THE TRISOMY SYNDROMES
Trisomy or monosomy for autosomes other than 13, 18, 21, and 22 are almost invariably lethal and are frequently seen in spontaneous abortions.
Trisomy 13 (Patau Syndrome)
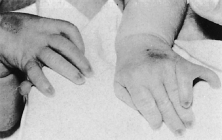
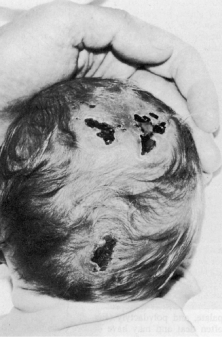
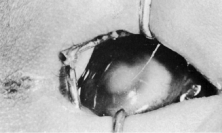
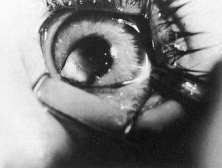
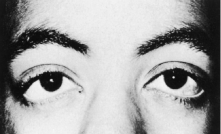
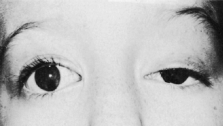
Trisomy 13 occurs among newborns at a frequency of 1 in 20,000.11 High maternal age is a factor predisposing to trisomy 13, as it is for other trisomies. About 45% of affected infants die before the age of 1 month with only 5% reaching the age of 3 years, and there are only 5 reports of patients surviving beyond the first decade.12 Wide phenotypic variability in trisomy 13 is the rule and may be secondary to the variable allelic content of the three homologous chromosomes. Partial trisomy for different parts of chromosome 13 has been described. Patients with these partial trisomies have only some of the features of trisomy 13. A clinical chromosomal map has been constructed by matching common clinical features with common regions of chromosome involvement. Clinical features of trisomy 13 include severe mental retardation resulting from holoprosencephaly, cleft lip and palate, and polydactyly (Fig. 1). These patients are often deaf and may have capillary hemangiomas and scalp defects (Fig. 2). The heel is often prominent and the bottom of the foot has the shape of the runners on a rocking chair.13 The ocular features in trisomy 13 range from anophthalmia to microphthalmia. A fleshy pterygium-like corneal opacification may be seen (Fig. 3), and iris and choroidal colobomas are common (Fig. 4). Hypertelorism occurs in almost all cases.14
|
|
|
|
|
|
|
|
Trisomy 18 (Edward's Syndrome)
Trisomy 18 occurs among newborns at a frequency of 1 in 6000.15 High maternal age is a factor predisposing to trisomy 18, and about 90% of affected infants die before the age of 6 months with only about 5% reaching the age of 1 year. A nearly diagnostic feature for this syndrome is the positioning of the thumb and fingers in a tightly clenched fist. The thumb is flexed inside the fist with the index finger overriding the third digit. Infants with trisomy 18 have a characteristic constellation of clinical features that includes mental retardation, hypertonicity, cardiac defects, low-set malformed ears, and flexion deformities.16 Ocular malformations are commonly found in trisomy 18 patients with hypertelorism described in 87% of patients, and prominent epicanthal folds, microphthalmia/anophthalmia, and ptosis seen in about one third of cases. Other less commonly described findings include globe abnormalities such as corneal opacity, anterior segment abnormalities, cataract, and optic nerve pits and colobomas.17
Trisomy 21 (Down Syndrome)
Down syndrome, the most frequent form of mental retardation caused by a microscopically demonstrable chromosomal aberration, is characterized by well-defined and distinctive phenotypic features and natural history; Down syndrome occurs in 1/800 newborns.18 Trisomy 21 is the most common of all the trisomy syndromes and is the result of aneuploidy involving the smallest human chromosome; 75% of these embryos are aborted spontaneously. Incidence increases with maternal age; the risk of having a liveborn with Down syndrome at maternal age 30 is 1 in 1000 and at maternal age 40 is 9 in 1000.19
