clouding may occur; this may be evident grossly or only on slit-lamp examination.87 The patient initially reported by Sly as having clear corneas subsequently developed progressive corneal clouding.82,88 In their postmortem study of this patient, Vogler and associates found vacuolated cytoplasm in nonpigmented ciliary epithelium, in corneal fibrocytes, and in lens epithelium; the retinal pigment epithelium was not vacuolated.88