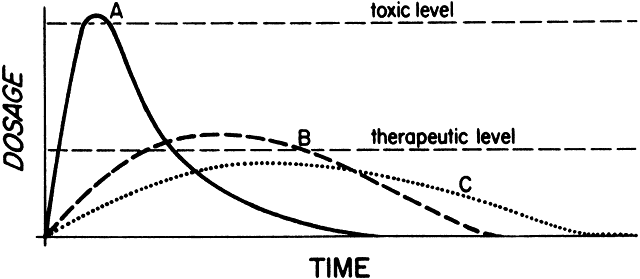
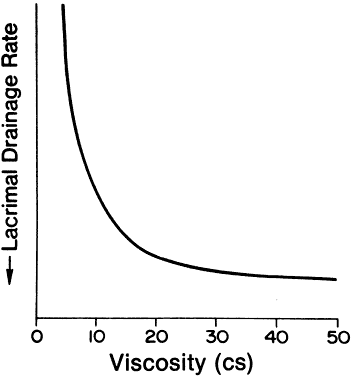
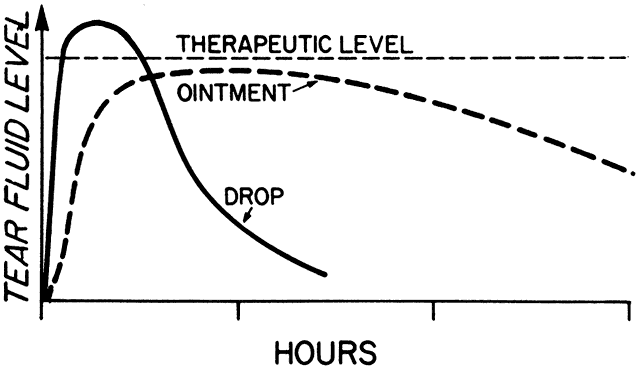
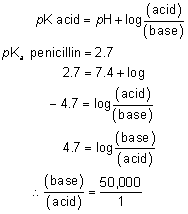1. Francke DE: Pharmacokinetic and biopharmaceutic terminology (editorial). Drug Intell Clin Pharm 11:304, 1977 2. Zathurecky L: Progress in developing a standard terminology in biopharmaceutics and pharmacokinetics. Drug Intell Clin Pharm 11:281, 1977 3. Consensus Workshop: Is there a need for more precise definitions of bioavailability? Eur
J Clin Pharmacol 40: 123, 1991 4. Smolen VF, Murdock HR, Williams EJ: Bioavailability analysis of chlorpromazine in humans from pupillometric
data. J Pharmacol Exp Ther 195:404, 1975 5. Kuehn PB, Jhawar AK, Wiegand WA et al: Pharmacokinetics of chlorpromazine-induced miotic response in rabbits. J Pharm Sci 65:1593, 1976 6. Jacob JT, Plein EM: Factors affecting dissolution rate of medicaments from tablets. II. Effect
of binder concentration, tablet hardness, and storage conditions on
the dissolution rate of phenobarbital from tablets. J Pharm Sci 57: 802, 1968 7. Lovering EG, Mainville CA, Rowe ML: Drug permeation through membranes. V. Interaction of diazepam with common
excipients. J Pharm Sci 65:207, 1976 8. Baugh R, Calvert RT, Fell JT: Stability of phenylbutazone in presence of pharmaceutical colors. J Pharm Sci 66:733, 1977 9. Levy G: Comparison of dissolution and absorption rates of different commercial
aspirin tablets. J Pharm Sci 50: 388, 1961 10. Fencher JH: Particle size of drugs and its relationship to absorption and activity. J Pharm Sci 57:1825, 1968 11. Reinhold JG, Phillips FJ, Flippin HF: A comparison of the behavior of microcrystalline sulfadiazine with that
of ordinary sulfadiazine in man. Am J Med Sci 210:141, 1965 12. Glazko AH, Kinkel AW, Alegnani WC et al: An evaluation of the absorption characteristics of different chloramphenicol
preparations in normal human subjects. Clin Pharmacol Ther 9:472, 1968 13. Juncher H, Raaschou F: The solubility of oral preparations of penicillin. V. Antibiot Med Clin Ther 4:497, 1957 14. Mesley J, Clements RL: Infrared identification of barbiturates with particular reference to the
occurrence of polymorphism. J Pharm Pharmacol 20:341, 1968 15. Summers VP, Carles JE, Enever RP: The polymorphism of aspirin. J Pharm Pharmacol 22:615, 1970 16. Umeda T, Ohnishi N, Yokoyama T et al: Physicochemical properties and isothermal transition of acetazolamide polymorphs. Chem Pharm Bull 33:3422, 1985 17. Poole J: Physicochemical factors influencing the absorption of the anhydrous and
trihydrate forms of ampicillin. Curr Ther Res 10:292, 1968 18. Poole JW: Drug formulation and biologic availability. Semin Drug Treatment 1:148, 1971 19. Koch-Weser J: Drug therapy: Bioavailability of drugs (second of two parts). N Engl J Med 291:503, 1974 20. US Food and Drug Administration: Holders of Approved New Drug Applications