The characteristic face of a child with trisomy 21 exhibits epicanthus, oblique palpebral fissures, a protruding tongue, a flat nasal bridge, and low malformed ears. Individuals with Down syndrome have specific major congenital malformations (found in 30% to 40% of patients) such as those involving the heart, particularly the atrioventricular canal, and the gastrointestinal tract. Patients with Down syndrome also have between 10 to 20 times greater risk of leukemia than the chromosomal normal population. In particular, there is an increase of 200 to 400 times the incidence of acute megakaryocytic leukemia. The affected child is mentally retarded and has shortened anteroposterior (AP) skull diameter; short stature; a typical dermatoglyphic pattern that consists of a simian line, distal palmar axial triradius, and a high frequency of ulnar loops; and a life expectancy that is reduced to 71% at 30 years. In the most common form of trisomy 21, there are three free copies of chromosome 21; in about 5% of patients one copy is translocated to another acrocentric chromosome, most often chromosome 14 or 21.20
Eye findings can be extensive and include Brushfield spots, which are circumferentially distributed white spots on the iris. Brushfield spots are a hallmark of Down syndrome and are found in one third of patients. Similar lesions may be present in the chromosomally normal individuals in whom they are referred to as Wofflin nodules. Lens changes are present in the Down syndrome population with approximately 300-fold greater incidence than the general population (11%). Epicanthal folds with short or sparse eyelashes are present in more than 50% of patients. Other eye abnormalities include strabismus (57%), esotropia (90%) greater than exotropia (5%), and frequently associated A or V patterns. Extremes in refractive errors are prevalent in the form of myopia (23%), hyperopia (21%), and astigmatism (22%). Nystagmus has a reported incidence of 29% without specified type. Keratoconus develops in some 15% of Down syndrome patients, and nasolacrimal duct obstruction is present in 15% of patients.21
Trisomy 22
Trisomy 22 is a rare disorder with only 20 reported cases. It is lethal in the immediate postpartum period. Mean maternal age at the time of birth is 30.5 years.22 All of the patients have obvious mental and physical retardation. The most common congenital abnormalities include microcephaly, preauricular skin tags, congenital heart disease, micrognathia, a long philtrum, malformed ears, and a long nose. At least 50% of the patients had cleft palate, malposed thumb and long slender fingers, congenitally dislocated hips, and hypotonia. As many as 30% have hypoplastic or lowset nipples. Cryptorchism was noted in all males. The most common ocular malformations are chorioretinal colobomas and microphthalmia. Other less common features include strabismus and antimongoloid lid slant.23
PARTIAL TRISOMY AND PARTIAL DELETION SYNDROMES
Partial trisomies and monosomies can be viable depending on the size and location of the aberration. These can arise de novo or as a result of unbalanced segregation from a parent carrying a balanced rearrangement. These phenotypes are more variable than those of complete trisomies because the amount of unbalanced genetic material is variable. Nevertheless, several phenotypes emerge as characteristics of extra or missing chromosomal segments in particular regions. Before beginning the discussion of the partial trisomy and partial deletion syndromes, it should be stressed that the exact phenotype associated with a chromosomal deletion or duplication will depend on how much of the chromosome is affected and on which genes are deleted or duplicated. The clinical phenotypes discussed here are useful diagnostically, however, because patients with a deletion or duplication of one arm of a particular chromosome are likely to share certain clinical abnormalities. It is quite possible that two patients, both with a partial duplication or deletion of one arm of a chromosome, share no common clinical features if the affected chromosomal segments do not overlap.
Partial trisomies and partial deletion syndromes with consistent ocular findings are outlined in the following discussion. Partial trisomies or partial deletion syndromes without significant ocular findings or those with isolated reports of eye findings are not included.
3q Plus Syndrome (Cornelia de Lange Phenotype)
Trisomy of a segment of the distal end of chromosome 3 is uncommon. The exact site of duplication lies within the region 3q23 to 3q27 in one of the number 3 chromosomes resulting in a partial trisomy for the distal segment. In most cases, the partial trisomy is the result of a balanced translocation in one parent. Because of these findings, careful banding study should be carried out in all patients with the Cornelia de Lange phenotype. These patients share many of the clinical features described for Cornelia de Lange's syndrome: “carp” mouth, failure to thrive, profound mental retardation, a long philtrum, microcephaly, and congenital heart disease.24 Ocular features include most commonly long eyelashes, hypertelorism, and prominent epicanthal folds.25
4p Plus Syndrome
The clinical features of the 4p+ syndrome are sufficiently consistent to permit clinical diagnosis at birth. In the young child, the face is round, there is a short nose with aplasia the nasal root, and the glabella protrudes over a hypoplastic nasal root. The upper lip is elongated and projects slightly; the chin is pointed and recessed. The ears are low set and usually abnormal. Skeletal deformities of the extremities may be present. Other findings include microcephaly and kyphoscoliosis. Height is usually considerably below normal, and obesity is common. Retardation is severe and may be complicated by seizures.26 Ocular features include iris and choroidal colobomas.27
4p Minus Syndrome
The 4 p- syndrome has an incidence of 1/50,000 live births.31 Children are feeble and hypotonic at birth. Major systemic manifestations include developmental delay (100%), growth retardation (90%), cleft lip and palate, congenital heart disease, and seizures. Simian creases, cryptorchism, and hypospadias are less common.28 Ocular abnormalities play a major role in the phenotypic findings and include strabismus (50%), iris coloboma (30%), and epicanthal folds in addition to hypertelorism, which is a marked feature in patients with the 4p- syndrome (70%).29,30
4q Plus Syndrome
Microcephaly is a consistent feature in the 4q+ syndrome, and the nasal crest continues with a prominent receding forehead. The upper lip is short, and when the mouth is closed both lips protrude in a very distinct manner. Duplication or absence of digits is common. Growth retardation is common. Mental retardation is severe and is frequently complicated by seizures. The ocular features include narrow palpebral fissures, ptosis, epicanthus, and corneal leukomas.31
5p Plus Syndrome
5p+ syndrome is an extremely rare syndrome, which is lethal before 6 months of age. Major manifestations include macrocephaly, low-set posteriorly rotated ears, cleft palate, and short philtrum. Ocular manifestations include primarily hypertelorism and telecanthus.32
5p Minus Syndrome (Cri du Chat Syndrome)
The mewing cat cry of babies with 5p- syndrome, from which the French name for this disorder is derived, becomes less pronounced with increasing age. Patients are unusually chafe as infants, and virtually all affected babies are of low birth weight, exhibit slow growth, and have microcephaly. IQ is severely affected and is usually in the 20 to 30 range. Hypertelorism and a large prominent glabella are present in more than 90% of patients. An antimongoloid lid fissure is present in more than 80% of patients, and epicanthal folds, as well as strabismus, are also common. Aside from the catlike cry and retarded growth, the ocular features are the most consistent findings.33
6p Minus Syndrome
Major malformations in the 6p- syndrome include craniosynostosis (70%) and congenital heart disease (90%), which are hallmarks of the disease. Eye abnormalities occur in about 65% of patients, and microphthalmia, anterior segment dysgenesis, iris colobomas, and prominent epicanthal folds have been reported.34
6q Minus Syndrome
Systemic manifestations of the 6q- syndrome include developmental delay, umbilical hernia, palmar creases, microcephaly, short stature, congenital heart disease, and ectopic kidneys. Prominent ophthalmic findings include downslanting small palpebral fissure (82%) and prominent epicanthal folds (58%).35
7q Plus Syndrome
There are two clinically distinct phenotypes involving duplications of different portions of the long arm of chromosome 7. The first syndrome is caused by deletion of the terminal portion of the long arm and is characterized by low birth weight, growth and mental retardation, cleft palate, micrognathia, a small nose, small palpebral fissures, and hypertelorism. The other 7q+ phenotype is caused by a more proximal duplication in the long arm. The clinical picture for this includes retarded development, hypertelorism, strabismus, and low-set ears. In both, hypertelorism is a prominent ocular feature.27
Trisomy 8 (Warkany Syndrome)
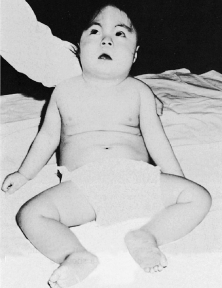