for Drugs Presenting Actual or Potential Bioequivalence Problems, publication
No. (FDA) 76-3006. Rockville, MD, US Department of Health, Education
and Welfare, 1976 21. Ellis PP, Price PK, Kelmenson R et al: Effectiveness: Of generic acetazolamide. Arch Ophthalmol 100:1920, 1982 22. Schoenwald RD, Garabedian ME, Yakatan GJ: Decreased bioavailability of sustained release acetazolamide dosage forms. Drug Dev Indust Pharm 4:599, 1978 23. Straughn AB, Gollamudi R, Meyer MC: Relative bioavailability of acetazolamide tablets. Biopharm Drug Dispos 3:75, 1982 24. Syversen GB, Morgan JP, Weintraub M et al: Acetazolamide-induced interference with primidone absorption. Arch Neurol 34:80, 1977 25. Edelman BA, Contractor AM, Shangraw RF: The stability of hypodermic tablets of nitroglycerin packed in dispensing
containers. J Am Pharm Assoc 11:30, 1971 26. Mitra AK, Mikkelson TJ: Ophthalmic solution buffer systems. I. The effects of buffer concentration
on the ocular absorption of pilocarpine. Int J Pharm 10:219, 1982 27. Boberg-Ans J, Grove-Rasmussen KV, Hammarlund ER: Buffering technique for obtaining increased physiologic responses from
alkaloidal eye drops. Br J Ophthalmol 43:670, 1959 28. Wang ESN, Hammarlund ER: Corneal absorption reinforcement of certain mydriatics. J Pharm Sci 59:1559, 1970 29. Smith SA, Smith SE, Lazare R: An increased effect of pilocarpine on the pupil by application of the drug
in oil. Br J Ophthalmol 62:314, 1978 30. Saari M, Koskela P, Masar S: Effect of vehicle on pilocarpine-induced ocular hypotension. Acta Ophthalmol 56: 489, 1978 31. Riegelman S, Vaughan DG: A rational basis for the preparation of ophthalmic solutions. Surv Ophthalmol 3:471, 1958 32. Apel P, Horsch W: Untersuchungen zur Vorbereitung der industriellen Produktion von Pilocarpin: Augentropfen. Pharmazie 31:657, 1976 33. Hass JS, Merrill DL: The effect of methylcellulose on responses to solutions of pilocarpine. Am J Ophthalmol 54: 21, 1962 34. Blaug SM, Canada AT Jr: Relationship of viscosity and prolongation of contact time in methylcellulose
containing ophthalmic solutions. Am J Hosp Pharm 22:662, 1965 35. Chrai SS, Robinson JR: Ocular evaluation of methylcellulose vehicle in albino rabbits. J Pharm Sci 63:1218, 1974 36. Adler CA, Maurice DM, Paterson ME: The effect of viscosity of the vehicle on the penetration of fluorescein
into the human eye. Exp Eye Res 11:34, 1971 37. Smolen VF, Schoenwald RD: Drug absorption analysis from pharmacological data. III. Influence of polymers
and pH on transcorneal biophasic availability and mydriatic response
of tropicamide. J Pharm Sci 63:1582, 1974 38. Waltmann SR, Patrowicz TC: Effects of hydroxypropylmethylcellulose and polyvinyl alcohol on ocular
penetration of topical fluorescein in man. Invest Ophthalmol 9:966, 1970 39. Bach PC, Riddel G, Miller C et al: The influence of vehicles on neomycin sulfate prevention of experimental