Trisomy 8 is associated with total duplication of one of the medium-sized autosomes. Most of the cases described have been mosaics; that is, they have both normal and abnormal chromosomal cell lines. Most patients are retarded with an IQ that ranges from 10 to 80. Skeletal abnormalities are frequently present including accessory ribs, vertebral defects, absent patella, and a distinctive toe posture with digits two and three having an equal length. Dermatoglyphics show deep furrows in the palms and soles.36 Hypertelorism was noted in 12.5% of patients, and a pronounced antimongoloid lid slant has been observed (33%) (Fig. 5). Less frequent findings related to the eye include ptosis, strabismus, corneal opacity, microphthalmia, coloboma, and heterochromia.36
|
|
9p Minus Syndrome
In the reported cases of 9p- syndrome, the patients' features are similar enough to allow clinical description. Trigonocephaly, microcephaly, a long upper lip, down-turned corners of the mouth, and a significant protrusion of antihelices are systemic features37 (Fig. 6). The ocular findings include glaucoma, enlarged and highly arched eyebrows, prominent eyes, epicanthal folds, and a mongoloid lid slant.38,39
|
|
9q Plus Syndrome
In the reported cases of 9q+ syndrome, only global delay is a consistent feature. Strabismus, epicanthus, and deep-set eyes are reported ocular findings.40
10p Plus Syndrome
The most common systemic findings associated with this syndrome include hypotonia, frontal bossing, abnormal nares, and clubfoot, all of which occur in greater than 60% of cases.41 Ocular findings are uncommon with this syndrome and include microphthalmia and coloboma.42
10q Plus Syndrome
10q+ syndrome is characterized by a high prominent forehead, an oval face, growth retardation, microcephaly, a small nose with a depressed nasal bridge and anteverted nostrils, a carp-shaped mouth with a prominent upper lip, close-set ears, a short neck, and undescended testicles. Skeletal defects involving the digits of the hands and feet, as well as cardiac defects, are occasionally present. Antimongoloid lid slant, hypertelorism, arched and widespread eyebrows, blepharophimosis, and microphthalmia and ptosis have also been described.43
10q Minus Syndrome
In 10q- syndrome, developmental delay is the rule with associated midfacial hypoplasia, prominent receding forehead, and hypotonia. Various dermatologic lesions are associated with this syndrome, although lesions frequently do not present until puberty. As with many partial deletion syndromes, hypertelorism and midfacial hypoplasia are prevalent ocular findings.44
11p Plus Syndrome
The developmental abnormalities in the few cases of the 11p+ syndrome that have been described are due primarily to disturbance of craniofacial development. Systemic findings include a high, prominent forehead with frontal upsweep of hair, wide glabella, a broad flat nasal bridge, and cleft lip or palate. Psychomotor retardation and hypotonia are the rule for this disorder. Ocular findings include supraorbital ridges antimongoloid palpebral fissures, as well as strabismus and nystagmus.45
11p Minus Syndrome
Wilms' tumor, aniridia, genitourinary abnormalities, and mental retardation (WAGR) syndrome is associated with a deletion in the short arm of chromosome 11 involving PAX6 at 11p11.3.46 Systemic manifestations of WAGR syndrome include hypotonia, midline brain abnormalities, and hearing deficits. The heterogeneity in the aniridia deletion syndrome is secondary to small variations in the amount of genetic material deleted in patients. Both males and females are affected with the 11p- syndrome, and nearly all cases have been sporadic. Gonadoblastoma with aniridia has been reported in children with the 11p- syndrome. The fact that dominantly inherited aniridia generally is not associated with Wilms' tumor or other abnormalities is not surprising, because linkage studies have shown that the locus for the mild familial form of aniridia is on chromosome 1.47 Wilms' tumor is usually associated with the sporadic form of aniridia. Aniridia is the most consistent eye finding, although others including cataracts, foveal hypoplasia, nystagmus, and glaucoma are also prevalent.48
11q Minus Syndrome (Jacobsen Syndrome)
This uncommon disorder has an incidence of less than 1 in 100,000 births.49 Developmental delay and trigonocephaly are the most common systemic abnormalities occurring in greater than 95% of patients afflicted with the syndrome.50 Micrognathia, carp-shaped mouth, cardiac abnormalities, and skeletal abnormalities are also common. Commonly reported ocular findings include hypertelorism, ptosis, and prominent epicanthal folds.51,52
13q Plus Syndromes
When the entire chromosome is duplicated, the well-described trisomy 13 syndrome occurs. There are at least two groups of patients with different clinical pictures who show partial duplication of chromosome 13.53 In the first group, the trisomy consists of duplication of the proximal one third to one half of the long arm of chromosome 13. The clinical features are nonspecific and include psychomotor retardation, microcephaly, low-set ears, microsomia, micrognathia, and clinodactyly with little ocular involvement. When the distal one third to two thirds of the long arm is present in triplicate, many of the features of complete trisomy 13 are present. In this group, unlike in the complete trisomy syndrome, deafness, eye malformations, cleft palate, harelip, and heart defects are not observed.
13q Minus Syndrome
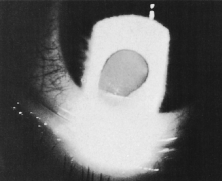
Chromosome 13 is of particular interest to ophthalmologists because when a portion of the long arm is deleted, retinoblastoma may occur. A review of patients with partial deletion of chromosome 13 showed that the presence of hypoplastic thumbs indicates involvement of the distal segment of the long arm of chromosome 13. Other clinical features of the distal segment deletion included mental retardation, microcephaly, and hypertelorism in approximately 85% of cases.54 Additional findings included a protruding maxilla, epicanthal folds, renal abnormalities, heart disease, metacarpal fusion, aplastic thumbs, microphthalmia, coloboma, and large low-set ears. A number of the patients with 13q minus syndrome had retinoblastoma subsequently found to be associated with a deletion that included a band in the proximal portion of the long arm.55 Although most of the children whose retinoblastoma is associated with the deletion of the long arm of chromosome 13 have other congenital abnormalities, some appear to be normal except for a mild developmental delay. The severity of the associated abnormalities depends on the amount of chromosomal material lost (Fig. 7). Among patients with retinoblastoma, approximately 3% have a chromosome 13 deletion. Because both unilateral and bilateral retinoblastoma have been observed in patients with deletion of chromosome 13, it may be virtually impossible to determine clinically whether a chromosomal deletion might be present. For these reasons, when observing a new patient with retinoblastoma, ophthalmologists should search diligently for evidence of other developmental abnormalities. Even in the absence of any systemic developmental defects, however, one cannot be certain that the chromosome deletion does not exist.56,57
|
|
14q Plus (Proximal and Distal) Syndrome
The 14q plus (distal) syndrome is one of two partial trisomy syndromes involving the long arm of chromosome 14. Mental retardation is severe. Other abnormalities include microcephaly, a high forehead, low-set ears, a highly arched or cleft palate, a protruding upper lip, camptodactyly, micrognathia, congenital heart disease, and 12th rib hypoplasia.58 Ocular features include epicanthal folds and antimongoloid slant. Prominent features of the 14q plus (proximal) syndrome include motor and mental retardation, seizures, hypotonia, low anterior hairline, low-set ears, a prominent philtrum, long tapered fingers, kyphosis, and a short neck. Ocular features include antimongoloid lid slant, small palpebral fissures, microphthalmia, strabismus, and ptosis.58
15q Minus Syndrome
The Prader-Willi syndrome and Angelman syndrome are associated with deletions of the long arm of chromosome 15. Systemic findings in Prader-Willi syndrome include hypotonia, developmental delay, and small extremities, as well as hypogonadism and cryptorchidism. Angelman syndrome has the systemic findings of Prader-Willi syndrome with more significant developmental delay including seizures.59 Ocular findings are not a large part of these syndromes.