ocular infection in rabbits. Am J Ophthalmol 69:659, 1970 40. Krishna N, Brow F: Polyvinyl alcohol as an ophthalmic vehicle: Effect on regeneration of corneal
epithelium. Am J Ophthalmol 57:99, 1964 41. Swanson AA, Jeter DJ, Cregor CR: The influence ophthalmic vehicle on 3H thymidine in normal rabbit corneas. Ophthalmologica 156:425, 1968 42. Patton TF, Robinson JR: Ocular evaluation of polyvinyl-alcohol vehicle in rabbits. J Pharm Sci 64:1312, 1975 43. Lawrence CA: Chemical preservatives in ophthalmic solutions. Am J Ophthalmol 39:385, 1955 44. Smolen VF, Clerenger JM, Williams EJ et al: Biophasic ability of ophthalmic carbachol. I. Mechanisms of cationic polymer
and surfactant promoted miotic activity. J Pharm Sci 62:958, 1973 45. Tonjum AM: Permeability of rabbit corneal epithelium to horseradish peroxidase after
the influence of benzalkonium chloride. Acta Ophthalmol 53:335, 1975 46. O'Brien CS, Swan KC: Carbaminoylcholine chloride in the treatment of glaucoma simplex. Arch Ophthalmol 27:253, 1942 47. Ginsburg M, Robson JM: Further investigations on the action of detergents on the eye. Br J Ophthalmol 33:574, 1949 48. von Sallmann L, Meyer K: Penetration of penicillin in the eye. Arch Ophthalmol 31:1, 1944 49. Green KG, Tonjurn A: Influence of various agents on corneal permeability. Am J Ophthalmol 72:897, 1971 50. Keller N, Moore D, Carper D et al: Increased corneal permeability induced by the dual effects of transient
tear film acidification and exposure to benzalkonium chloride. Exp Eye Res 30:203, 1980 51. Krogh A, Lund CG, Pedersen-Bjergaard K: The osmotic concentration of human lacrimal fluid. Acta Physiol Scand 10:88, 1945 52. Riegelman ER, Vaughan DG Jr et al: Compounding ophthalmic solutions. J Am Pharm Assoc 16:742, 1955 53. Sokoloski TD, Higuchi T: Kinetics of degradation in solution of epinephrine by molecular oxygen. J Pharm Sci 51: 172, 1962 54. Higuchi T, Schracter LC: Reactivity of bisulfite with a number of pharmaceuticals. J Am Pharm Assoc 48:535, 1959 55. Haddad MJ, Moyer NJ, Riley FC Jr: Mydriatic effect of phenylephrine hydrochloride. Am J Ophthalmol 70:729, 1970 56. deVries H, Beijersbergen van Henegouwen GMJ, Huf FA: Photochemical decomposition of chloramphenicol in a 0.25% eyedrop and in
a therapeutic intraocular concentration. Int J Pharm 20:265, 1984 57. Ball SF, Schneider E: Cost of beta-adrenergic receptor blocking agents for ocular hypertension. Arch Ophthalmol 110:654, 1992 58. Schwartz JS, Christensen RE, Lee DA: Comparison of timolol maleate and levobunolol: Doses and volume per bottle. Arch Ophthalmol 107:17, 1989 59. Gray RH: The influence of drop size on pupil dilation. Eye 5:615, 1991 60. Sieg JW, Robinson JR: Vehicle effects on ocular bioavailability. I. Evaluation of fluorometholone. J Pharm Sci 64: 931, 1975 61. Mindel JS, Goldberg J, Tavitian HO: Similarity of the intraocular pressure response to different corticosteroid
esters when compliance is controlled. Trans Am Acad Ophthalmol 86:99, 1979 62. Howard SA, Mauger JW, Phusanti L: Dissolution profiles for multisized prednisolone acetate suspensions. J Pharm Sci 66:557, 1977 63. Schoenwald RD, Stewart P: Effect of particle size on ophthalmic bioavailability of dexamethasone
suspensions in rabbits. J Pharm Sci 69:391, 1980 64. Zimmer A, Kreuter J: Microspheres and nanoparticles used in ocular delivery systems. Adv Drug Del Rev 16:61, 1995 65. Riegelman S: Pharmacokinetic factors affecting epidermal penetration and percutaneous
absorption. Clin Pharmacol Ther 16:873, 1974 66. Hendrickson RO, Hanna C: Use of drugs in ointment for routine mydriasis. Ann Ophthalmol 9:333, 1977 67. Cable MK, Hendrickson RO, Hanna C: Evaluation of drugs in ointment for mydriasis and cycloplegia. Arch Ophthalmol 96:84, 1978 68. Goldberg I, Ashburn FS Jr, Kass MA et al: Efficacy and patient acceptance of pilocarpine gel. Am J Ophthalmol 88:843, 1979 69. Johnson DH, Epstein DL, Allen RC et al: A one-year multicenter clinical trial of pilocarpine gel. Am J Ophthalmol 97:723, 1984 70. Zignani M, Tabatabay C, Gurny R: Topical semi-solid drug delivery: Kinetics and tolerance of ophthalmic
hydrogels. Adv Drug Del Rev 16:51, 1995 71. Longwell A, Birss S, Keller N et al: Effect of topically applied pilocarpine on tear film pH. J Pharm Sci 65:1654, 1976 72. Urbanyi T, Piedmont A, Willis E et al: Simultaneous determination of pilocarpine and isopilocarpine in pharmaceutical
preparations by liquid chromatography. J Pharm Sci 65:257, 1976 73. Pollack IP, Quigley HA, Harbin TS: The Ocusert pilocarpine system: Advantages and disadvantages. South Med J 69:1296, 1976 74. Hennig J: Pilocarpintropfen-Ocusert-Pilocarpine: Eine vergleicherde Untersuchung. Klin Monatsbl Augenheilkd 169:112, 1976 75. Podos SM, Becker B, Asseff C et al: Pilocarpine therapy with soft contact lenses. Am J Ophthalmol 73:336, 1972 76. Kaufman HE, Uotila MH, Gasset AR et al: The medical uses of soft contact lenses. Trans Am Acad Ophthalmol Otolaryngol 75:361, 1971 77. Krohn DL, Breitfeller JM: Quantitation of pilocarpine delivery across isolated rabbit cornea by noncross-linked
high viscosity polymer gel. Invest Ophthalmol 15:324, 1976 78. McCarey BE, Schmidt FH, Wilkinson KD et al: Gentamicin diffusion across hydrogel bandage lenses and its kinetic distribution
on the eye. Curr Eye Res 3:977, 1984 79. Busin M, Spitznas M: Sustained gentamicin release by presoaked medicated bandage contact lenses. Ophthalmology 95:796, 1988 80. Jain MR: Drug delivery through soft contact lenses. Br J Ophthalmol 72:150, 1988 81. Reidy JJ, Limberg M, Kaufman HE: Delivery of fluorescien to the anterior chamber using the corneal collagen
shield. Ophthalmololgy 97:1201, 1990 82. Schaeffer HE, Krohn DL: Liposomes in topical drug delivery. Invest Ophthalmol Vis Sci 21:220, 1982 83. Stratford RE, Yang DC, Redell MA et al: Ocular distribution of liposome-encapsulated epinephrine and insulin in