16p Minus Syndrome
This rare deletion syndrome is most commonly associated with developmental delay and alpha thalassemia. Other systemic findings include capillary hemangiomas and midfacial hypoplasia. As with many other deletion syndromes, hypertelorism and prominent epicanthal folds are common eye findings.60
17p Minus Syndrome (Smith-Magenis Syndrome)
The most characteristic physical findings of Smith-Magenis syndrome include short stature, midfacial hypoplasia, and brachydactyly, as well as developmental delay and profound mental retardation.61 Ocular findings are inconsistent and include most commonly prominent epicanthal folds, with scattered reports of Brushfield type spots and high myopia.62
18p Minus Syndrome
Clinical features of the 18p- syndrome include mental retardation, growth delay, and abnormal ears (see Fig. 7). Myopia, antimongoloid lid fissures, cataract, and uveal colobomas are reported to be present27 (Fig. 8).
|
|
18q Minus Syndrome (DeGrouchy Syndrome)
DeGrouchy syndrome is perhaps the most common of all the partial trisomy and partial deletion syndromes. More than 100 cases have been studied and reported. Systemic features include low birth weight, profound mental retardation, developmental delay, short stature, microcephaly, midface dysplasia, carp-shaped mouth, widely spaced nipples, long tapering fingertips, and irregular external ears.63 Conspicuous skin dimples are present over the sides of the patella and the back of the hands. Ocular abnormalities include nystagmus (30%) and bilateral optic atrophy (29%), as well as strabismus, glaucoma, pigmentary retinal degeneration, and ocular colobomas.27,64
20p Plus Syndrome
Patients with partial trisomy of the short arm of chromosome 20 are almost always thought to be normal at birth. The development of prominent cheeks and a short chin characterizes the facies. Telecanthus is present in 50% of cases, and there is a mongoloid like lid slant in 75% of cases.45
20p Minus Syndrome
Prominent physical findings in this condition include triangular chin with prominent forehead and long straight nose. Systemic associations are cholestasis, hemivertebrae, and pulmonary tree stenosis. Ocular features such as hypertelorism and deep-set eyes, as well as anterior segment digenesis including posterior embryotoxon and iris adhesions, are common.45 Alagille syndrome is believed to be a variant of 20p minus syndrome involving a single gene site, 20p11.23–12.65
22q minus Syndrome (DiGeorge Syndrome)
DiGeorge syndrome occurs in 1/4000 live births. The hallmark systemic findings include conotruncal heart anomalies, hypoplastic thymus, and variable developmental delays. Ophthalmic findings have been well studied and include posterior embryotoxon (69%), tortuous retinal vessels (58%), and upper lid hooding (41%) among others.66
The Cat-Eye Syndrome (Supernumerary Chromosome 22q)
The cat-eye syndrome is associated with a supernumerary marker derived from chromosome 22q. However, in many of the reported cases, the marker is present in only a portion of the patient's cells. Because mosaicism is sometimes transmitted through generations, Mendelian factors may be important in its causation.67 Although variability of clinical features is enormous, the usual triad necessary for the diagnosis of the cat-eye syndrome is made up of uveal coloboma, an imperforate anus, and renal malformations. The disorder was named the cat-eye syndrome because of the characteristic vertical iris colobomas that are frequently present. Additional systemic findings are preauricular skin pits, fistulas, or tags and congenital heart disease.68 Although the disorder has been described with a normal karyotype, the usual finding is the presence of a small, extra submetacentric chromosome. Variable features of the syndrome may be present in other family members. The exact nature of the extra fragment in all cases is not agreed on, and it is clear that the extra fragment is mitotically unstable and may retard cell division during the critical period of embryogenesis when the fetal fissure would normally be closing.
Several reports suggest that the coloboma, which is a consistent feature of trisomy 13, might be contributed to the cat-eye syndrome by the portion of chromosome 13 that is near the centromere. That portion of the cat-eye syndrome that is similar to trisomy 22 might be contributed by the portion of chromosome 22 near the centromere. A balanced translocation carrier state may explain those families in which generations seem to be skipped.69
The minimal criteria for making the diagnosis of cat-eye syndrome are as follows: (1) combination of two major features, coloboma and anal atresia, with or without associated abnormalities; (2) combination of one major feature, coloboma or anal atresia, with at least one of the most frequently associated minor anomalies, such as preauricular skin tags or renal anomalies; (3) the presence of one major feature plus several less frequently found abnormalities, such as antimongoloid lid slants, congenital heart disease, and skeletal anomalies; and (4) a combination of five or more minor specific features. There is some overlap in clinical features between trisomy 22 and the cat-eye syndrome.70