the albino rabbit. Curr Eye Res 2:377, 1983 84. Smolin G, Okumoto M, Feiler S et al: Idoxuridine-liposome therapy for herpes simplex keratitis. Am J Ophthalmol 91:220, 1981 85. Lee VHL, Urrea PT, Smith RE et al: Ocular drug bioavailability from topically applied liposomes. Surv Ophthalmol 29:335, 1985 86. Norley SG, Sendele D, Huang L et al: Inhibition of herpes simplex virus replication in the mouse cornea by drug
containing immunoliposomes. Invest Ophthalmol Vis Sci 28: 591, 1987 87. Khoobehl B, Peyman GA, McTurnan WG et al: Externally triggered release of dye and drugs from liposomes into the eve. Ophthalmology 95:950, 1988 88. Smith TJ, Pearson A, Blandford DL et al: Intravitreal sustained-release ganciclovir. Arch Ophthalmol 110:255, 1992 89. Bundgaard H, Johansen M: Pro-drugs as drug delivery systems: Bioreversible derivatization of phenytoin, acetazolamide, chlorzoxazone and various other NH- acidic compounds
by N-aminomethylation to effect enhanced dissolution rates. Int J Pharm 7:129, 1980 90. Bodor N, Shek E, Higuchi T: Improved delivery through biological membranes. I. Synthesis and properties
of 1-methyl-1, 6-dihydropyridine-2-carbaldoxime, a pro-drug of N-methylpyridinium-2-carbaldoxime chloride. J Med Chem 19:102, 1976 91. Bundgaard H, Falch E, Larsen C et al: Pilocarpic acid esters as novel sequentially labile pilocarpine pro-drugs
for improved ocular delivery. J Med Chem 28:979, 1985 92. Greenblatt DJ, Shader RI, Koch-Weser J: Slow absorption of intramuscular chlordiazepoxide. N Engl J Med 291: 116, 1974 93. Hillestad L, Hansen T, Melsom H et al: Diazepam metabolism in normal man. Clin Pharmacol Ther 16:479, 1974 94. MacDonald R Jr, Keller K, Blatt MM et al: Sterility and concentration of pilocarpine solutions. Am J Ophthalmol 68:1099, 1969 95. Mathews A, Nunes A, Brochmann-Hanssen E: Hydrolysis and epimerization kinetics of pilocarpine in aqueous solution. J Pharm Sci 63:716, 1974 96. Kreienbaum MA, Page DP: Stability of pilocarpine hydrochloride and pilocarpine nitrate ophthalmic
solutions submitted by U.S. hospitals. Am J Hosp Pharm 43:109, 1986 97. Becker B, Shaffer RN: Diagnosis and Therapy of the Glaucomas, p 417. 2nd
ed. St. Louis, CV Mosby, 1965 98. Watson JR, Lawrence RC: Selective GLC determination of epinephrine, isoproterenol, and phenylephrine
in pharmaceutical dosage form. J Pharm Sci 66:560, 1977 99. Frank J, Chafetz L: IR spectrophotometric assay of carbachol solutions. J Pharm Sci 66:439, 1976 100. Kay ML, Yanoff M, Katowitz JA: Development of sympathetic uveitis in spite of corticosteroid therapy. Am J Ophthalmol 75:90, 1974 101. Campagna FA, Cureton G, Mirigan RA et al: Inactive prednisone tablets U.S.P. XVI. J Pharm Sci 52:605, 1963 102. Levy G, Hall NA, Nelson E: Studies on inactive prednisone tablets U.S.P. XVI. Am J Hosp Pharm 21:402, 1964 103. Healey LA: Generic equivalents. Arthritis Rheum 16:775, 1973 104. Mindel JS: Bioavailability and generic prescribing. Surv Ophthalmol 21:262, 1976 |